

Abstract
The upstream Gγ-globin gene cAMP response element (G-CRE) was previously shown to play a role in drug-mediated fetal hemoglobin induction. This effect is achieved via p38 mitogen activated protein kinase (MAPK)-dependent CREB1 and ATF-2 phosphorylation and G-CRE trans-activation. Since this motif is also a predicted consensus binding site for cJun we extended our analysis to determine the ability of cJun to trans-activate γ-globin through the G-CRE. Using chromatin immunoprecipitation assays we showed comparable in vivo cJun and CREB1 binding to the G-CRE region. Protein-protein interactions were confirmed between cJun/ATF-2 and CREB1/ATF-2 but not between CREB1 and cJun. However, we observed cJun and CREB1 binding to the G-CRE in vitro by electrophoretic mobility shift assay. Promoter pull-down assay followed by sequential western blot analysis confirmed co-localization of cJun, CREB1, and ATF-2 on the G-CRE. To show functional relevance, enforced expression studies with pLen-cJun and a Gγ-promoter (-1500 to +36) luciferase reporter were completed; we observed a concentration-dependent increase in luciferase activity with pLen-cJun similar to that produced by CREB1 enforced expression. Moreover, the G/A mutation at -1225 in the G-CRE abolished cJun trans-activation. Finally, enforced cJun expression in K562 cells and normal primary erythroid progenitors enhanced endogenous γ-globin gene expression. We concluded from these data indicate that cJun activate the Gγ-globin promoter via the G-CRE in a manner comparable to CREB1 and propose a model for γ-globin activation based on DNA-protein interactions in the G-CRE.
List of key words: cJun, CREB1, γ-globin, cAMP response element, ATF-2
Introduction
Human β-globin disorders, such as sickle cell anemia are relatively common genetic diseases that affect millions of people worldwide. A myriad of clinical symptoms including vaso-occlusion, pain, stroke, and other complications occur in this population. However increased fetal hemoglobin production (20-30%) can ameliorate disease severity [1]. Therefore, understanding γ-globin gene regulation and developing strategies to reverse its silencing after birth has important therapeutic implications.
The β-globin locus is comprised of five functional genes–ε, Gγ, Aγ, δ and β–that are arranged and expressed developmentally in the order they appear in the locus. The γ-globin genes are normally expressed at high levels during fetal-stage development [2-4] in the presence of an optimal trans-factor environment. To define molecular mechanisms of γ-globin gene regulation, drug-mediated fetal hemoglobin induction has been investigated as a strategy to develop treatments for sickle cell disease and β-thalassemia.
Previous work from our laboratory demonstrated a role for the -1222 Gγ-globin cAMP response element (G-CRE) in drug-mediated gene activation [5]. The transcription factors CRE binding protein 1 (CREB1) and activating transcription factor-2 (ATF-2) bind the G-CRE to activate γ-globin expression after treatment with sodium butyrate (NaB) or trichostatin A (TSA) via p38 MAPK signaling. The palindromic G-CRE consensus, 5′-TGACGTCA-3′ is also an overlapping binding site for cJun and other ATF family members [6]. This group of trans-factors forms homo- or heterodimers such as CREB1/ATF-2 when bound to DNA to achieve context-mediated gene activation or repression in various promoters [7]. In our current study we sought to explicate the role of cJun in γ-globin regulation through interaction with other trans-factors bound to the G-CRE.
cJun functions as a proto-oncogene and is a key player in survival, stress response and apoptotic pathways [8]. cJun binds the AP-1 consensus site (5′-TGAGTCA-3′) as a heterodimer with cFos, JunB, JunD and ATF family members including ATF-2, ATF-3 and ATF-4 [9,10]. cJun also binds the CRE primarily combined with ATF-2 in the interferon-β [11] and tumor necrosis factor-α [12] promoters. Moreover, overlapping tandem AP-1/NF-E2 sites in hypersensitive site 2 of the β-locus control region were previously characterized as enhancers in γ-globin gene expression [13] however a possible negative role has also been implicated when cJun interacts with NF-E2 [14]. Context dependent regulation of γ-globin expression by cJun after drug induction has been investigated by others [15-18]. Increased cJun mRNA levels via post transcriptional stabilization were observed in cells treated with hydroxyurea [15]. By contrast, other studies demonstrated that cJun might act as an inhibitor of HDAC inhibitor-induced erythroid differentiation [17,18].
A controversy remains over the role of cJun in γ-globin gene regulation therefore additional data are required. In the current study we investigated the ability of cJun to activate γ-globin expression. We demonstrated cJun binding to the G-CRE region in vivo and its ability to trans-activate the Gγ-globin promoter. Furthermore, cJun co-localized with CREB1 and ATF-2 to the G-CRE however protein-protein interaction was only detected between cJun and ATF-2. Mutating the G-CRE abolished the ability of cJun and CREB1 to trans-activate the γ-globin promoter. Alternative models for G-CRE function are discussed.
Material
Cell culture
K562 cells were maintained in Iscove's Modified Dulbecco's medium containing 10% fetal bovine serum (Atlanta Biologicals, Atlanta GA), supplemented with penicillin (100U/ml) and streptomycin (0.1mg/ml) at 37°C and 5% CO2. For drug studies, cells were treated for 48 hrs with 2mM NaB or 0.5μM TSA purchased from Sigma (St. Louis, MO).
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed in K562 cells as previously described [5,19] using the Upstate (Lake Placid, NY) protocol per the manufacturers' instructions. Briefly, 40 million cells from different conditions were crosslinked with 1% formaldehyde. After cell lysis, nuclei were sonicated (Sonicator 3000; Misonix, Farmingdale, NY) for 8-10 pulses at 12W and output level 5, on ice to generate 500-600 bp fragments. Sonicated DNA was purified by phenol chloroform extraction and then immunoprecipitation reactions performed with the following antibodies (Santa Cruz Biotechnology, Santa Cruz, CA): phosphorylated CREB1 (pCREB1, sc-7978), total CREB1 (t-CREB1, sc-240), p-cJun (sc-16312) and t-cJun (sc-1694). TFIID (sc-204X), human IgG (Sigma) and no antibody reactions were set up as controls. The DNA-protein complexes were collected in elution buffer and reverse crosslinking achieved by heating samples at 65°C. Chromatin was precipitated by ethanol and used for quantitative PCR (qPCR).
Reverse transcription qPCR (RT-qPCR) Analysis
qPCR was performed on an iCycler (Bio-Rad, Hercules, CA) using the Sybergreen iQ Supermix (Bio-Rad) and 10 pM of gene-specific primers (Table 1). To confirm in vivo cJun and CREB1 binding to the G-CRE, primers located at nucleotides -1350 to -1100 were used; TFIID binding at -53 to +69 in the proximal γ-globin promoter was analyzed as a positive control (Table 1).
Table 1. Summary of primers used for qPCR analysis.
| G-CRE | Forward: 5′ CGGCGGCTGGCTAGGGATGAA 3′ |
| Reverse: 5′ CTGTGAAATGACCCATGGCG 3′ | |
| γ-TATA box | Forward:5′AAGCCTTACACAGGATTATGAAGTCTG 3′ |
| Reverse: 5′ ACATGGCAGGAAGTATTCATGCTG 3′ | |
| γ-globin | Forward: 5′-GGCAACCTGTCCTCTGCCTC-3′ |
| Reverse: 5′-GAAATGGATTGCCAAAACGG-3′ | |
| β-globin | Forward: 5′ CTCATGGCAAGAAAGTGCTCG 3′ |
| Reverse: 5′ AATTCTTTGCCAAAGTGATGGG 3′ | |
| GAPD | Forward: 5′-GAAGGTGAAGGTCGGAGT-3′ |
| Reverse: 5′-GAAGATGGTGATGGGATTTC-3′ | |
| cJun | Forward: 5′- CCCAAGAACGTGACAGATGA-3′ |
| Reverse: 5′- CACTGTCTGAGGCTCCTCCT-3′ | |
| CREB1 | Forward: 5′- ATTACCCAGGGAGGAGCAAT-3′ |
| Reverse: 5′- TGGTTGCTGGGCACTAAGAT -3′ |
To quantify globin gene expression levels, total RNA was isolated using RNA Stat-60™ (TEL-TEST “B” Inc., Friendswood, TX) and used for reverse transcription (RT)-qPCR analysis as previously published [5]. Briefly, cDNA was generated from total RNA using the Improm-II reverse transcriptase system and oligo (dT)15 primers (Promega, Madison, WI). Levels of γ-globin, β-globin and glyceraldehyde-3-phosphate dehydrogenase (GAPD) were quantified in K562 cells and primary erythroid progenitors. Standard curves were generated using serial 10-fold dilutions of Topo7 plasmids carrying a γ-globin cDNA (Topo7-γ-globin), Topo7-β-globin and Topo7-GAPD. Globin mRNA levels were normalized to GAPD (γ/GAPD, β/GAPD) and γ/β-globin mRNA ratios were calculated by dividing γ/GAPD by β/GAPD.
Western blot
Cellular protein extracts were prepared from two million K562 cells from the various conditions using lysis buffer (Promega, Madison, WI). Extracts (50-100 μg) were loaded on 10% sodium dodecyl sulfate-polyacrylamide gels (SDS-PAGE) and transferred to nitrocellulose membrane for western blot analysis as previously published [5]. Membranes were blocked in 5% bovine serum albumin for 30 min and then incubated with primary antibodies (pCREB1, t-CREB1, p-cJun and t-cJun) at 1:500 to 1:1000 dilutions overnight at 4°C. Actin antibody (Chemicon Millipore, Billerica, MA; MAB1501,) was used as a loading control. Horseradish peroxidase-conjugated secondary antibodies including mouse-anti-goat and goat-anti-mouse purchased from Pierce (Rockford, IL; #31400 and #31430) and goat-anti-rabbit (sc-2004, Santa Cruz Biotech.) were used and proteins were detected using the ECL system (Amersham, Piscataway NJ). Band intensities were quantified using the ChemiDoc System (Bio-Rad).
Protein immunoprecipitation (IP)
Nuclear extracts (300-400 μg) prepared from K562 cells as previously published [19] were pre-cleared with protein A agarose beads (Santa Cruz) in IP buffer containing 10 mM HEPES, pH7.9, 25 mM KCl, 500 μM EDTA, 500 μM DTT, 2.5% glycerol and protease inhibitors. The supernatants were then incubated individually with t-cJun, t-CREB1, t-ATF-2 (sc-242) or human IgG control for one hr. Protein A agarose beads were added to precipitate immune complexes overnight at 4°C. Precipitated agarose beads were heated at 95°C for 5 min and proteins were resolved on a 9% SDS-PAGE gel followed by western blotting to characterize specific protein-protein interactions.
Electrophoretic Mobility Shift Assay (EMSA)
EMSA was performed with 8 μg of K562 nuclear extracts incubated for 20 min with wild type and mutant G-CRE oligonucleotide probes; G-CRE: 5′-AGAGTTTCTGACGTCATAATCTACCAAG-3′ (the G-CRE is underlined and bases shown in lower case are for mutant probes), G-m1: 5′-AGAGTTTCTGACaTCATAATCTACCAAG-3′, G-m2: 5′-AGAGTTTCTGtgGTCATAATCTACCAAG-3′, and G-m3s: 5′-AGAGTTTCtctctgtaTAATCTACCAAG-3′. All probes were end-labeled using T4 kinase and [γ-32P]ATP and binding reactions were performed in gel shift binding buffer containing poly [dI:dC] (Promega). Competition studies to confirm protein bands specificity were performed with 100-fold excess of the corresponding unlabelled oligonucleotide. Supershift reactions were performed by adding t-cJun or t-CREB1 antibody 20 min prior to the addition of radiolabeled G-CRE probe. DNA-protein complexes were resolved in 4% native polyacrylamide gels.
Promoter pull-down assay
This method was adopted from previously published protocols [20,21] with some modifications. Oligonucleotides were synthesized and biotinylated at the 5′-end (Sigma-Aldrich, Woodland, TX): G-CRE: 5′biotin-CCAGAGTTTCTGACGTCATAATCTACCAAGG 3′ and G-m2: 5′biotin-CCAGAGTTTCTGtgGTCATAATCTACCAAGG 3′ (the G-CRE is underlined and the mutated nucleotides are lower case). Single-stranded sense and anti-sense oligonucleotides were annealed to generate double stranded probes for pull-down reactions. Briefly, 300 μg of K562 nuclear extract was incubated with 2 μg of probe and 10 μg of salmon sperm DNA in pull-down buffer (10 mM HEPES, pH7.9, 25 mM KCl, 500 μM EDTA, 500 μM DTT, 2.5% glycerol and protease inhibitors) on ice for 90 min. Streptavidin agarose beads were added to the pull down reaction at the end of incubation. The samples were mixed overnight at 4°C and then pelleted beads were washed with 500 μl of the pull-down buffer and then heated at 95°C for 5 min. The proteins were resolved on SDS-PAGE gels followed by electro-transfer to nitrocellulose. Each membrane was analyzed by sequential western blotting with t-CREB1, t-ATF2 and t-cJun antibody (Santa Cruz Biotechnology); membranes were stripped in between blots to demonstrate co-localization of trans-factors on the G-CRE probe.
Transient transfections
Transient transfections were performed as previously described [5,19]. Four reporter constructs were analyzed including GγLuc: carrying the wild-type Gγ-promoter (-1500 to +36) cloned into the pGL3-Basic luciferase reporter plasmid (Promega) and three mutant reporters GγLuc(m1): carrying a -1225 G/A mutation; GγLuc(m2) carrying the -1227 AC/TG mutation; and GγLuc(m3s) carrying a scrambled G-CRE sequence 5′-TCTATGTA-3′. Briefly, 10μg of each reporter construct was co-transfected with 2.5μg of pSV-β-galactosidase plasmid (Promega) to monitor transfection efficiency, into 10 million K562 cells using a GenePulser (Bio-Rad) at 260 V and 975 μF. For dose response studies, K562 cells were transfected with 10-50μg of expression vectors pCMV and pCMV-CREB1 (Clontech, Mountainview, CA), or pLen and pLen-cJun, (a generous gift from Dr. Gail Breen, University of Texas at Dallas). pLen was generated from pLen-cJun by restriction digestion with BamHI to remove cJun and then was circularized to generate the empty vector control. β-Galactosidase and luciferase activity were quantified using the β-Galactosidase Enzyme Assay and Luciferase Assay systems respectively (Promega) per the manufacturer's protocol. Luciferase activity was measured on a TD-20/20 Luminometer (Turner Biosystems, San Diego, CA). All values were normalized by β-galactosidase activity and total protein measured by Bradford assay (BioRad).
cJun and CREB1 K562 stable lines
Ten micrograms of BamH1 or EcoR1 linearized pCMV, pCMV-CREB1, pLen and pLen-cJun plasmid were transfected by electroporation into 10 million K562 cells in serum-free medium. Cells were transferred to Iscove's Modified Dulbecco's Medium with 10% fetal bovine serum for three days and then 500 μg/mL G418 (Sigma) was added to select for resistant cell pools. After 21 days the resistant cells were maintained in 200 μg/ml G418 and enforced expression of cJun and CREB1 was confirmed by western blot analysis.
Expression vector copy number was calculated by a recently published qPCR-based method [19] using genomic DNA isolated with a FlexiGene DNA kit (Qiagen, Valencia, CA). The copy number of the pCMV, pCMV-CREB1, pLen, and pLen-cJun vectors along with endogenous GAPD as control was measured in stable clones. The primers used for gene copy number are shown in Table 1. The relative copy number (Q) of target gene versus the GAPD gene was calculated using the following equation:
Where Ns is the ratio of the copy number of target gene divided by GAPD and Nk is the analogous calculation for K562 cells. NsT is the copy number of target gene in stable lines and NkT the copy number of target genes in K562 cells; NsG and NkG represent the copy number of GAPD in stable lines and K562 cells respectively.
Enforced transcription factor expression in primary erythroid cells
For primary erythroid progenitor studies, human peripheral blood mononuclear cells were purchased from Carter BloodCare (Fort Worth, TX) in accordance with guidelines of the Institutional Review Board at the University of Texas at Dallas. Erythroid progenitors were generated using the two-phase liquid cultures system [22]. During phase 1, cells were grown in Iscove's Modified Dulbecco's Medium supplemented with 30% fetal bovine serum and 50 ng/mL each, of granulocyte-monocyte colony-stimulating factor, Interleukin-3, and stem cell factor. To initiate phase 2 on day 7, the medium was changed and erythropoietin (3U/mL) and stem cell factor (50ng/ml) were added. Erythroid progenitors were transfected in triplicate on day 11 for 48 hrs and 72 hrs with 5 μg of vector (pCMV, pCMV-CREB1, pLen and pLen-cJun). The CD34-Nucleofector kit was used to perform transfections per manufacturer's instructions (Amaxa Inc., Gaithersburg, MD) on a Nucleofector device (Amaxa Inc.). Total RNA was isolated for RT-qPCR analysis and viability was monitored using 2% trypan blue stain. The primers used for qPCR analysis are shown in Table 1.
Statistical analysis
The data were reported as the mean ± standard error of the mean (SEM) from at least five independent experiments conducted under different conditions including untreated K562 cells or erythroid progenitors as steady state controls. Data were analyzed by a two-tailed student's t test and values of p<0.05 were considered statistically significant. Statistical analyses were performed using Microsoft Exel (Redmond, WA).
Results
We have previously demonstrated that the histone deacetylase inhibitors Sodium Butyrate and TSA induce γ-globin gene expression via p38 MAPK signaling [5]. Two downstream effectors of p38 were identified as CREB1 and ATF-2 which trans-activate the G-CRE. However, the G-CRE also contains a consensus site for cJun and other ATF family members. Therefore, we extended our investigation to determine whether cJun bind the G-CRE and/or interact with CREB/ATF family members to regulate γ-globin gene expression.
cJun bind the G-CRE region in vivo
ChIP assay was performed in K562 cells to investigate whether cJun bind the G-CRE. Two regions including the upstream G-CRE or TATA box were examined for chromatin enrichment by qPCR analysis (Fig. 1A). Immunoprecipitations of untreated K562 cells with p-cJun antibody produced 3-fold (p<0.05) chromatin enrichment compared to no antibody and IgG controls (Fig. 1B); 8.5-fold (p<0.05) enrichment was observed with pCREB1 antibody. We next performed drug inductions with agents know to activate γ-globin expression. Chromatin enrichment for pCREB1 precipitants was increased 13-fold and 11-fold by treatment with NaB and TSA respectively. However, chromatin enrichment for p-cJun precipitants decreased 31-57% with both drugs. We previously demonstrated that ATF-2 also interact in the G-CRE in vivo [5] therefore these studies were not repeated. To monitor γ-globin transcription for the different experimental conditions, TFIID binding to the TATA box region was quantified. A significant 23-fold (p<0.001) chromatin enrichment was observed with TFIID immunoprecipitation which increased further after drug inductions, confirming robust γ-globin gene transcription in K562 cells. Since there are no other AP-1 or CRE sites in the target region, these studies support cJun binding to the G-CRE in vivo. Moreover, the binding trends produced with NaB and TSA are consistent with competitive occupancy of the G-CRE by cJun and CREB after drug inductions.
Fig. 1. cJun and CREB1 bind G-CRE region in vivo.
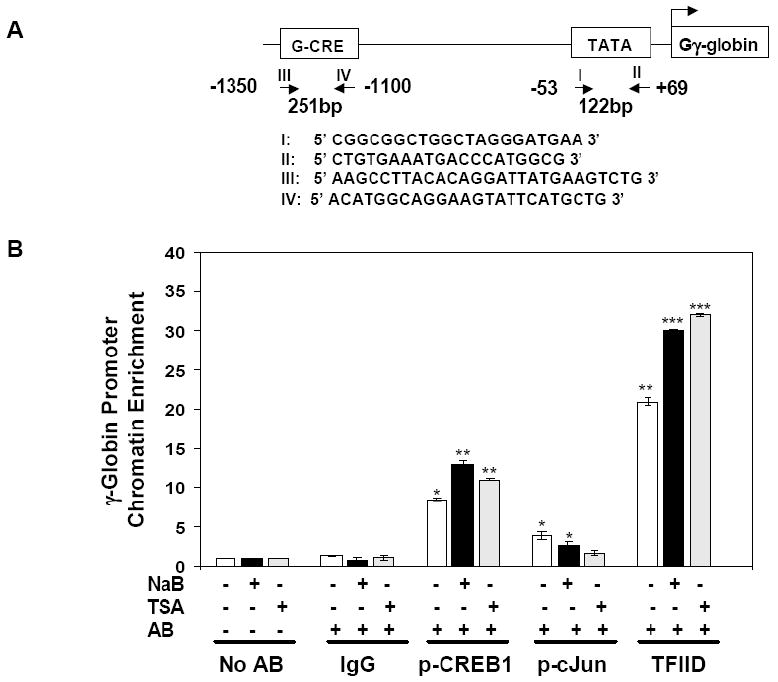
ChIP assays were performed in K562 cells to determine cJun and CREB1 binding to the Gγ-globin CRE (G-CRE) region in vivo. A) Two sets of primers shown in the diagram were used for quantitative PCR (qPCR) to verify chromatin enrichment in the Gγ-globin promoter. Primers I and II target the -53 to +69 region to confirm TFIID binding to the TATA box while primers III and IV were used to measure chromatin enrichment in the G-CRE region. B) ChIP assay quantitative results for untreated K562 cells (-) or after treatment with 2 mM NaB or 0.5 μM TSA (+) are shown in the graph. Data from qPCR analysis was plotted as fold change in chromatin enrichment for the immunoprecipitation reactions performed with the antibodies shown. The no antibody (No AB) and IgG samples were used as controls. Chromatin enrichment levels for No AB samples were normalized to one. Data are shown as the mean ± standard error of the mean (SEM), *p<0.05 was considered significant (**p<0.01 and ***p<0.001).
Treatment with histone deacetylase inhibitors mediates cJun phosphorylation
To further understand mechanisms of γ-gene regulation by cJun, western blots were performed to study changes in p-cJun levels after drug treatments. A 2-fold (p<0.05) increase in p-cJun was observed in K562 cells treated with NaB and TSA (Fig. 2A and C) compared to an 8-fold and 3-fold increase respectively in p-CREB1 (Fig. 2B and C). These studies suggest that cJun was activated by NaB and TSA to a lower degree than CREB1.
Fig. 2. cJun and CREB1 phosphorylation levels increase after drug treatment.
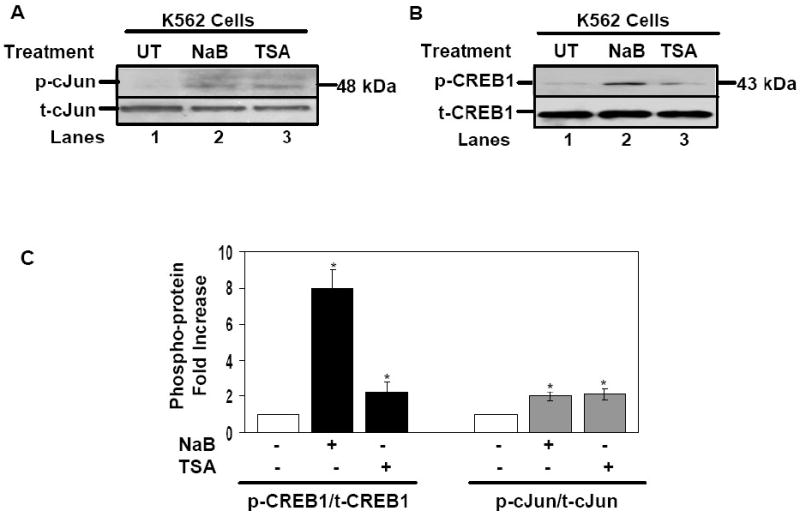
Protein extracts were isolated from K562 cells treated with NaB (2 mM) or TSA (0.5 μM) for 48 hrs (see Materials). A) Shown is the western blot performed with phosphorylated-cJun (p-cJun) and total-cJun (t-cJun) antibody. B) Western blot analysis results for p-CREB1 and t-CREB1 antibody is shown. C) Quantitative data obtained from the western blot gels using ChemiDoc software (Biorad) are shown in the graphs. Shown are p-CREB1/t-CREB1 (black bars) and p-cJun/t-cJun (gray bars) protein levels in the absence (-) or presence (+) of drug treatments. Data are shown as the mean ± SEM, *p<0.05 was considered significant.
cJun undergoes protein-protein interaction with ATF-2
The data generated for cJun support its ability to bind the G-CRE similar to CREB1 and ATF-2. Therefore, our next set of experiments involved IP to determine whether cJun interacts with CREB1 and/or ATF-2 to form complexes in erythroid cells. Nuclear extracts from K562 cells were IP with cJun, CREB, ATF-2 or human IgG antibodies and then western blot was performed with the same antibodies to detect the presence of these proteins in the immuno-complexes. Western blot with CREB1 antibody produced the predicted 43 kDa band with t-ATF-2 IP samples, but no band was observed for t-cJun IP samples (Fig. 3A). However western blot with t-ATF-2 antibody produced a 72 kDa band for both CREB1 and cJun IP samples. As expected, no protein band was observed with control IgG reactions. These data support the ability of ATF-2 to undergo protein-protein interaction with cJun and CREB1 however interaction between CREB1 and cJun was not observed in K562 cells. Other investigators demonstrated a lack of CREB and cJun interaction in NIH3T3, HeLa, and other cell types [23-26].
Fig. 3. cJun co-localized to the G-CRE with CREB1 and ATF-2.
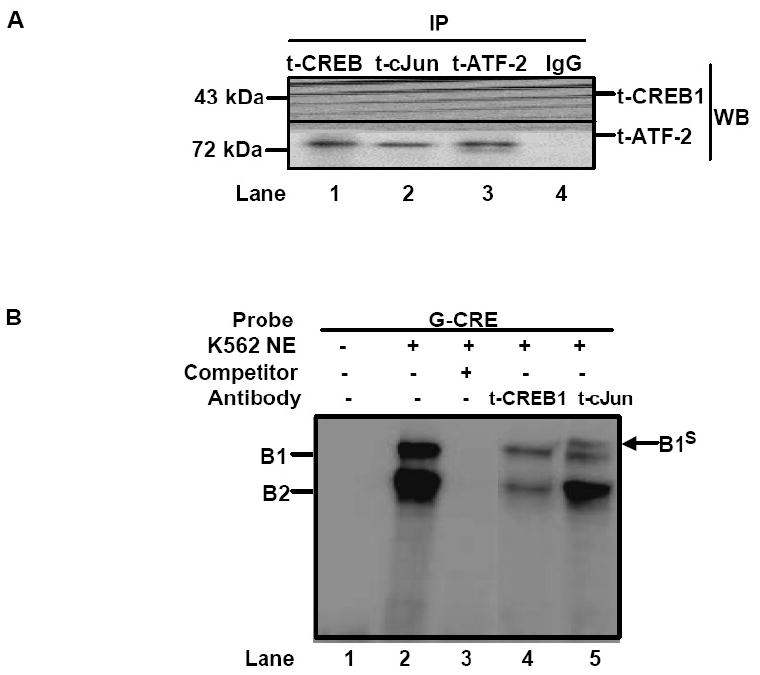
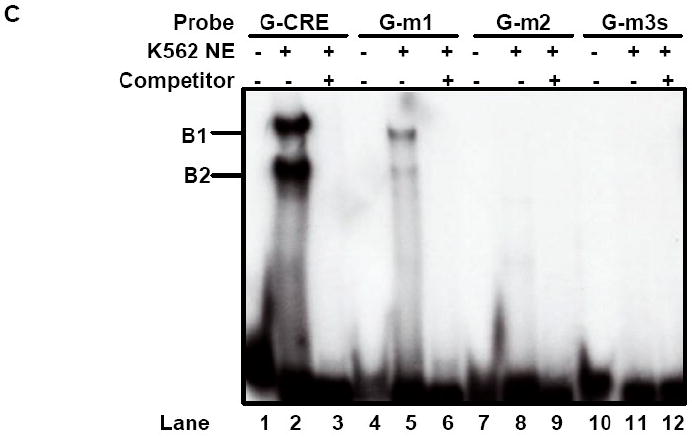
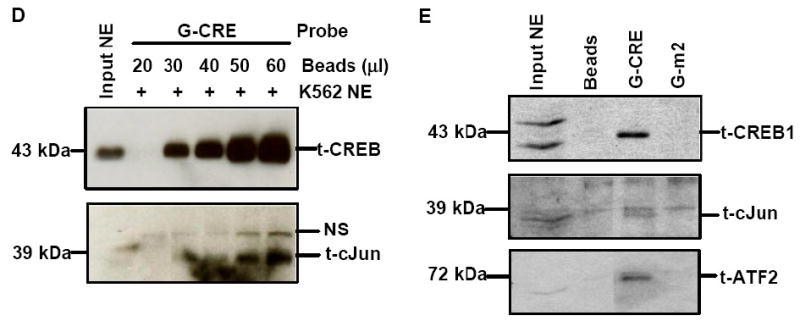
A) Immunoprecipitation (IP) reactions were performed with 200-300 μg of K562 nuclear extracts and t-CREB1, t-cJun, t-ATF-2 and IgG antibodies and then western blot (WB) was performed with separate membranes and the antibody shown to detect protein-protein interaction (See Materials). Note the presence of a protein band when the IP and WB antibody are identical serving as a positive control. IP with IgG served as a negative control and did not produce a protein band as expected. B) Electrophoretic mobility shift assay (EMSA) was performed with K562 cell nuclear extracts (NE) and the G-CRE probe. Different reactions in the absence (Lane 1) or presence of 8μg of K562 NE (Lanes 2-5) are shown. Supershift assays were performed with t-CREB1 and t-cJun antibodies. C) EMSA was performed under the same conditions as described in Panel B except reactions with the mutant probes G-m1 (G/A), G-m2 (AC/TG) and G-m3 with a scrambled G-CRE (tctctgta) were included. Competition reactions were completed in the presence of NE and cold oligonucleotide (++) to determine specificity of binding. D) Promoter pull-down reactions were performed with 2 μg of 5′biotinylated G-CRE probe and 300 μg of nuclear extract prepared from K562 cells. Increasing amounts of streptavidin beads (Beads) were analyzed to test efficiency of complex pull-down. The proteins bound to the G-CRE were detected by western blotting with t-CREB1 and t-cJun antibody (see Materials). Nuclear extract (Input NE) was used as a positive control. A non-specific (NS) protein band was observed on the t-cJun western blot. E) Promoter pull-down was performed with 5′biotinylated G-CRE and G-m2 (AC/TG) mutant probes as described in Panel D except 20 μl of streptavidin agarose beads were used in all reactions. Western blot analysis was also performed with total ATF-2 (t-ATF-2) antibody. A negative control reaction run in the absence of biotinylated probes (Beads) is shown.
cJun, CREB1 and ATF-2 co-localize to the G-CRE in vitro
While ChIP assay demonstrated in vivo cJun binding in the upstream Gγ-globin region, EMSA was performed to gain evidence that cJun directly binds the G-CRE. Nuclear extract from K562 cells was incubated with a G-CRE probe in the presence and absence of self competitor or t-CREB1 and t-cJun antibody. Two complexes, B1 and B2 were established at baseline, which were completely abolished with cold self competitor (Fig. 3B, Lanes 2 and 3). To determine if specific transcription factors are present in the complexes, supershift assays were performed. Reactions with t-CREB1 antibody markedly decreased the intensity of both complexes (Lane 4) however t-cJun antibody produced a supershifted B1s complex with a concomitant decrease in B1 (Lane 5). These data confirms that cJun and CREB1 binds the G-CRE in vitro.
We next performed EMSA with mutant probes to interrogate the nucleotides required to establish stable DNA-protein complexes. Experiments were performed with the G-m1 probe carrying a G/A mutation at -1225, G-m2 with an AC/TG mutation at -1227, and G-m3s with a scrambled G-CRE sequence (Materials). The single base mutation in G-m1 markedly reduced binding in the B1 and B2 complexes (Fig. 3C, Lanes 2 and 5) while the G-m2 and G-m3s probes completely abolished binding (Lanes 7-12).
A second in vitro method, promoter pull-down combined with western blot analysis was developed to determine if the target trans-factors co-localize to the G-CRE, which could not be established using EMSA. This assay takes advantage of the high binding affinity between streptavidin-conjugated agarose beads and biotinylated DNA probes allowing the pull-down of DNA-bound protein complexes. We first performed dose response studies to test whether increased amounts of streptavidin beads could produce greater quantities of protein-DNA complexes detected by western blot analysis. As shown in Fig. 3D, using 300 μg of K562 nuclear extract in each reaction and 2 μg of biotinylated G-CRE probe, we observed a steady increased in CREB1 protein on western blot at a dose range of 20 μl and 40 μl of streptavidin beads. A similar dose-dependent increase in c-Jun protein binding was detected.
Subsequently we performed promoter pull-down with wild-type G-CRE and G-m2 probes carrying the -1227 AC/TG mutation. From a single pull down reaction with the G-CRE probe followed by western blotting of the same membrane processed sequentially with the three antibodies, we detected CREB1, ATF2 and c-Jun protein (Fig. 3E); no band was detected for beads-only reactions. By contrast, the G-m2 mutation completely abolished binding for the three proteins. These data clearly demonstrate that cJun, CREB1, and ATF-2 co-localize simultaneously on the G-CRE and that optimal binding depends on an intact G-CRE.
cJun and CREB1 trans-activates the γ-globin promoter in a competitive pattern
To investigate the functional relevance of cJun binding to the G-CRE, co-transfection studies were performed in K562 cells with the GγLuc reporter plasmid carrying the wild-type Gγ-globin promoter (Fig. 4A) and the pLen-cJun expression vector. A dose-dependent increase in GγLuc luciferase activity was produced with 10-50 μg of pLen-cJun expression vector (Fig. 4B). At the 40 μg concentration luciferase activity increased 350-fold (p<0.001). A similar but less robust dose-dependent increase in luciferase activity was demonstrated with the pCMV-CREB1 expression vector (Fig. 4C). Since the ChIP assay data suggested competitive binding to the G-CRE region after drug inductions, we next performed studies where the cJun and CREB1 expression vectors were co-transfected. cJun enforced expression (30μg) produced an 85-fold increase in luciferase activity with the GγLuc reporter compared to an 11.2-fold increase produced by pCMV-CREB1 (Fig. 4D). When both vectors were transfected together an intermediate 29-fold increase in luciferase activity was observed. We concluded that cJun is a strong trans-activator of the Gγ-globin promoter and that CREB1 and cJun probably compete for functional interaction in the G-CRE.
Fig. 4. cJun and CREB1 compete for trans-activation of the Gγ-globin promoter.
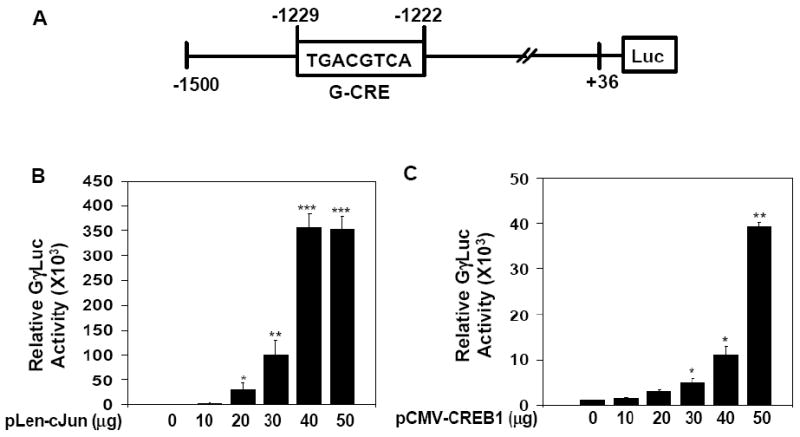
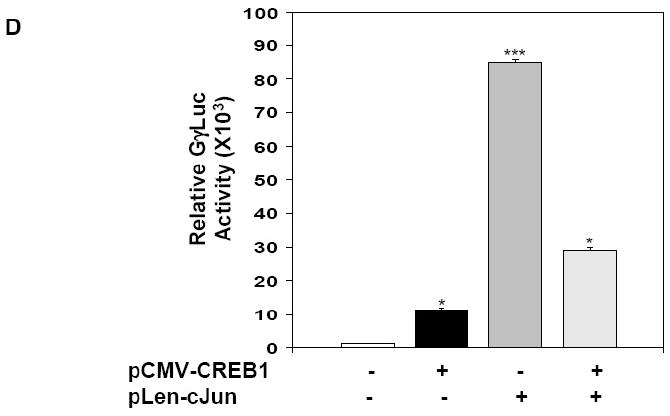
A) Shown is a schematic diagram of the GγLuc plasmid carrying 1536 bp of the wild type Gγ-globin promoter linked to the luciferase reporter gene. B) Transient transfection assays were performed in K562 cells using the GγLuc reporter co-transfected with increasing concentrations of the cJun expression vector, pLen-cJun. Shown is luciferase activity after subtracting luciferase values obtained for transfections performed with the pLen empty vector control. Data are shown as the mean ± SEM, *p<0.05 was considered significant (**p<0.01; ***p<0.001). C) Similar experiments with the pCMV-CREB1 expression vector and corresponding empty vector control pCMV were performed. Shown are the luciferase quantitative data. D) Co-transfection experiments were performed with 30 μg each of pCMV-CREB1 (black bar) or pLen-cJun (gray bar) vectors alone (+) or both vectors combined (striped bar; ++). Shown is the relative luciferase activity for the different conditions.
To confirm the requirement of the G-CRE for Gγ-globin promoter trans-activation by cJun, we tested three mutant GγLuc reporters. A single base pair mutation in GγLuc(m1) produced a 40% decrease in luciferase activity compared to wild-type GγLuc (Fig. 5A). When two bases were mutated in GγLuc(m2) or the G-CRE was scrambled in GγLuc(m3s), a 95% decrease in luciferase activity was observed. These studies confirmed that the G-CRE is required for basal Gγ-promoter transcription. Next we performed cJun enforced expression studies in this genetic reporter system. pLen-cJun trans-activated GγLuc 52-fold (p<0.001) which was greater than 95% abolished in the three mutant reporters (Fig. 5B). By contrast, pCMV-CREB1 stimulated luciferase activity in the GγLuc and GγLuc(m1) reporters, however luciferase activity was also abolished for the other two mutant reporters. These data suggests that the ability of cJun to interact with the G-CRE is less tolerant of sequence variation than that of CREB1.
Fig. 5. An intact G-CRE is required for Gγ-globin promoter trans-activation.
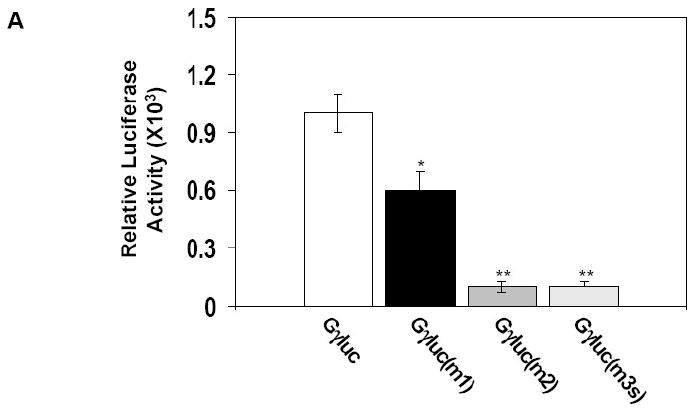
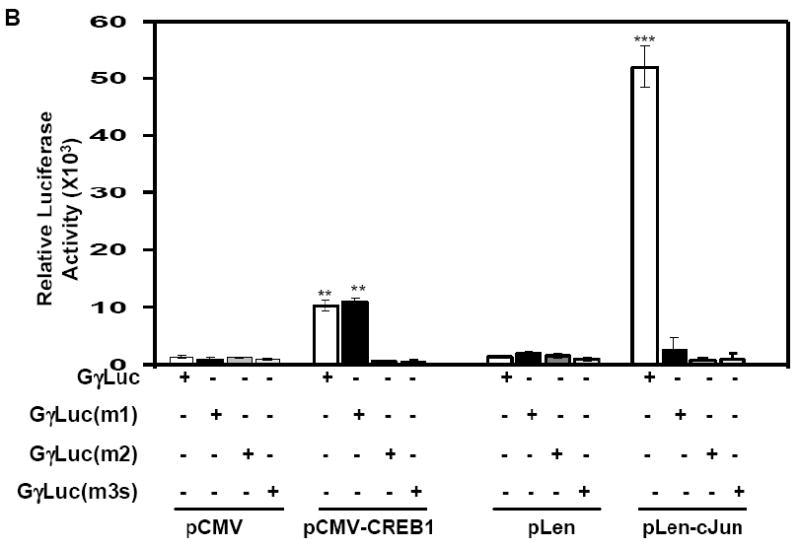
A) Transient transfection assays were performed with wild type (Gγluc) and mutant Gγluc(m1), Gγluc(m2) and Gγluc(m3) reporter constructs in K562 cells (see Materials). B) Co-transfection studies were performed with the same reporter plasmids tested in Panel A and the pLen-cJun or pCMV-CREB1 expression vectors. The corresponding empty vectors pLen and pCMV were used as respective controls.
Stable enforced cJun expression enhances endogenous γ-globin transcription
We established K562 stable lines with the pLen-cJun and pCMV-CREB1 expression vectors to determine if the effects produced by transient transfections could be substantiated for endogenous γ-globin. Enforced cJun expression was confirmed by western blot analysis in three independent stable lines (Fig. 6A). Increased cJun levels were associated with a 2.0-fold (p<0.05) increase in γ/GAPD mRNA compared to empty vector pLen stable lines (Fig. 6B). Likewise enforced CREB1 expression activated γ/GAPD 1.5-fold (p<0.05) compared to empty vector pCMV control lines (Fig. 6C and D). We concluded that cJun and CREB1 have the ability to trans-activate endogenous γ-globin expression in K562 cells independent of drug inductions.
Fig. 6. Stable enforced cJun expression enhances γ-globin transcription.
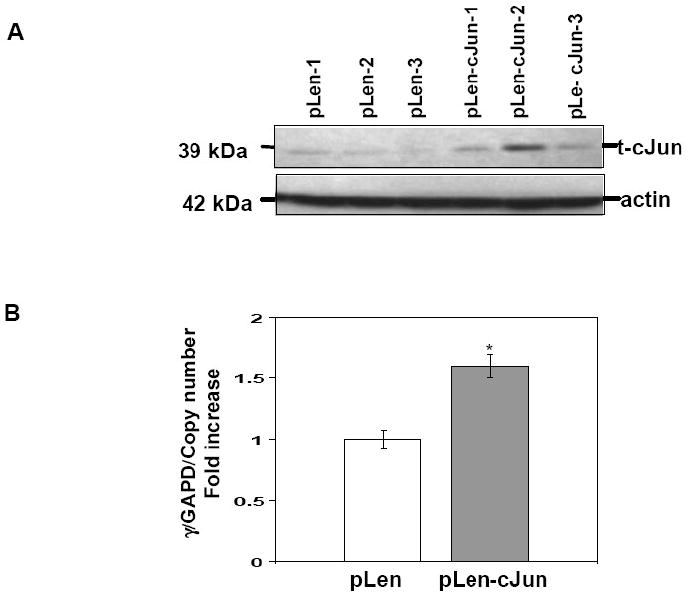
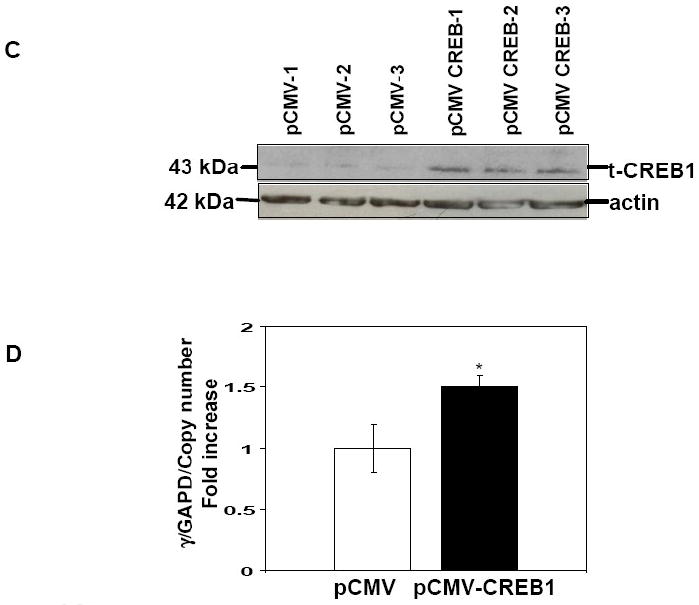
Stable lines were established in K562 cells using the pLen-cJun and pCMV-CREB1 expression vectors and corresponding empty vector controls (see Material). A) Western blot was performed with protein extracts isolated from the three stable lines with t-cJun antibody to confirm stable enforced expression. Membranes were stripped and probed with actin antibody as an internal control. B) The effect of enforced cJun expression on endogenous γ-globin gene expression was measured by qPCR analysis with γ-globin gene specific primers (Table 1). Shown are the γ/GAPD mRNA ratios corrected for pLen-cJun vector copy number. C) Western blot was performed with protein isolated from the three stable lines and t-CREB1 antibody to confirm stable enforced t-CREB1 expression. D) Shown are the γ/GAPD mRNA ratios corrected for pCMV-CREB1 vector copy number.
Enforced cJun expression in primary erythroid cells activates γ-globin transcription
A final set of experiments were completed to test the ability of cJun to stimulate γ-globin in normal erythroid progenitors grown in a two-phase liquid culture system. Progenitors were transfected with pLen-cJun or pCMV-CREB1 on day 11 for 48 hrs and 72 hrs (see Materials). After 72 hrs in culture, enforced cJun expression increased the γ/β mRNA ratio 2-fold (p<0.05) which was comparable to the 2-fold and 1.8 fold increase in γ/β mRNA produced by enforced CREB1 expression at 48 hrs and 72 hrs respectively (Fig. 7). These studies confirm that cJun and CREB1 trans-activate γ-globin in normal erythroid progenitors.
Fig. 7. Enforced cJun and CREB1 expression increase γ-globin transcription in primary erythroid cells.
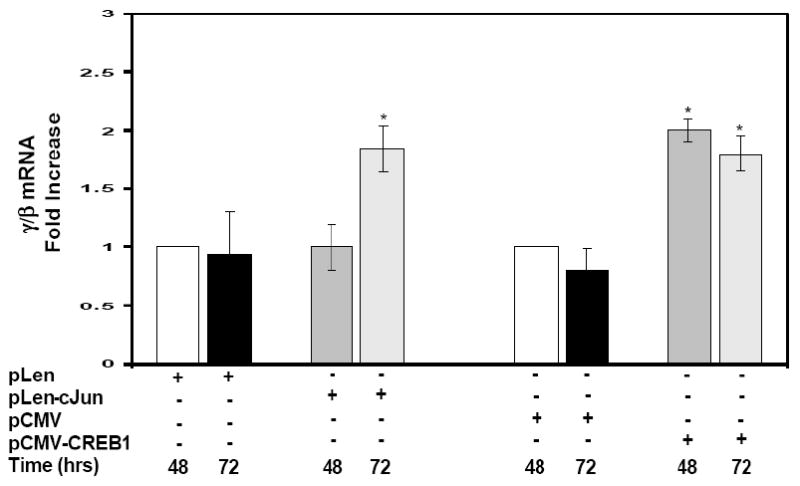
Erythroid progenitors were grown from peripheral blood mononuclear cells in a two-phase liquid culture system (see Materials). Enforced pLen-cJun and pCMV-CREB1 expression was performed on day 11 in culture for 48 hrs and 72 hrs. The empty pLen and pCMV vectors were used as controls for non-specific effects. RNA was isolated at the times indicated for qPCR analysis.
Discussion
The CRE, which contain the conserved palindrome TGACGTGA, and AP-1 phorbol 12-O-tetradecanoate 13 acetate responsive element TGA(C/G)TCA (AP-1/TRE), are two major classes of regulatory elements that control transcription through a variety of extracellular signals [27, 28]. Members of the CREB and ATF family recognize the CRE while the AP-1/TRE is recognized by the AP-1 family members Fos and Jun [10, 28]. Both group of proteins share the basic leucine-zipper motif, but they have discrete transcriptional activities, and interact with distinct DNA-binding proteins that contribute to their regulatory function [27-30].
CREB/ATF and Fos/Jun proteins form selective homo- or heterodimers that bind the CRE and TRE respectively to produce variable gene responses [28, 29, 31, 32]. In general, CREs have enhancer properties in that they regulate transcription in a position- or orientation-independent manner. CREB1 is activated upon phosphorylation at Ser133 by several kinases and interacts with CREB binding protein [33-35], a key molecule involved in communication between DNA-binding proteins and the basal transcriptional machinery [36]. Likewise, Jun family members function as homo- or heterodimers among themselves and members of the Fos and CREB/ATF families [28, 37, 38]. The differences between the CRE and AP-1/TRE consensus sequence is only one nucleotide and thus overlap and/or crosstalk affecting transcription can occur [32, 39].
We previously demonstrated a role for p-CREB1 and p-ATF-2 in Gγ-globin induction by NaB and TSA through p38 MAPK signaling and G-CRE trans-activation [5]. To expand on these findings in the current study we demonstrated that cJun binds the G-CRE region in vivo at steady state. Furthermore, EMSA and promoter pull-down studies confirm co-localization of cJun, CREB1 and ATF-2 on the G-CRE. Our findings in erythroid cells are in agreement with published work in the trophoblast α-subunit [7] and corticotrophin releasing hormone genes [40] where the CRE binds CREB, CREM, ATF-1, ATF-2, Fos and cJun. These data implicate the CRE as a target for gene regulation by multiple signaling pathways including p38 MAPK [41], cAMP [42] and cJun N-terminal kinase [43].
p38 MAPK signaling has been implicated in mechanisms for γ-globin induction by several histone deacetylase inhibitors [3, 5, 41, 44], hydroxyurea [45] and thalidomide more recently [46]. Previous studies also demonstrated a role for cyclic guanosine monophosphate and cAMP in γ-globin gene induction. Kuroyanagi et al. [47] demonstrated that cAMP activation inhibited γ-globin gene expression in K562 cells but induced expression in adult erythroblasts. The c-Myb proto-oncogene was shown to be involved in the cAMP-mediated differential regulation of γ-globin gene expression. However, it is believed that the cJun N-terminal kinase signaling pathway does not play a role in drug-mediated fetal hemoglobin induction [41].
cJun partners with a wide variety of basic leucine-zipper proteins and functions as an oncogene to control cell growth and proliferation [48, 49]. Although cJun/cFos heterodimers bind AP-1/TRE sites, cJun also binds the CRE as a heterodimer with ATF-2 [49]. Our IP data show ATF-2/CREB1 and ATF-2/cJun protein-protein interactions similar to published data however cJun and CREB1 did not form heterodimers in erythroid cells. Hence, we performed molecular studies to elucidate the role of cJun in Gγ-globin gene regulation.
To gain insights into the ability of cJun to trans-activate Gγ-globin, luciferase co-transfection studies were completed. We observed dose-dependent Gγ-promoter activation with enforced cJun expression to a higher degree than produced by CREB1. Interestingly when both factors were combined, an intermediate effect was produced suggesting cJun and CREB1 compete for binding to the G-CRE and/or other binding partners. Mutations in the G-CRE produced a profound effect with almost complete loss of luciferase activity with the -1225 A/G GγLuc(m1) reporter for cJun. Although cJun trans-activation was abolished, CREB1 was able to activate GγLuc(m1) suggesting differences in protein binding affinities. A similar inhibition of CRE function was demonstrated in the mouse, bovine, and rat α-subunit genes [50].
The G-CRE is located between -1650 and -1150 in the Gγ-promoter where multiple GATA-1 and Sp1 sites were identified with functional cis-acting properties [50]. Four PreGγ frameworks were characterized by Pissard and colleagues based on point mutations in the G-CRE (-1225 G/A), -1280 GATA-1 and -1450 TATA binding protein motif. PreGγ framework 1 associated with the G-CRE mutation (G/A) was associated with lower fetal hemoglobin levels in sickle cell patients carrying a Benin β-globin gene cluster haplotype [50]. These data suggest the G-CRE may have a physiologic role in γ-globin gene expression.
Our data support a mechanism of γ-globin activation either by a CREB1/ATF-2 or cJun/ATF-2 heterodimers bound to the G-CRE. The role of AP-1 proteins in globin regulation has been clearly defined by studies of the tandem AP-1/NF-E2 binding site of 5′ hypersensitive site 2 in the β-globin cluster locus control region [13, 51]. This region contains an 18-bp sequence that is inducible in K562 cells [13] and its enhancer activity is mediated by Jun and Fos homodimers, or a NF-E2 protein complex composed of p45 NF-E2 and p18 Maf [52, 53]. Francastel et al. [14] identified cJun as a positive regulator of NF-E2 but when associated with p18 Maf, NF-E2-mediated transcriptional activation was inhibited in mouse erythroleukemia cells [14]. We observed enhanced γ-globin gene expression with enforced cJun expression in K562 stable lines and primary erythroid cells. Whether cJun produces endogenous γ-globin activation via the G-CRE and/or locus control region requires further investigation.
The convergence of multiple signaling pathways is probably required to bring about gene responses via CREs. CREB1/ATF-2, cJun/ATF-2 or homodimer complexes most likely bind the G-CRE to modulate γ-globin expression. Published studies have shown that ATF-2 requires cJun as a nuclear importer and is required to stabilize ATF-2 in the nucleus [54]. Other regulatory factors such as CREB binding protein can facilitate the formation of enhanceosome complexes [55] which have been shown to modulate CREB- and cFos/cJun-mediated transcription of the steroidogenic acute regulatory gene [56]. The data generated herein and published studies allowed us to generate a model for protein interactions in the G-CRE. Our IP data support at least two possible scenarios contingent on the specificity of protein binding to the G-CRE. First, CREB1/CREB1 or CREB1/ATF-2 dimers compete for ATF-2/cJun binding to the G-CRE to dampen γ-globin transcription. Alternatively, adaptor proteins might facilitate formation of a CREB1/ATF-2/cJun enhanceosome complex. CREB binding protein has been shown to interacts with histone acetylating proteins to mediate chromatin remodeling or recruitment of TATA factor IIB to enhance gene transcription [57]. The functional interaction and/or competition between the basic leucine-zipper DNA-binding proteins and the G-CRE could represent crosstalk between various signaling molecules involved in globin gene activation. Molecular approaches such as chromatin conformation capture, sequential ChIP analysis and mass spectrometry will be required to explicate components of the enhanceosome complex and function of the G-CRE in globin gene regulation.
Acknowledgments
This work was supported by grant HL69234 from the National Heart Lung and Blood Institute to and funding through the Center for Applied Biology at the University of Texas at Dallas to Betty S. Pace, MD.
We sincerely thank Xiao Yao, PhD for assistance with the chromatin immunoprecipitation assays and Wei Li, PhD for guidance with primary erythroid cell cultures.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Stamatoyannopoulos G. Control of globin gene expression during development and erythroid differentiation. Exp Hematol. 2005;3:259–71. doi: 10.1016/j.exphem.2004.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chakalova L, Carter D, Debrand E, et al. Developmental regulation of the beta-globin gene locus. Prog Mol Subcell Biol. 2005:183–206. doi: 10.1007/3-540-27310-7_8. [DOI] [PubMed] [Google Scholar]
- 3.Pace BS, Zein S. Understanding mechanisms of gamma-globin gene regulation to develop strategies for pharmacological fetal hemoglobin induction. Dev Dyn. 2006;7:1727–37. doi: 10.1002/dvdy.20802. [DOI] [PubMed] [Google Scholar]
- 4.Mahajan MC, Karmakar S, Weissman SM. Control of beta globin genes. J Cell Biochem. 2007;4:801–10. doi: 10.1002/jcb.21507. [DOI] [PubMed] [Google Scholar]
- 5.Sangerman J, Lee MS, Yao X, et al. Mechanism for fetal hemoglobin induction by histone deacetylase inhibitors involves gamma-globin activation by CREB1 and ATF-2. Blood. 2006;10:3590–9. doi: 10.1182/blood-2006-01-023713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jorgensen JS, Nilson JH. AR suppresses transcription of the alpha glycoprotein hormone subunit gene through protein-protein interactions with cJun and activation transcription factor 2. Mol Endocrinol. 2001;9:1496–504. doi: 10.1210/mend.15.9.0690. [DOI] [PubMed] [Google Scholar]
- 7.Heckert LL, Schultz K, Nilson JH. The cAMP response elements of the alpha subunit gene bind similar proteins in trophoblasts and gonadotropes but have distinct functional sequence requirements. J Biol Chem. 1996;49:31650–6. doi: 10.1074/jbc.271.49.31650. [DOI] [PubMed] [Google Scholar]
- 8.Szabo E, Riffe ME, Steinberg SM, et al. Altered cJUN expression: an early event in human lung carcinogenesis. Cancer Res. 1996;2:305–15. [PubMed] [Google Scholar]
- 9.Chiu R, Boyle WJ, Meek J, et al. The c-Fos protein interacts with c-Jun/AP-1 to stimulate transcription of AP-1 responsive genes. Cell. 1988;4:541–52. doi: 10.1016/0092-8674(88)90076-1. [DOI] [PubMed] [Google Scholar]
- 10.Rauscher FJ, 3rd, Cohen DR, Curran T, et al. Fos-associated protein p39 is the product of the jun proto-oncogene. Science. 1988;4855:1010–6. doi: 10.1126/science.3130660. [DOI] [PubMed] [Google Scholar]
- 11.Madireddi MT, Dent P, Fisher PB. AP-1 and C/EBP transcription factors contribute to mda-7 gene promoter activity during human melanoma differentiation. J Cell Physiol. 2000;1:36–46. doi: 10.1002/1097-4652(200010)185:1<36::AID-JCP3>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 12.Tsai EY, Yie J, Thanos D, Goldfeld AE. Cell-type-specific regulation of the human tumor necrosis factor alpha gene in B cells and T cells by NFATp and ATF-2/JUN. Mol Cell Biol. 1996;10:5232–44. doi: 10.1128/mcb.16.10.5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ney PA, Sorrentino BP, McDonagh KT, Nienhuis AW. Tandem AP-1-binding sites within the human beta-globin dominant control region function as an inducible enhancer in erythroid cells. Genes Dev. 1990;6:993–1006. doi: 10.1101/gad.4.6.993. [DOI] [PubMed] [Google Scholar]
- 14.Francastel C, Augery-Bourget Y, Prenant M, et al. c-Jun inhibits NF-E2 transcriptional activity in association with p18/maf in Friend erythroleukemia cells. Oncogene. 1997;7:873–7. doi: 10.1038/sj.onc.1200902. [DOI] [PubMed] [Google Scholar]
- 15.Adunyah SE, Chander R, Barner VK, et al. Regulation of c-jun mRNA expression by hydroxyurea in human K562 cells during erythroid differentiation. Biochim Biophys Acta. 1995;2:123–32. doi: 10.1016/0167-4781(95)00079-v. [DOI] [PubMed] [Google Scholar]
- 16.Safaya S, Ibrahim A, Rieder RF. Augmentation of gamma-globin gene promoter activity by carboxylic acids and components of the human beta-globin locus control region. Blood. 1994;11:3929–35. [PubMed] [Google Scholar]
- 17.Solomon WB, Lin CH, Palma J, et al. Suppression of a cellular differentiation program by phorbol esters coincides with inhibition of binding of a cell-specific transcription factor (NF-E2) to an enhancer element required for expression of an erythroid-specific gene. J Biol Chem. 1993;7:5089–96. [PubMed] [Google Scholar]
- 18.Huang HM, Liu JC. c-Jun blocks cell differentiation but not growth inhibition or apoptosis of chronic myelogenous leukemia cells induced by STI571 and by histone deacetylase inhibitors. J Cell Physiol. 2009;3:568–74. doi: 10.1002/jcp.21627. [DOI] [PubMed] [Google Scholar]
- 19.Yao X, Kodeboyina S, Liu L, et al. Role of STAT3 and GATA-1 interactions in gamma-globin gene expression. Exp Hematol. 2009;8:889–900. doi: 10.1016/j.exphem.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Samten B, Townsend JC, Weis SE, et al. CREB, ATF, and AP-1 transcription factors regulate IFN-gamma secretion by human T cells in response to mycobacterial antigen. J Immunol. 2008;3:2056–64. doi: 10.4049/jimmunol.181.3.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deng WG, Zhu Y, Montero A, Wu KK. Quantitative analysis of binding of transcription factor complex to biotinylated DNA probe by a streptavidin-agarose pulldown assay. Anal Biochem. 2003;1:12–8. doi: 10.1016/j.ab.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 22.Fibach E, Rachmilewitz EA. The two-step liquid culture: a novel procedure for studying maturation of human normal and pathological erythroid precursors. Stem Cells. 1993:36–41. doi: 10.1002/stem.5530110608. [DOI] [PubMed] [Google Scholar]
- 23.Muchardt C, Li C, Kornuc M, Gaynor R. CREB regulation of cellular cyclic AMP-responsive and adenovirus early promoters. J Virol. 1990;9:4296–305. doi: 10.1128/jvi.64.9.4296-4305.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thiel G, Al Sarraj J, Vinson C, et al. Role of basic region leucine zipper transcription factors cyclic AMP response element binding protein (CREB), CREB2, activating transcription factor 2 and CAAT/enhancer binding protein alpha in cyclic AMP response element-mediated transcription. J Neurochem. 2005;2:321–36. doi: 10.1111/j.1471-4159.2004.02882.x. [DOI] [PubMed] [Google Scholar]
- 25.Benbrook DM, Jones NC. Heterodimer formation between CREB and JUN proteins. Oncogene. 1990;3:295–302. [PubMed] [Google Scholar]
- 26.Benbrook DM, Jones NC. Different binding specificities and transactivation of variant CRE's by CREB complexes. Nucleic Acids Res. 1994;8:1463–9. doi: 10.1093/nar/22.8.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sassone-Corsi P, Ransone LJ, Verma IM. Cross-talk in signal transduction: TPA-inducible factor jun/AP-1 activates cAMP-responsive enhancer elements. Oncogene. 1990;3:427–31. [PubMed] [Google Scholar]
- 28.Hai T, Curran T. Cross-family dimerization of transcription factors Fos/Jun and ATF/CREB alters DNA binding specificity. Proc Natl Acad Sci U S A. 1991;9:3720–4. doi: 10.1073/pnas.88.9.3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dwarki VJ, Montminy M, Verma IM. Both the basic region and the ‘leucine zipper’ domain of the cyclic AMP response element binding (CREB) protein are essential for transcriptional activation. Embo J. 1990;1:225–32. doi: 10.1002/j.1460-2075.1990.tb08099.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Manna PR, Eubank DW, Stocco DM. Assessment of the role of activator protein-1 on transcription of the mouse steroidogenic acute regulatory protein gene. Mol Endocrinol. 2004;3:558–73. doi: 10.1210/me.2003-0223. [DOI] [PubMed] [Google Scholar]
- 31.Millhouse S, Kenny JJ, Quinn PG, et al. ATF/CREB elements in the herpes simplex virus type 1 latency-associated transcript promoter interact with members of the ATF/CREB and AP-1 transcription factor families. J Biomed Sci. 1998;6:451–64. doi: 10.1007/BF02255935. [DOI] [PubMed] [Google Scholar]
- 32.Rutberg SE, Adams TL, Olive M, et al. CRE DNA binding proteins bind to the AP-1 target sequence and suppress AP-1 transcriptional activity in mouse keratinocytes. Oncogene. 1999;8:1569–79. doi: 10.1038/sj.onc.1202463. [DOI] [PubMed] [Google Scholar]
- 33.Chrivia JC, Kwok RP, Lamb N, et al. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;6449:855–9. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- 34.Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999:821–61. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- 35.Richards JS. New signaling pathways for hormones and cyclic adenosine 3′,5′-monophosphate action in endocrine cells. Mol Endocrinol. 2001;2:209–18. doi: 10.1210/mend.15.2.0606. [DOI] [PubMed] [Google Scholar]
- 36.Vo N, Goodman RH. CREB-binding protein and p300 in transcriptional regulation. J Biol Chem. 2001;17:13505–8. doi: 10.1074/jbc.R000025200. [DOI] [PubMed] [Google Scholar]
- 37.Kerppola TK, Curran T. Fos-Jun heterodimers and Jun homodimers bend DNA in opposite orientations: implications for transcription factor cooperativity. Cell. 1991;2:317–26. doi: 10.1016/0092-8674(91)90621-5. [DOI] [PubMed] [Google Scholar]
- 38.O'Shea EK, Rutkowski R, Kim PS. Mechanism of specificity in the Fos-Jun oncoprotein heterodimer. Cell. 1992;4:699–708. doi: 10.1016/0092-8674(92)90145-3. [DOI] [PubMed] [Google Scholar]
- 39.Masquilier D, Sassone-Corsi P. Transcriptional cross-talk: nuclear factors CREM and CREB bind to AP-1 sites and inhibit activation by Jun. J Biol Chem. 1992;31:22460–6. [PubMed] [Google Scholar]
- 40.King BR, Smith R, Nicholson RC. Novel glucocorticoid and cAMP interactions on the CRH gene promoter. Mol Cell Endocrinol. 2002;1-2:19–28. doi: 10.1016/s0303-7207(02)00218-6. [DOI] [PubMed] [Google Scholar]
- 41.Mabaera R, West RJ, Conine SJ, et al. A cell stress signaling model of fetal hemoglobin induction: what doesn't kill red blood cells may make them stronger. Exp Hematol. 2008;9:1057–72. doi: 10.1016/j.exphem.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 42.Roesler WJ, Vandenbark GR, Hanson RW. Cyclic AMP and the induction of eukaryotic gene transcription. J Biol Chem. 1988;19:9063–6. [PubMed] [Google Scholar]
- 43.Roberson MS, Ban M, Zhang T, Mulvaney JM. Role of the cyclic AMP response element binding complex and activation of mitogen-activated protein kinases in synergistic activation of the glycoprotein hormone alpha subunit gene by epidermal growth factor and forskolin. Mol Cell Biol. 2000;10:3331–44. doi: 10.1128/mcb.20.10.3331-3344.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Witt O, Sand K, Pekrun A. Butyrate-induced erythroid differentiation of human K562 leukemia cells involves inhibition of ERK and activation of p38 MAP kinase pathways. Blood. 2000;7:2391–6. [PubMed] [Google Scholar]
- 45.Park JI, Choi HS, Jeong JS, et al. Involvement of p38 kinase in hydroxyurea-induced differentiation of K562 cells. Cell Growth Differ. 2001;9:481–6. [PubMed] [Google Scholar]
- 46.Aerbajinai W, Zhu J, Gao Z, et al. Thalidomide induces gamma-globin gene expression through increased reactive oxygen species-mediated p38 MAPK signaling and histone H4 acetylation in adult erythropoiesis. Blood. 2007;8:2864–71. doi: 10.1182/blood-2007-01-065201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuroyanagi Y, Kaneko Y, Muta K, et al. cAMP differentially regulates gamma-globin gene expression in erythroleukemic cells and primary erythroblasts through c-Myb expression. Biochem Biophys Res Commun. 2006;3:1038–47. doi: 10.1016/j.bbrc.2006.03.203. [DOI] [PubMed] [Google Scholar]
- 48.Chinenov Y, Kerppola TK. Close encounters of many kinds: Fos-Jun interactions that mediate transcription regulatory specificity. Oncogene. 2001;19:2438–52. doi: 10.1038/sj.onc.1204385. [DOI] [PubMed] [Google Scholar]
- 49.van Dam H, Castellazzi M. Distinct roles of Jun: Fos and Jun: ATF dimers in oncogenesis. Oncogene. 2001;19:2453–64. doi: 10.1038/sj.onc.1204239. [DOI] [PubMed] [Google Scholar]
- 50.Pissard S, Beuzard Y. A potential regulatory region for the expression of fetal hemoglobin in sickle cell disease. Blood. 1994;1:331–8. [PubMed] [Google Scholar]
- 51.Sorrentino B, Ney P, Bodine D, Nienhius AW. A 46 base pair enhancer sequence within the locus activating region is required for induced expression of the gamma-globin gene during erythroid differentiation. Nucleic Acids Res. 1990;9:2721–31. doi: 10.1093/nar/18.9.2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Andrews NC, Kotkow KJ, Ney PA, et al. The ubiquitous subunit of erythroid transcription factor NF-E2 is a small basic-leucine zipper protein related to the v-maf oncogene. Proc Natl Acad Sci U S A. 1993;24:11488–92. doi: 10.1073/pnas.90.24.11488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Igarashi K, Kataoka K, Itoh K, et al. Regulation of transcription by dimerization of erythroid factor NF-E2 p45 with small Maf proteins. Nature. 1994;6463:568–72. doi: 10.1038/367568a0. [DOI] [PubMed] [Google Scholar]
- 54.Liu H, Deng X, Shyu YJ, et al. Mutual regulation of c-Jun and ATF2 by transcriptional activation and subcellular localization. Embo J. 2006;5:1058–69. doi: 10.1038/sj.emboj.7601020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hu PP, Harvat BL, Hook SS, et al. c-Jun enhancement of cyclic adenosine 3′,5′-monophosphate response element-dependent transcription induced by transforming growth factor-beta is independent of c-Jun binding to DNA. Mol Endocrinol. 1999;12:2039–48. doi: 10.1210/mend.13.12.0405. [DOI] [PubMed] [Google Scholar]
- 56.Manna PR, Stocco DM. Crosstalk of CREB and Fos/Jun on a single cis-element: transcriptional repression of the steroidogenic acute regulatory protein gene. J Mol Endocrinol. 2007;4:261–77. doi: 10.1677/JME-07-0065. [DOI] [PubMed] [Google Scholar]
- 57.Blobel GA. CREB-binding protein and p300: molecular integrators of hematopoietic transcription. Blood. 2000;3:745–55. [PubMed] [Google Scholar]