

Abstract
Prostate cancer is the second leading cause of cancer-related deaths in men in the United States. Our previous studies have shown that ligands for the nuclear type II [3H]estradiol binding site such as luteolin significantly inhibit prostate cancer cells in vitro and in vivo; however, the role of these ligands in cell growth and proliferation is poorly understood. In order to further elucidate the molecular mechanism through which luteolin exerts its effects on PC-3 cells, cRNA microarray analyses was performed on 38,500 genes to determine the genes altered by luteolin treatment. The expression of 3,331 genes was changed greater than 1.2-fold after luteolin treatment. Analysis of the altered genes identified two pathways that were significantly affected by luteolin. The Cell Cycle Pathway contained 22 down-regulated genes (including polo-like kinase 1, cyclin A2, cyclin E2 and proliferation cell nuclear antigen) and one up-regulated gene (cyclin-dependent kinase inhibitor 1B). In addition, 13 genes were down-regulated by luteolin in the RNA Transcription Pathway. Real-time polymerase chain reactions and western blots verified the observations from the microarray. In addition, two synthetic, chemically distinct type II ligands, ZN-2 and BMHPC, mimicked the effects of luteolin on gene expression at the mRNA and protein level in PC-3 cells. Finally, chromatin immunoprecipitation assays indicated that luteolin exerts its effects on genes by altering the acetylation state of promoter-associated histones. Taken together, the data suggest that type II ligands inhibit cell growth and proliferation through epigenetic control of key genes involved in cell cycle progression and RNA transcription.
Keywords: Bioflavonoid, Nuclear Type II [3H]Estradiol Binding Site, Cell Cycle, RNA Transcription
Introduction
Two classes of [3H]estradiol bindings sites have been described in the rat uterus, designated as types I and II [1, 2]. Type I sites are the classical estrogen receptors (ERα or ERβ), which bind [3H]estradiol and triphenylethylene antiestrogens [3, 4] with a high affinity (Kd < 1.0 nM) and a finite binding capacity (~20,000 sites per cell). ER’s are target tissue specific transcription factors [5]. Conversely, type II sites bind [3H]estradiol cooperatively with a relatively low affinity (Kd ~ 20 nM) and demonstrate different ligand binding specificities from ER [6]. The nuclear type II sites remain at a basal level (~2000 sites per cell) in non-estrogenized cells. Following estradiol administration, their level increases (>100,000 sites per cell nucleus) [2, 7] in correspondence with estradiol stimulation of rat uterine cellular hypertrophy and hyperplasia [2, 6, 8]. This induction of type II sites can be blocked estrogen antagonists including dexamethasone and progesterone [9]. Therefore, nuclear type II site stimulation is a key event in estrogenic control of proliferative pathways. This mechanism has been documented in the uterus and other tissues including rat [10-14] and human prostate [14] and human breast cancer [15-17].
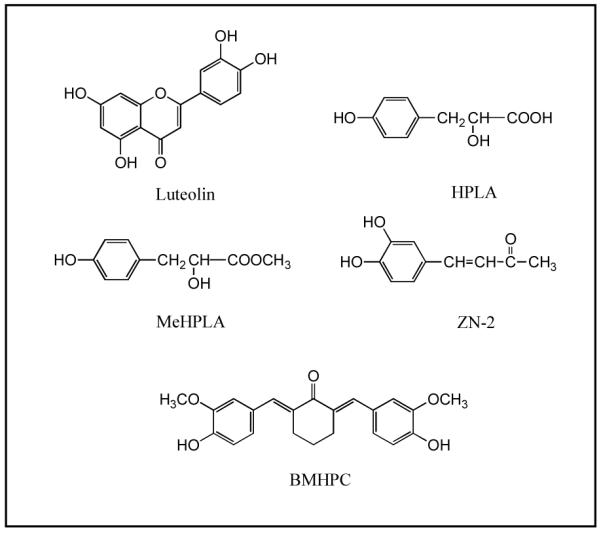
Although type II sites were initially identified on the basis of their [3H]estradiol binding characteristics, methyl p-hydroxyphenyllactate (MeHPLA, Fig 1) was identified as an endogenous ligand that binds the type II site with high affinity [18, 19]. Occupancy of type II sites by MeHPLA suppresses cellular proliferation in uterus, mammary gland or prostate [14, 15, 20, 21]. This suppression can be reversed by estradiol stimulation, which causes a specific dissociation of MeHPLA from the type II site [19, 20, 22]. In addition, estradiol stimulates an esterase, which hydrolyzes MeHPLA to the corresponding free acid (HPLA; p-hydroxyphenyllactic acid) in normal and malignant tissues [15, 21, 23]. The hydrolyzed ligand binds nuclear type II sites with very low affinity and is unable to inhibit cellular proliferation [19]. Malignant tissues are notably deficient in MeHPLA, which leads to a elevated level of unoccupied nuclear type II sites and loss of regulatory control [15, 16, 18, 19, 22]. Esterase-stable MeHPLA analogs and related compounds (Figure 1), such as 4-(3,4-dihydroxyphenyl)but-3-en-2-one (ZN-2) and 2,6-bis((3-methoxy-4-hydroxyphenyl)methylene)cyclohexanone (BMHPC), that bind to nuclear type II sites with high affinity also inhibit the proliferation of breast [19, 21, 24], pancreatic [25], prostatic [14], colorectal [26], ovarian cancer cells [27], lymphoblastoid cells [28], and leukemia [29] in vitro and in vivo. Therefore, nuclear type II sites represent a key control mechanism in mammalian tissues that respond to steroid hormones, bioflavonoids, and specific bioflavonoid or tyrosine metabolites such as MeHPLA. Protein purification studies led to the identification of the type II binding site as histone H4 [30-32]. The studies in this manuscript demonstrate that type II binding site ligands including luteolin, ZN-2 and BMHPC, control genes involved in cellular growth and division through what appears to be an epigenetic mechanism.
Figure 1. Type II [3H]Estradiol Binding Site Ligands.
Materials and Methods
PC-3 Cell Growth Conditions
PC-3 human prostate cancer cells were grown in T-150 flasks as previously described [14]. For microarray analyses, the cells were seeded into T-75 flasks containing 10 mls of DMEM-F12 media containing 10% fetal calf serum (FCS) and 1% penicillin-streptomycin and allowed to attach for 24 hours. At this time, the media was changed and the cells grown for 6 hours in growth media containing 2 μl of EtOH (controls) or luteolin in 2 μl of EtOH (5 μg/ml final concentration, the IC50 of luteolin for these cell culture conditions). The 6 hour time point was chosen as earlier PCR experiments showed regulation of cyclin D1 by luteolin (not shown). After incubation, 5 million cells for each treatment group were harvested for RNA isolation. In some experiments cells were treated with ZN-2 or BMHPC and collected at different times as indicated in the text and figure legends. Viable, attached cell numbers were monitored by hemocytometer counts based on trypan blue dye exclusion [24].
Whole Cell [3H]Estradiol Binding Assay
Type II sites in PC-3 cell were measured by whole cell binding assays as previously described [15, 33]. PC-3 cells were seeded in 24-well plates (~5000 cells/well) in the growth media described above and allowed to attach for 72 hours. The media was replaced with 2 ml of serum-free medium. The cells were incubated (37°C × 60 min) in the presence of 40 nM [3H]estradiol plus or minus 0.004–40 μM competitor (luteolin or ZN-2). Following incubation, the cells were cooled on ice and washed with Hank’s balanced salt solution to remove free ligand. Bound ligand was extracted with 2 ml of EtOH and quantified by liquid scintillation. Specific binding was determined by subtracting total cpm from non-specific cpm.
RNA Preparation
Cells from flasks of EtOH controls or treated cells were washed with PBS and collected with 0.25% trypsin-0.02%EDTA (4 mls). Following 5-minute incubation, the trypsin was inactivated by the addition of 10 mls of media containing 10% FCS. Approximately 5.0 × 106 cells from each flask were centrifuged (2000 rpm × 5 minutes) in RNAse/DNAse free tubes, resuspended in 1 ml of PBS and 4 mls of RNAlater (Qiagen) and stored at −20°C. The frozen cells were thawed on ice, collected by centrifugation and lysed by resuspension in 0.6 mls of RTL (Qiagen) containing β-mercaptoethanol. The lysed cells from various treatment groups were homogenized by centrifugation through Qiashredders (18,000 × g × 2 minutes). Pass through from the Qiashredders was diluted with an equal amount of 70% EtOH and loaded onto RNeasy spin columns. The column was washed with RW1 followed by RNAse-free DNAse digestion to remove residual DNA and further washed with RPE buffers according to the manufacturer’s instructions. Purified total RNA was eluted with 50 μl of RNAse-free water following 5 minute RT incubation. RNA integrity was verified on an Agilent 2100 Bioanalyzer by the Baylor College of Medicine Microarray Core.
Microarray Analyses
RNA from EtOH controls or luteolin treated PC-3 cells was subjected to oligo(deoxythymidine)-Reverse Transcription, in vitro transcription and biotin-labeling of cRNA (Enzo Biochem, Farmingdale, NY). The fragmented, biotin-labeled cRNA was hybridized to Human Genome U133 Plus 2.0 oligonucleotide arrays (Affymetrix, Santa Clara, CA) containing approximately 54,000 probe sets (38,500 genes). Each transcript is represented on the chip as 11 probe pairs and each pair contained a perfect match and mismatch. Microarrays were stained with strepavidin antibody and streptavidin-phycoerythrin in an Affymetrix Fluidics station. Arrays were scanned at 3 μM with a GeneArray scanner (Affymetrix). All experiments were performed in triplicate with independent pools of cRNA from EtOH controls and luteolin-treated PC-3 cells using six separate microarray chips. Following low-level quantification of the scanned data using GeneChip Operating System (GCOS, Affymetrix), data were further analyzed with dChip 2006 (Harvard) to adjust the arrays to a common baseline and to estimate expression using the PM-only model [34, 35]. All 6 GeneChips were normalized to the same baseline (in this case, the GeneChip with the median average intensity was luteolin Chip #1) and all were modeled together. Data quality was reviewed using present call rates from GCOS (average 38.25%, range 36.7% to 40.3%), ratios of 3′ to 5′ glyceraldehydes 3-phosphate dehydrogenase probe sets from GCOS (average 0.98: range 0.95 to 0.99) and array outlier rates from dChip (average 0.0505: range 0.006 to 0.145). Differentially expressed genes in the luteolin treated groups relative to EtOH controls were selected using a two-sample comparison with a lower boundary 90% confidence interval of fold change greater than 1.2 and a value difference between group means of >50. The medium number of detected genes in 50 permuted samples was used to estimate the false discovery rate. GenMAPP (Gene Map Annotator and Pathway Profiler, Version 2.1, Gladstone Institutes, University of California at San Francisco) identified differentially expressed gene pathways according to Gene Ontology function
Real-Time Quantitative Polymerase Chain Reaction (qPCR)
Pre-validated commercially available primers (Qiagen) for the Cell Cycle and Transcription Factor Signaling Pathway genes were used. qPCR was performed using the MyiQ SYBR Green Supermix (Bio-Rad) and quantified on MyiQ Single Color Real-Time PCR Detection System using MyiQ Optical System Software, version 2.0 (Bio-Rad). Validation of each primer pair was accomplished by generating standard serial dilution and melt curves on cDNA from PC-3 cells to ensure that reaction efficiencies of 90–110% and correlation coefficients of >0.995 are obtained. Melt curves demonstrating a single product with an appropriate melting temperature confirmed that primer dimerization was not problematic. Results from QPCR on triplicate pools of RNA from EtOH controls or luteolin, BMHPC or ZN-2 treated cells were normalized to 18S RNA and analyzed statistically by Instat (Graphpad Software, Inc) with a suitable t-test on the treatment means. Products of the optimized reactions were analyzed by agarose gel electrophoresis to ensure that the size of the amplicon corresponds to the data provided by Qiagen for each primer pair.
SDS PAGE Analyses
Cells were grown as described above in triplicate and treated with ligand for 24 hours. The cells lysed with Qiagen RTL buffer and pass through Qiashredders. Proteins from the extract were collected by precipitation with 4 volumes of ice cold acetone and centrifugation. The pellets were dried under nitrogen, re-dissolved in sample extraction buffer (0.05M Tris, pH 7.4, 8M urea, 1% SDS, 0.1% β-mercaptoethanol) containing 0.02% cetyltrimethylammonium bromide (CTAB; Aldrich, Milwaukee, WI) and diluted 2:1 with sample loading buffer (0.0625M Tris, pH 6.8, 20% glycerol + bromophenol blue). The solubilized samples were centrifuged and heated for 3 min at 90°C. The cooled samples were resolved on 4–12% NuPAGE Bis-Tris gradient gels using MES running buffer (Invitrogen, Carlsbad, CA).
Western Blot Analyses
The electrophoresed proteins were transferred to nitrocellulose membrane (Trans-Blot Transfer Medium, Bio-Rad, Hercules, CA), washed, and blocked in Tris-buffered saline (1X TBS) with 7.3% non-fat dried milk. Rinsed membranes were incubated 1–16 hours with primary antibody (1:100 to 1:1000 dilution) as described below. Primary antibodies suitable for Western blots for human cell cycle genes were purchased from Santa Cruz Biotechnologies (CDKN1B, PCNA, PLK1, β-actin) or Abcam (CCNA2, CCNE2). Following a 60 minute incubation with the appropriate species-specific horse-radish peroxidase-conjugated secondary antibody (Upstate), specific antigens were detected by the Visualizer Western Blot Detection Kit (Upstate) on X-OMAT Scientific Imaging Film (Eastman Kodak Company Rochester, New York). All blots were quantified with UNSCAN-IT Gel Software (Silk Scientific Software). In order for the protein bands to be normalized with β-actin, the nitrocellulose membrane was stripped with Stripping Buffer II (0.4M glycine, 0.2% SDS, Tween-20, pH 2.2) according to the manufacturer’s instructions prior to re-probing with β-actin antibody.
Chromatin Immunoprecipitation Assay
Following treatment of the PC-3 cells as described above, the monolayers were fixed with formaldehyde to crosslink protein to DNA. The chromatin was then digested (10–25′) with Enzymatic Shearing Cocktail (ChIP-IT Kit, Active Motif) to generate DNA fragments averaging 200 bp in length. Aliquots of the digested chromatin were pre-cleared with Protein A-Sepharose and incubated with (+Ab) or without (−Ab) anti-acetylated (Lys 5, 8, 12, 16) histone H4 antibody. Following incubation with Protein A-Sepharose beads and cross-link reversal, the immunoprecipitated DNA was de-proteinized and analyzed by PCR and qPCR. Primers for the promoter regions were as follows: PLK1: forward: 5′-GGTTTGGTTTCCCAGGCTAT-3′, reverse: 5′-GCTGGGAACGTTACAAAAGG-3′ and RB1: forward: 5′-CACAGCTCCCTCCCTTCCTTTC-3′, and reverse CTACCCAGAACCACCCCCTCCA-3′. PCR products from the Input DNA and DEPC water were additional controls
Results
Type II Site Ligands Regulation of PC-3 Prostate Cancer Cells
In an attempt to define genes regulated in human prostate cancer cells by type II sites, the effects of ligands including luteolin, BMHPC and a recently synthesized novel type II ligand, ZN-2, on gene expression in PC-3 cells were examined. Previous studies with luteolin [33], and BMHPC [14] showed a direct correlation between the occupancy of type II sites by these compounds and the inhibition of malignant cell proliferation. Both ligands showed similar binding affinities for type II sites and cell inhibition dose response curves. We also evaluated the type II site binding and cell inhibition properties of ZN-2. Whole cell binding assays demonstrated that luteolin and ZN-2 bind to type II sites with approximately equivalent affinities (Fig 2A) even though ZN-2 inhibited PC-3 cell proliferation at 10-fold lower concentrations than luteolin (Fig 2B). This observation likely reflects different solubility and cell uptake properties of luteolin and ZN-2. Nevertheless, the three type II site ligands were suitable for studies to evaluate changes in gene expression that correlate with type II site occupancy and the inhibition of cellular proliferation.
Figure 2. Luteolin and ZN-2 Competition for Type II Sites and PC-3 Cell Inhibition.
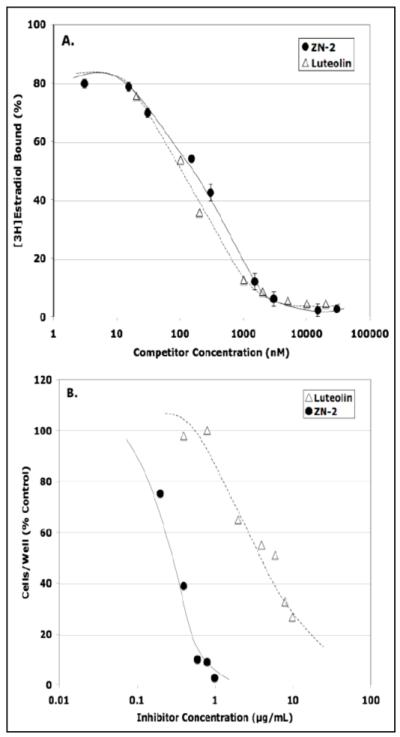
Panel A. PC-3 cells were incubated (4°C × 60 min) with 40 nM [3H]estradiol plus or minus the indicated concentrations (0.004 to 40 μM) ZN-2 (black circles) or luteolin (open triangles). Specifically bound [3H]estradiol (~30,000 cpm) was determined by subtracting nonspecific cpm from the total amount of [3H]estradiol bound in the absence of competitor. Panel B. PC-3 cells were plated and allowed to attach for 48 hours. At this time (Day 0), the media was changed and the cells treated with the indicated concentrations of luteolin or ZN-2. Cell number was determined by trypan blue exclusion on Day 6.
Microarray Hybridization Analysis of Luteolin Effects on Gene Expression in PC-3 Prostate Cancer Cells
Genes regulated by type II ligands were identified by comparing the mRNA expression pattern of PC-3 human prostate cancer cells treated with EtOH or luteolin. Microarray analysis utilizing the HG_U133 Plus 2.0 chips was performed as described in Materials and Methods. Of the 38,500 genes represented on the array, 37–40% of the genes were expressed in the PC-3 cells. Genes expressed differentially between EtOH controls and luteolin treated cells were identified in dChip using a lower boundary of a 90% confidence interval, a fold change greater than 1.2, an absolute difference between group means greater than 50 and a p-value less than 0.05 (Fig 3, Supplementary Table 1). Further analysis of the differentially expressed genes with GenMAPP identified 32 cell regulatory pathways (Supplementary Table 2) significantly modulated by luteolin treatment. The cell cycle (p<0.001) and RNA transcription (p<0.002) pathways were most significantly affected by luteolin treatment. Twenty-three genes in the Cell Cycle Pathway (Fig 4, Table 1) were up-regulated (pink) or down-regulated (blue) in PC-3 cells following luteolin treatment. Only cyclin-dependent kinase inhibitor 1B (p27; CDKN1B) was up-regulated. A high number of down-regulated as compared to up-regulated genes is probably due to luteolin being an inhibitor of cell growth up not a promoter of apoptosis. Several other key genes in the pathway including polo-like kinase 1 (PLK1), cyclin A2 (CCNA2), cell division cycle 25A (CDC25A), cyclin E2 (CCNE2) and proliferating cell nuclear antigen (PCNA) were down-regulated by luteolin treatment relative to the EtOH controls.
Figure 3. Number of significantly increased and decreased PC-3 cell genes 6 hours following luteolin treatment.
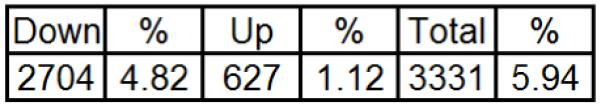
Figure 4. GenMAPP Diagram of Cell Cycle Pathway Genes Significantly Increased or Decreased by Luteolin Treatment.
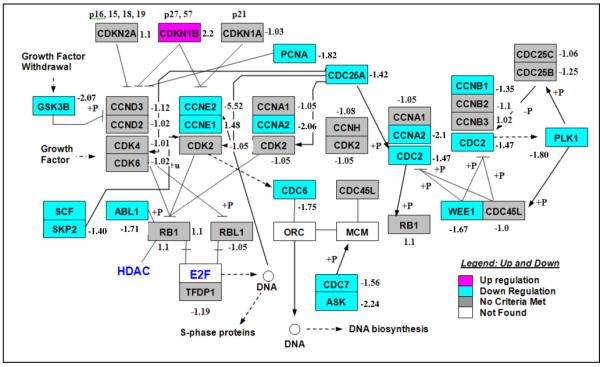
The results represent mean fold-change in gene expression in response to luteolin treatment relative to EtOH control. D-Chip output was analyzed by GenMAPP to generate the diagram. Significantly (p<0.05) up-regulated genes are in pink and down-regulated genes are in blue. Genes shown in gray were not significantly affected. The fold-change relative to EtOH control is indicated next to each gene in the GenMAPP diagram.
Table 1. Genes in the GenMAPP Cell Cycle pathway significantly increased or decreased in 6 hours following treatment of PC-3 cells with luteolin.
| AffyID | Gene Title | Gene Symbol |
Entrez Gene |
Fold Change |
Adjusted P Value |
|---|---|---|---|---|---|
| 202123_s_at | v-abl Abelson murine leukemia viral oncogene homolog 1 | ABL1 | 25 | −1.71 | 0.0005804 |
| 204244_s_at | activator of S phase kinase | ASK | 10926 | −2.24 | 0.0000095 |
| 203755_at | BUB1 budding uninhibited by benzimidazoles 1 homolog beta (yeast) | BUB1B | 701 | −1.64 | 0.0004463 |
| 201456_s_at | BUB3 budding uninhibited by benzimidazoles 3 homolog (yeast) | BUB3 | 9184 | −1.38 | 0.0031299 |
| 213226_at | Cyclin A2 | CCNA2 | 890 | −2.06 | 0.0000037 |
| 213523_at | cyclin E1 | CCNE1 | 898 | −1.48 | 0.0010471 |
| 205034_at | cyclin E2 | CCNE2 | 9134 | −5.52 | 0.0000013 |
| 203213_at | Cell division cycle 2, G1 to S and G2 to M | CDC2 | 983 | −1.47 | 0.0012987 |
| 204696_s_at | cell division cycle 25A | CDC25A | 993 | −1.42 | 0.0000672 |
| 203967_at | CDC6 cell division cycle 6 homolog (S. cerevisiae) | CDC6 | 990 | −1.75 | 0.0000692 |
| 209112_at | cyclin-dependent kinase inhibitor 1B (p27, Kip1) | CDKN1B | 1027 | 2.20 | 0.0000107 |
| 205394_at | CHK1 checkpoint homolog (S. pombe) | CHEK1 | 1111 | −1.69 | 0.0001134 |
| 204817_at | extra spindle poles like 1 (S. cerevisiae) | ESPL1 | 9700 | −1.43 | 0.0008915 |
| 226191_at | Glycogen synthase kinase 3 beta | GSK3B | 2932 | −2.07 | 0.0000330 |
| 222037_at | MCM4 minichromosome maintenance deficient 4 (S. cerevisiae) | MCM4 | 4173 | −1.62 | 0.0006883 |
| 205085_at | origin recognition complex, subunit 1-like (yeast) | ORC1L | 4998 | −1.50 | 0.0000122 |
| 204853_at | origin recognition complex, subunit 2-like (yeast) | ORC2L | 4999 | −1.53 | 0.0001676 |
| 211212_s_at | origin recognition complex, subunit 5-like (yeast) | ORC5L | 5001 | −2.01 | 0.0000576 |
| 201202_at | proliferating cell nuclear antigen | PCNA | 5111 | −1.82 | 0.0008122 |
| 202240_at | polo-like kinase 1 (Drosophila) | PLK1 | 5347 | −1.80 | 0.0000329 |
| 210567_s_at | S-phase kinase-associated protein 2 (p45) | SKP2 | 6502 | −1.40 | 0.0026516 |
| 212533_at | WEE1 homolog (S. pombe) | WEE1 | 7465 | −1.67 | 0.0017875 |
| 222985_at | tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, | YWHAG | 7532 | −1.41 | 0.0023041 |
Luteolin treatment also down-regulated a number of genes involved in RNA transcription (Fig 5 and Table 2) including general transcription factor 2B (GTF2B), general transcription factor 2H1 (GTF2H1), general transcription factor 2H2 (GTF2H2), RNA polymerase I polypeptide A (POLR1A), RNA polymerase II, polypeptide C (POLR2C), RNA polymerase I polypeptide D (POLR1D), RNA polymerase III polypeptide K (POLR3K) and TATA box binding protein (TBP)-associated factors 5, 7 and 9 (TAF5, TAF7, TAF9). A number of the luteolin regulated genes in both pathways were selected for further analysis by qPCR.
Figure 5. GenMAPP Diagram of RNA Transcription Pathway Genes Significantly Increased or Decreased by Luteolin Treatment.
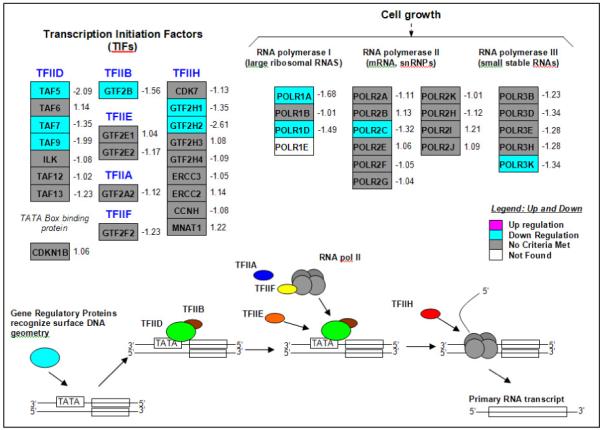
The results represent mean fold-change in gene expression in response to luteolin treatment relative to EtOH control. D-Chip output was analyzed by GenMAPP to generate the diagram. Significantly (p<0.05) up-regulated genes are in pink and down-regulated genes are in blue. Genes shown in gray were not significantly affected. The fold-change relative to EtOH control is indicated next to each gene in the GenMAPP diagram.
Table 2. Genes in the GenMAPP RNA transcription pathway significantly increased or decreased in 6 hours following treatment of PC-3 cells with luteolin.
| AffyID | Gene Title | Gene Symbol |
Entrez Gene |
Fold Change |
Adjusted P Value |
|---|---|---|---|---|---|
| 208066_s_at | general transcription factor IIB | GTF2B | 2959 | −1.56 | 0.000023 |
| 202453_s_at | general transcription factor IIH, polypeptide 1, 62kDa | GTF2H1 | 2965 | −1.35 | 0.000042 |
| 223758_s_at | general transcription factor IIH, polypeptide 2, 44kDa | GTF2H2 | 2966 | −2.61 | 0.000151 |
| 206038_s_at | nuclear receptor subfamily 2, group C, member 2 | NR2C2 | 7182 | −1.57 | 0.000313 |
| 209119_x_at | nuclear receptor subfamily 2, group F, member 2 | NR2F2 | 7026 | −1.62 | 0.000438 |
| 222704_at | polymerase (RNA) I polypeptide A, 194kDa | POLR1A | 25885 | −1.68 | 0.000863 |
| 218258_at | polymerase (RNA) I polypeptide D, 16kDa | POLR1D | 51082 | −1.49 | 0.001944 |
| 208996_s_at | polymerase (RNA) II (DNA directed) polypeptide C, 33kDa | POLR2C | 5432 | −1.32 | 0.001959 |
| 218866_s_at | polymerase (RNA) III (DNA directed) polypeptide K, 12.3 kDa | POLR3K | 51728 | −1.34 | 0.002802 |
| 210053_at | TAF5 RNA polymerase II, TATA box binding protein (TBP)-associated factor, 100kDa | TAF5 | 6877 | −2.70 | 0.003161 |
| 201023_at | TAF7 RNA polymerase II, TATA box binding protein (TBP)-associated factor, 55kDa | TAF7 | 6879 | −1.35 | 0.004727 |
| 203893_at | TAF9 RNA polymerase II, TATA box binding protein (TBP)-associated factor, 32kDa | TAF9 | 6880 | −1.99 | 0.012936 |
| 204254_s_at | vitamin D (1,25- dihydroxyvitamin D3) receptor | VDR | 7421 | −2.44 | 0.014324 |
Evaluation of Luteolin Effects on RNA Transcription and Cell Cycle Genes in PC-3 Cells by qPCR
To further confirm the microarray data (Figs 4 and 5) presented above, PC-3 cells were treated with luteolin or EtOH (controls) for 6 hours as described for the microarray studies. qPCR experiments on cDNA made from the total RNA from these cells were performed. Studies of selected cell cycle genes (Fig 6A) show that luteolin treatment stimulated CDKN1B gene expression, as indicated by the microarray analysis (Table 1) and inhibited the expression of PLK1, CCNA2, CCNE2 and PCNA. Results from the qPCR analyses on selected genes in the RNA transcription pathway are shown in Fig 6B. The data confirmed the microarray studies showing that luteolin treatment significantly reduced the expression of a number of genes involved in RNA transcription including POLR1A, POLR1D, POLR3K, TAF7, GTF2B, GTF2H1 and GTF2H2. It is likely that CDC25A and POLR2C gene expression was also suppressed, but not quite enough to achieve significance at this time (6 hours) after luteolin treatment. These data leave little doubt that luteolin has very significant effects on genes involved in RNA transcription. Thus, luteolin treatment of PC-3 cells directly affected the transcription of a number of transcription factors and cell cycle genes likely directly involved in the inhibition of cell proliferation by luteolin and related type II site ligands. Furthermore, qPCR analysis of RNA from PC-3 cells treated with BMHPC (Fig 7A) or ZN-2 (Fig 7B) showed nearly identical patterns of cell cycle gene expression as those seen with luteolin. These studies validated the microarray analyses and demonstrate that three chemically distinct type II site ligands exhibit remarkably similar effects on gene transcription in these molecular pathways controlling gene transcription and cellular proliferation.
Figure 6. qPCR Analysis of Alteration of Cell Cycle or RNA Transcription Gene Expression by Luteolin.
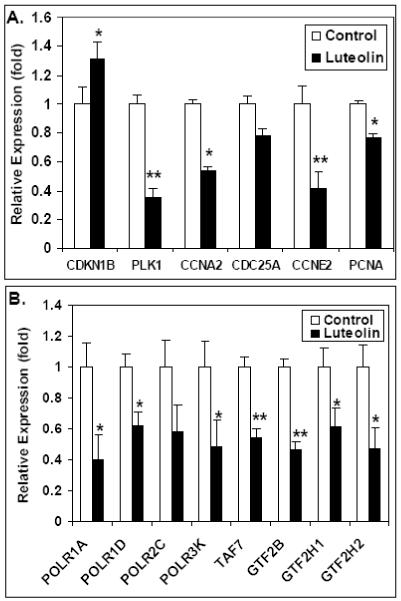
PC-3 cells were treated for 6 Hours with 2 μl EtOH (controls) or luteolin in 2 μl EtOH (final concentration of 5 μg/ml). RNA was extracted, cDNA prepared with iScript reverse transcriptase and analyzed by qPCR as described in Methods (Panel A, Cell Cycle genes; Panel B, RNA Transcription genes). The results represent the mean ± SEM for three independent RNA sets normalized to 18S RNA. *P<0.05, **p<0.001
Figure 7. qPCR Analysis of Alteration of Cell Cycle Gene Expression by BMHPC or ZN-2.
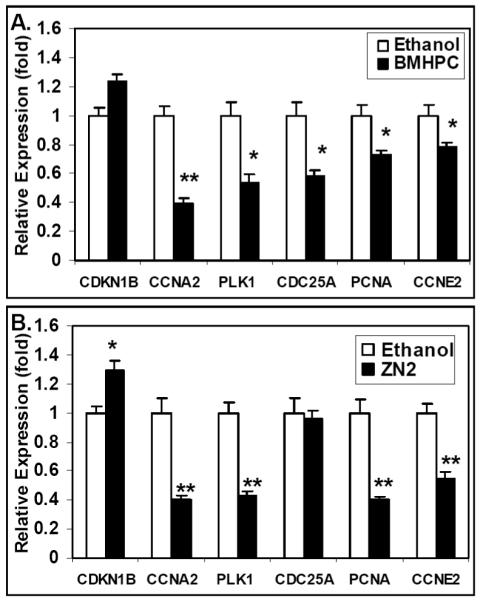
PC-3 cells were treated for 6 hours with 2μl EtOH (controls) or BMHPC (Panel A) (final concentration of 5 μg/ml) or ZN-2 (Panel B) (final concentration of 1 μg/ml) in 2 μl EtOH RNA was extracted, cDNA prepared with iScript reverse transcriptase and analyzed by qPCR as described in Methods. The results represent the mean ± SEM for three independent RNA sets normalized to 18S RNA. *P<0.05, **p<0.001
Western Blot Analysis of PC-3 Cells Treated with Type II Site Ligands
In order to further confirm the affects of luteolin, ZN-2 and BMHPC on gene expression in PC-3 cells, a series of western blots was performed. PC-3 cells were treated with ligand (triplicate samples) for 24 hours prior to harvest. The cells were lysed and protein extracted from the lysate. The protein was subjected to western blot analysis with antibodies against PLK1, CCNE2, CCNA2, PCNA and CDKN1B (Fig 8A) with β-actin as a control. The immunoblot bands were scanned with Un-Scan-It gel to determine the individual intensity of each band. As expected, PLK1, CCNE2, CCNA2 and PCNA show diminished protein levels while the CDKN1B protein expression was increased by treatment with any of the three ligands. The data (Fig 8B) clearly demonstrate that the effects of luteolin, ZN-2 and BMHPC on gene expression translate into corresponding changes in the level of protein translation.
Figure 8. Western Blot Analyses of Cell Cycle Protein Expression Altered by Type II Ligands.

Panel A. Protein extracted from PC-3 cells treated for 24 hours with luteolin, Zn-2 or BMHPC were subjected to western blot analysis with EtOH-treated cells as a control. Panel B. Bands were analyzed by UN-SCAN-IT gel to determine band intensity, normalized to β-actin and graphed according to the change in fold expression from the EtOH control. **P<0.01
Luteolin Modification of Histone H4 Acetylation at the PLK1 Promoter
As the type II site was identified as histone H4, we developed methodology for analyzing the acetylation state of histone H4 associated with the PLK1 promoter. This study is particularly important in view of the possibility that type II site ligands may regulate gene expression via an epigenetic mechanism involving interactions with, and potential modification of the acetylation, methylation or phosphorylation of the histone H4 (type II site). PC-3 cells treated with EtOH (control) or luteolin as described above were harvested and chromatin prepared as described in Methods. The enzymatically digested chromatin consisting of 200 base pair fragments (Fig 9A) was immunoprecipitated with anti-acetyl histone H4 antibodies and analyzed by PCR with primers against the PLK1 promoter (Fig 9B). Luteolin treatment caused a significant reduction in the intensity of the PCR signal. Quantitation of the signal with Un-Scan-It gel and normalization of the signal to the Input DNA revealed a 50% reduction in signal of luteolin treated cells compared to control (Fig 9C). This was subsequently confirmed with qPCR (Fig 9D). In contrast, a gene (RB1) that was not significantly regulated by luteolin in the microarray studies (See Fig 4) showed no change in the acetylation state of the histone H4 associated with the promoter region of RB1 either by PCR (Fig 9B and 9C) or qPCR (Fig 9D). These findings suggest that luteolin inhibition of PLK1 gene expression occurred as a result from an alteration in the binding of histone acetyltransferases and/or histone deacetylases.
Figure 9. Chromatin Immunoprecipitation Assay of the PLK1 and RB1 Promoters by Anti-Histone H4 Antibodies.

(Panel A). The gel contains a 100 bp molecular weight ladder (MWL), non-digested chromatin, sheared DNA from 10′, 15′, 20′ and 25′ digests, a DEPC water control and molecular weight markers (MWM). The 20′ digests yielded 100–200 bp DNA fragments suitable for ChIP assays. In panel B (ChIP Assay), DNA cross-linked chromatin from EtOH controls or luteolin treated PC-3 cells pre-incubated with (+Ab) or without (−Ab) anti-acetylated (Lys 5, 8, 12, 16) histone H4 antibodies (Upstate). Following incubation with Protein A-Sepharose beads, and cross-link reversal, immunoprecipitated DNA was de-proteinized and subjected to PCR analysis using primers against the PLK1 and RB1 promoters. PCR products from the input DNA (1:10 dilution) are shown as additional controls. The results from 3 PCR reactions (as in Panel B) for EtOH controls and luteolin treated PC-3 cells were quantified with Un-Scan-IT (Silk Scientific Software) and analyzed with Instat (Panel C). In Panel D, the PLK1 and RB1 promoters present in triplicates of samples such as those shown in Panel C was also quantified by qPCR. Data in the EtOH controls and luteolin treated samples were normalized to the Input DNA for the +Ab samples. No signal was detected in the −Ab samples, and thus, the data were not plotted. The data in panels B-D show that treatment of PC-3 cells with luteolin for 6 hours significantly reduced the acetylation state of the histones associated with the PLK1 promoter, but not the RB1 promoter.
Discussion
Benign prostate hyperplasia (BPH) is a major cause of surgery for men over the age of 60 [36, 37] and prostate cancer is the second leading cause of cancer-related deaths for American males [38]. BPH and prostate cancer are androgen-dependent diseases typically treated with antiandrogens (Flutamide, cyproterone acetate) to block androgen binding, 5-α reductase inhibitors (4-hydroxyandrostenedione, Finasteride) to block the conversion of testosterone to dihydrotesterone or gonadotropin-releasing hormone agonists, which inhibit androgen synthesis [39, 40]. These androgen-modulatory drugs reduce prostate size, but have numerous side affects including gynecomastia, erectile dysfunction, impaired libido, bone pain and hot flashes [41-43]. Similarly, the prostate is also an estrogen target tissue, containing estrogen receptors [44]. Therefore, antiestrogens (tamoxifen; an ER antagonist) and aromatase inhibitors (Atamestane) are used to treat BPH and prostate cancer [36, 45] as well. The former possess intrinsic estrogenic activity [36, 46] and the latter can increase androgen levels, which can stimulate the prostate. Consequently, new therapeutic agents for treatment of BPH and prostate cancer are desirable.
It has been known for decades that the consumption of legumes, fruits and vegetables is associated with a reduced risk of prostate, colon and breast cancer in humans [47]. Japanese men have an 8-fold lower incidence of prostate cancer compared to American men [48]. Although this protective effect has characteristically been ascribed to phytoestrogens (isoflavonoids) in the diet, [49], it is well known that sustained exposure to impeded (low affinity) estrogens can hyperestrogenize target tissues and promote mammary cancer [50]. Thus, the precise role of phytoestrogens in BPH and prostate cancer remains unresolved. Alternatively, dietarily-derived flavonoids (luteolin and quercetin) are antiestrogenic in vitro and in vivo [20]. These compounds bind to type II sites (histone H4) with high affinity, but not to ERα or ERβ [24] in ER positive and ER negative cell lines and they lack estrogenic activity [15, 19, 20]. The endogenous ligand (MeHPLA) for type II site is also a bioflavonoid metabolite, and thus it is not surprising that bioflavonoids including luteolin and quercetin bind to type II sites with high affinity, block estradiol stimulation of uterine growth, inhibit MCF-7 cancer cell proliferation in vitro and the growth of mammary tumors in mice [20]. These concepts have been confirmed by the published evidence that type II sites are ubiquitous and have a demonstrated role in the regulation of malignant cell proliferation in breast [17], colorectal [26], pancreatic [25], ovarian [27] and bladder [51] cancer. Likewise, luteolin and BMHPC reduce the weight of the normal prostate in male mice [14, 33] and inhibit human prostatic cancer cell proliferation in vitro and in vivo [14, 33] and this is likely to be the case for ZN-2 as well. These properties are consistent with the regulation of cell cycle and RNA transcription factor genes by luteolin, BMHPC and ZN-2 in PC-3 cancer cells presented here.
In this manuscript, we have identified several gene pathways through which luteolin, ZN-2 and BMHPC regulate PC-3 prostate cancer cell proliferation. The three compounds exhibit a profound inhibition of PC-3 cellular proliferation (Fig 2B and [14, 33]). Microarray analysis of luteolin treated PC-3 cells showed significant regulation of genes in the Cell Cycle (Fig 4, Table 1) and RNA Transcription (Fig 5, Table 2) Pathways. These observations were confirmed by qPCR (Figs 6A and 6B). Regulation of the cell cycle genes was also seen in cells treated with ZN-2 or BMHPC (Fig 7) and by western blot analysis (Fig 8). The genes regulated in these pathways by the type II site ligands were consistent with the inhibitory effects of these compounds on cell proliferation. The luteolin-stimulated gene, CDKN1B, is a cyclin dependent kinase inhibitor that arrests cell cycle progression [52, 53]. Thus, the induction of this gene by luteolin is consistent with the inhibitory effects of this type II site ligand on malignant cell proliferation in vitro and in vivo [14, 33]. The down-regulated cyclins (A2 and E2) and CDC25A are involved in the regulation of progression through the cell cycle [54]. PLK1 is involved in entry into mitosis, spindle formation and exit from mitosis [55, 56]. PCNA interacts with p53-controlled proteins and has multiple functions related to cellular proliferation [57]. Therefore, up-regulation of CDKN1B and the down-regulation of these other genes by luteolin, BMHPC or ZN-2 lead to an inhibition of the cell cycle. Furthermore, as luteolin also down-regulates genes involved in RNA transcription, both in the initiation of transcription through the general transcription factors (GTF2B, GTF2H1 and GTF2H2) and TATA box binding protein (TBP)-associated factors (TAF’s 5, 7, 9) and by the reduction of components of polymerases I (POLR1A, POLR1D), II (POLR2C) and III (POLR3K), it is likely that over time, luteolin treatment leads to a reduction of gene expression in multiple gene pathways. The exact nature of the relationship between the cell cycle and transcription factor pathways remains to be established. Due to the early six hour time point, it is likely that luteolin regulates the two pathways independent of one another. The ChIP data (Figure 9) indicates that luteolin may directly regulate the cell cycle genes.
The studies described in this manuscript have identified genetic targets involved in the mechanism of action of type II site ligands and their binding domain on histone H4 that regulate normal and malignant cell growth and proliferation [14, 15, 19, 24]. The previous work in our laboratory with rat uterine nuclear extract [32], calf thymus histone [31] and recombinant histone [30] identified the type II site as a histone H4. Histone H4, along with histones H2A, H2B and H3, exists in the cell as part of the nucleosome, which binds and packages DNA [58, 59]. Normally, DNA is bound tightly by the nucleosome and is therefore inaccessible. An important step in gene regulation of gene transcription at the nucleosome level is the recruitment of histone acetyltransferases (HATs) by transcription factors [60, 61]. Histone acetylation precedes and facilitates DNA access by the transcription machinery. In addition, other post-translational modifications, such as methylation, phosphorylation and ubiquitination, can lead to gene silencing or apoptosis [62]. Thus, type II site ligands such as luteolin, BMHPC and ZN-2 may be directly involved in the post-translational modification of the histones.
The identification of specifically regulated genes provides targets that will aid in the identification of specific modifications of histone H4 associated with the binding of and/or treatment with type II ligands. As seen in Figure 9, treatment of PC-3 cells with luteolin directly affects the acetylation state of lysines 5, 8, 12 and 16 of histone H4 associated with the PLK1 promoter. These four lysines are typically acetylated in active euchromatin and hypoacetylated in heterochromatin [63]. In particular, acetylation of lysines 8 and 16 tend to be associated with transcription, while acetylation of lysines 5 and 12 are typically associated with histone disposition [64]. Therefore, it is not surprising that luteolin down-regulation of PLK1 correlates with a lower acetylation state of the promoter. Utilizing ChIP assays and the knowledge of what promoters are affected will allow us to identify other post-translational nucleosome modifications associated with binding of the type II ligands. We believe type II sites and their respective ligands including luteolin, BMHPC, ZN-2 and dietarily-derived bioflavonoids that bind to type II sites with high affinity represent a universal epigenetic mechanism involved in the regulation of cell cycle progression, RNA transcription and normal and malignant cell proliferation. Studies on the mechanism of action of luteolin, BMHPC and ZN-2 may lead to the development of a novel class of drugs for the treatment of a variety of diseases such as BPH or prostate cancer that are devoid of side effects typically associated with anti-cancer agents.
Supplementary Material
Acknowledgements
This research is supported by grants from the National Institute for Environmental Health Sciences (ES-09964), and the National Cancer Institute (CA 35480 and CA 128932).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Eriksson H, Upchurch S, Hardin JW, Peck EJ, Jr., Clark JH. Heterogeneity of estrogen receptors in the cytosol and nuclear fractions of the rat uterus. Biochem Biophys Res Commun. 1978;81(1):1–7. doi: 10.1016/0006-291x(78)91622-4. [DOI] [PubMed] [Google Scholar]
- 2.Markaverich BM, Clark JH. Two binding sites for estradiol in rat uterine nuclei: relationship to uterotropic response. Endocrinology. 1979;105(6):1458–62. doi: 10.1210/endo-105-6-1458. [DOI] [PubMed] [Google Scholar]
- 3.Kuiper GG, Enmark E, Pelto-Huikko M, Nilssonand S, Gustafsson JA. Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci U S A. 1996;93(12):5925–30. doi: 10.1073/pnas.93.12.5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tremblay GB, Tremblay A, Copeland NG, Gilbert DJ, Jenkins NA, Labrieand F, Giguere V. Cloning, chromosomal localization, and functional analysis of the murine estrogen receptor beta. Mol Endocrinol. 1997;11(3):353–65. doi: 10.1210/mend.11.3.9902. [DOI] [PubMed] [Google Scholar]
- 5.McKenna NJ, Lanzand RB, O’Malley BW. Nuclear receptor coregulators: cellular and molecular biology. Endocr Rev. 1999;20(3):321–44. doi: 10.1210/edrv.20.3.0366. [DOI] [PubMed] [Google Scholar]
- 6.Markaverich BM, Williams M, Upchurchand S, Clark JH. Heterogeneity of nuclear estrogen-binding sites in the rat uterus: a simple method for the quantitation of type I and type II sites by [3H]estradiol exchange. Endocrinology. 1981;109(1):62–9. doi: 10.1210/endo-109-1-62. [DOI] [PubMed] [Google Scholar]
- 7.Markaverich BM, Roberts RR, Alejandroand M, Clark JH. The effect of low dose continuous exposure to estradiol on the estrogen receptor (type I) and nuclear type II sites. Endocrinology. 1984;114(3):814–20. doi: 10.1210/endo-114-3-814. [DOI] [PubMed] [Google Scholar]
- 8.Markaverich BM, Upchurch S, McCormack SA, Glasserand SR, Clark JH. Differential stimulation of uterine cells by nafoxidine and clomiphene: relationship between nuclear estrogen receptors and type II estrogen binding sites and cellular growth. Biol Reprod. 1981;24(1):171–81. doi: 10.1095/biolreprod24.1.171. [DOI] [PubMed] [Google Scholar]
- 9.Markaverich BM, Upchurchand S, Clark JH. Progesterone and dexamethasone antagonism of uterine growth: a role for a second nuclear binding site for estradiol in estrogen action. J Steroid Biochem. 1981;14(2):125–32. doi: 10.1016/0022-4731(81)90164-3. [DOI] [PubMed] [Google Scholar]
- 10.Ho SM, Yu M. Selective increase in type II estrogen-binding sites in the dysplastic dorsolateral prostates of noble rats. Cancer Res. 1993;53(3):528–32. [PubMed] [Google Scholar]
- 11.Ho SM, Viccioneand T, Yu M. In vitro and in vivo inhibition of nuclear type II estrogen binding sites in the dorsolateral prostate of noble rats. J Steroid Biochem Mol Biol. 1993;46(4):489–95. doi: 10.1016/0960-0760(93)90103-4. [DOI] [PubMed] [Google Scholar]
- 12.Ho SM, Yu M. Hormonal regulation of nuclear type II estrogen binding sites in the dorsolateral prostate of noble rats. J Steroid Biochem Mol Biol. 1995;52(3):233–8. doi: 10.1016/0960-0760(94)00170-q. [DOI] [PubMed] [Google Scholar]
- 13.Ho SM, Leav I, Merk FB, Yu M, Kwanand PW, Ziar J. Induction of atypical hyperplasia, apoptosis, and type II estrogen-binding sites in the ventral prostates of Noble rats treated with testosterone and pharmacologic doses of estradiol-17 beta. Lab Invest. 1995;73(3):356–65. [PubMed] [Google Scholar]
- 14.Markaverich BM, Alejandro MA. Type II [3H]estradiol binding site antagonists: inhibition of normal and malignant prostate cell growth and proliferation. Int J Oncol. 1998;12(5):1127–35. doi: 10.3892/ijo.12.5.1127. [DOI] [PubMed] [Google Scholar]
- 15.Markaverich BM, Gregory RR, Alejandro M, Kittrell FS, Medina D, Clark JH, Varmaand M, Varma RS. Methyl p-hydroxyphenyllactate and nuclear type II binding sites in malignant cells: metabolic fate and mammary tumor growth. Cancer Res. 1990;50(5):1470–8. [PubMed] [Google Scholar]
- 16.Markaverich BM, Schauweker TH, Gregory RR, Varma M, Kittrell FS, Medinaand D, Varma RS. Nuclear type II sites and malignant cell proliferation: inhibition by 2,6-bis-benzylidenecyclohexanones. Cancer Res. 1992;52(9):2482–8. [PubMed] [Google Scholar]
- 17.Scambia G, Ranelletti FO, Benedetti Panici P, Piantelli M, Bonanno G, De Vincenzo R, Ferrandina G, Pierelli L, Capelliand A, Mancuso S. Quercetin inhibits the growth of a multidrug-resistant estrogen-receptor-negative MCF-7 human breast-cancer cell line expressing type II estrogen-binding sites. Cancer Chemother Pharmacol. 1991;28(4):255–8. doi: 10.1007/BF00685531. [DOI] [PubMed] [Google Scholar]
- 18.Markaverich BM, Roberts RR, Finneyand RW, Clark JH. Preliminary characterization of an endogenous inhibitor of [3H]estradiol binding in rat uterine nuclei. J Biol Chem. 1983;258(19):11663–71. [PubMed] [Google Scholar]
- 19.Markaverich BM, Gregory RR, Alejandro MA, Clark JH, Johnsonand GA, Middleditch BS. Methyl p-hydroxyphenyllactate. An inhibitor of cell growth and proliferation and an endogenous ligand for nuclear type-II binding sites. J Biol Chem. 1988;263(15):7203–10. [PubMed] [Google Scholar]
- 20.Markaverich BM, Roberts RR, Alejandro MA, Johnson GA, Middleditchand BS, Clark JH. Bioflavonoid interaction with rat uterine type II binding sites and cell growth inhibition. J Steroid Biochem. 1988;30(1–6):71–8. doi: 10.1016/0022-4731(88)90078-7. [DOI] [PubMed] [Google Scholar]
- 21.Markaverich BM, Gregory RR, Alejandro MA, Varma RS, Johnsonand GA, Middleditch BS. Estrogen regulation of methyl p-hydroxyphenyllactate hydrolysis: correlation with estrogen stimulation of rat uterine growth. J Steroid Biochem. 1989;33(5):867–76. doi: 10.1016/0022-4731(89)90234-3. [DOI] [PubMed] [Google Scholar]
- 22.Markaverich BM, Roberts RR, Alejandroand MA, Clark JH. An endogenous inhibitor of [3H]estradiol binding to nuclear type II estrogen binding sites in normal and malignant tissues. Cancer Res. 1984;44(4):1515–9. [PubMed] [Google Scholar]
- 23.Maybruck WM, Markaverich BM. Partial purification and characterization of methyl-p-hydroxyphenyllactate esterase in rat uterine cytosol. Steroids. 1997;62(3):321–30. doi: 10.1016/s0039-128x(96)00227-9. [DOI] [PubMed] [Google Scholar]
- 24.Markaverich BM, Shoularsand K, Alejandro MA. Nuclear type II [3H]estradiol binding site ligands: inhibition of ER-positive and ER-negative cell proliferation and c-Myc and cyclin D1 gene expression. Steroids. 2006;71(10):865–74. doi: 10.1016/j.steroids.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 25.Carbone A, Ranelletti FO, Rinelli A, Vecchio FM, Lauriola L, Piantelliand M, Capelli A. Type II estrogen receptors in the papillary cystic tumor of the pancreas. Am J Clin Pathol. 1989;92(5):572–6. doi: 10.1093/ajcp/92.5.572. [DOI] [PubMed] [Google Scholar]
- 26.Piantelli M, Ricci R, Larocca LM, Rinelli A, Capelli A, Rizzo S, Scambiaand G, Ranelletti FO. Type II oestrogen binding sites in human colorectal carcinoma. J Clin Pathol. 1990;43(12):1004–6. doi: 10.1136/jcp.43.12.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scambia G, Ranelletti FO, Panici PB, Piantelli M, Bonanno G, De Vincenzo R, Ferrandina G, Rumi C, Laroccaand LM, Mancuso S. Inhibitory effect of quercetin on OVCA 433 cells and presence of type II oestrogen binding sites in primary ovarian tumours and cultured cells. Br J Cancer. 1990;62(6):942–6. doi: 10.1038/bjc.1990.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scambia G, Ranelletti FO, Benedetti Panici P, Piantelli M, Rumi C, Battaglia F, Larocca LM, Capelliand A, Mancuso S. Type-II estrogen binding sites in a lymphoblastoid cell line and growth-inhibitory effect of estrogen, anti-estrogen and bioflavonoids. Int J Cancer. 1990;46(6):1112–6. doi: 10.1002/ijc.2910460627. [DOI] [PubMed] [Google Scholar]
- 29.Larocca LM, Teofili L, Maggiano N, Piantelli M, Ranellettiand FO, Leone G. Quercetin and the growth of leukemic progenitors. Leuk Lymphoma. 1996;23(1–2):49–53. doi: 10.3109/10428199609054801. [DOI] [PubMed] [Google Scholar]
- 30.Shoulars K, Rodriguez MA, Crowley J, Turk J, Thompsonand T, Markaverich BM. Reconstitution of the type II [3H]estradiol binding site with recombinant histone H4. J Steroid Biochem Mol Biol. 2006;99(1):1–8. doi: 10.1016/j.jsbmb.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 31.Shoulars K, Rodrigues MA, Crowley JR, Turk J, Thompsonand T, Markaverich BM. Nuclear type II [3H]estradiol binding sites: a histone H3-H4 complex. J Steroid Biochem Mol Biol. 2005;96(1):19–30. doi: 10.1016/j.jsbmb.2004.12.047. [DOI] [PubMed] [Google Scholar]
- 32.Shoulars K, Brown T, Alejandro MA, Crowleyand J, Markaverich BM. Identification of nuclear type II [(3)H]estradiol binding sites as histone H4. Biochem Biophys Res Commun. 2002;296(5):1083–90. doi: 10.1016/s0006-291x(02)02042-9. [DOI] [PubMed] [Google Scholar]
- 33.Markaverich B, Alejandro M. Bioflavonoids, type II [3H]estradiol binding sites and prostatic cancer cell proliferation. Int J Oncol. 1997;11:1311–1319. doi: 10.3892/ijo.11.6.1311. [DOI] [PubMed] [Google Scholar]
- 34.Li C, Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci U S A. 2001;98(1):31–6. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li C, Hung Wong W. Model-based analysis of oligonucleotide arrays: model validation, design issues and standard error application. Genome Biol. 2001;2(8) doi: 10.1186/gb-2001-2-8-research0032. RESEARCH0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Habenicht UF, el Etreby MF. Rationale for using aromatase inhibitors to manage benign prostatic hyperplasia. Experimental studies. J Androl. 1991;12(6):395–402. [PubMed] [Google Scholar]
- 37.Kirby RS. The clinical assessment of benign prostatic hyperplasia. Cancer. 1992;70(1 Suppl):284–90. doi: 10.1002/1097-0142(19920701)70:1+<284::aid-cncr2820701316>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 38.Howe HL, Wingo PA, Thun MJ, Ries LA, Rosenberg HM, Feigaland EG, Edwards BK. Annual report to the nation on the status of cancer (1973 through 1998), featuring cancers with recent increasing trends. J Natl Cancer Inst. 2001;93(11):824–42. doi: 10.1093/jnci/93.11.824. [DOI] [PubMed] [Google Scholar]
- 39.Geller J. Overview of benign prostatic hypertrophy. Urology. 1989;34(4 Suppl):57–63. doi: 10.1016/0090-4295(89)90235-5. discussion 87-96. [DOI] [PubMed] [Google Scholar]
- 40.Tutrone RF, Jr., Ball RA, Ornitz DM, Lederand P, Richie JP. Benign prostatic hyperplasia in a transgenic mouse: a new hormonally sensitive investigatory model. J Urol. 1993;149(3):633–9. doi: 10.1016/s0022-5347(17)36169-4. [DOI] [PubMed] [Google Scholar]
- 41.Labrie F. Medical castration with LHRH agonists: 25 years later with major benefits achieved on survival in prostate cancer. J Androl. 2004;25(3):305–13. doi: 10.1002/j.1939-4640.2004.tb02791.x. [DOI] [PubMed] [Google Scholar]
- 42.Shapiro E. Embryologic development of the prostate. Insights into the etiology and treatment of benign prostatic hyperplasia. Urol Clin North Am. 1990;17(3):487–93. [PubMed] [Google Scholar]
- 43.Vogelzang NJ, Chodak GW, Soloway MS, Block NL, Schellhammer PF, Smith JA, Jr., Caplanand RJ, Kennealey GT, Zoladex Prostate Study Group Goserelin versus orchiectomy in the treatment of advanced prostate cancer: final results of a randomized trial. Urology. 1995;46(2):220–6. doi: 10.1016/s0090-4295(99)80197-6. [DOI] [PubMed] [Google Scholar]
- 44.Carter HB, Coffey DS. The prostate: an increasing medical problem. Prostate. 1990;16(1):39–48. doi: 10.1002/pros.2990160105. [DOI] [PubMed] [Google Scholar]
- 45.Leav I, Ho SM, Ofner P, Merk FB, Kwanand PW, Damassa D. Biochemical alterations in sex hormone-induced hyperplasia and dysplasia of the dorsolateral prostates of Noble rats. J Natl Cancer Inst. 1988;80(13):1045–53. doi: 10.1093/jnci/80.13.1045. [DOI] [PubMed] [Google Scholar]
- 46.Jordan VC, Murphy CS. Endocrine pharmacology of antiestrogens as antitumor agents. Endocr Rev. 1990;11(4):578–610. doi: 10.1210/edrv-11-4-578. [DOI] [PubMed] [Google Scholar]
- 47.Steinmetz KA, Potter JD. Food-group consumption and colon cancer in the Adelaide Case-Control Study. I. Vegetables and fruit. Int J Cancer. 1993;53(5):711–9. doi: 10.1002/ijc.2910530502. [DOI] [PubMed] [Google Scholar]
- 48.Parkin DM, Muir CS. Cancer Incidence in Five Continents. Comparability and quality of data. IARC Sci Publ. 1992;(120):45–173. [PubMed] [Google Scholar]
- 49.Adlercreutz H, Markkanenand H, Watanabe S. Plasma concentrations of phyto-oestrogens in Japanese men. Lancet. 1993;342(8881):1209–10. doi: 10.1016/0140-6736(93)92188-y. [DOI] [PubMed] [Google Scholar]
- 50.Clark JH, Markaverich BM. The agonistic and antagonistic actions of estriol. J Steroid Biochem. 1984;20(4B):1005–13. doi: 10.1016/0022-4731(84)90011-6. [DOI] [PubMed] [Google Scholar]
- 51.Larocca LM, Giustacchini M, Maggiano N, Ranelletti FO, Piantelli M, Alciniand E, Capelli A. Growth-inhibitory effect of quercetin and presence of type II estrogen binding sites in primary human transitional cell carcinomas. J Urol. 1994;152(3):1029–33. doi: 10.1016/s0022-5347(17)32649-6. [DOI] [PubMed] [Google Scholar]
- 52.Bemis DL, Capodice JL, Desai M, Buttyanand R, Katz AE. A concentrated aglycone isoflavone preparation (GCP) that demonstrates potent anti-prostate cancer activity in vitro and in vivo. Clin Cancer Res. 2004;10(15):5282–92. doi: 10.1158/1078-0432.CCR-03-0828. [DOI] [PubMed] [Google Scholar]
- 53.Yim D, Singh RP, Agarwal C, Lee S, Chiand H, Agarwal R. A novel anticancer agent, decursin, induces G1 arrest and apoptosis in human prostate carcinoma cells. Cancer Res. 2005;65(3):1035–44. [PubMed] [Google Scholar]
- 54.Douglas RM, Haddad GG. Genetic models in applied physiology: invited review: effect of oxygen deprivation on cell cycle activity: a profile of delay and arrest. J Appl Physiol. 2003;94(5):2068–83. doi: 10.1152/japplphysiol.01029.2002. discussion 2084. [DOI] [PubMed] [Google Scholar]
- 55.van Vugt MA, van de Weerdt BC, Vader G, Janssen H, Calafat J, Klompmaker R, Wolthuisand RM, Medema RH. Polo-like kinase-1 is required for bipolar spindle formation but is dispensable for anaphase promoting complex/Cdc20 activation and initiation of cytokinesis. J Biol Chem. 2004;279(35):36841–54. doi: 10.1074/jbc.M313681200. [DOI] [PubMed] [Google Scholar]
- 56.Eckerdt F, Strebhardt K. Polo-like kinase 1: target and regulator of anaphase-promoting complex/cyclosome-dependent proteolysis. Cancer Res. 2006;66(14):6895–8. doi: 10.1158/0008-5472.CAN-06-0358. [DOI] [PubMed] [Google Scholar]
- 57.Paunesku T, Mittal S, Protic M, Oryhon J, Korolev SV, Joachimiakand A, Woloschak GE. Proliferating cell nuclear antigen (PCNA): ringmaster of the genome. Int J Radiat Biol. 2001;77(10):1007–21. doi: 10.1080/09553000110069335. [DOI] [PubMed] [Google Scholar]
- 58.Cheung P, Allisand CD, Sassone-Corsi P. Signaling to chromatin through histone modifications. Cell. 2000;103(2):263–71. doi: 10.1016/s0092-8674(00)00118-5. [DOI] [PubMed] [Google Scholar]
- 59.Fischle W, Wangand Y, Allis CD. Histone and chromatin cross-talk. Curr Opin Cell Biol. 2003;15(2):172–83. doi: 10.1016/s0955-0674(03)00013-9. [DOI] [PubMed] [Google Scholar]
- 60.Klochendler-Yeivin A, Yaniv M. Chromatin modifiers and tumor suppression. Biochim Biophys Acta. 2001;1551(1):M1–10. doi: 10.1016/s0304-419x(01)00021-x. [DOI] [PubMed] [Google Scholar]
- 61.Archer SY, Hodin RA. Histone acetylation and cancer. Curr Opin Genet Dev. 1999;9(2):171–4. doi: 10.1016/s0959-437x(99)80026-4. [DOI] [PubMed] [Google Scholar]
- 62.Wang Y, Fischle W, Cheung W, Jacobs S, Khorasanizadehand S, Allis CD. Beyond the double helix: writing and reading the histone code. Novartis Found Symp. 2004;259:3–17. discussion 17–21, 163–9. [PubMed] [Google Scholar]
- 63.Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389(6649):349–52. doi: 10.1038/38664. [DOI] [PubMed] [Google Scholar]
- 64.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403(6765):41–5. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.