

Abstract
Histone deacetylase inhibitors (HDACi) are endowed with plethora of biological functions including anti-proliferative, anti-inflammatory, anti-parasitic, and cognition-enhancing activities. Parsing the structure–activity relationship (SAR) for each disease condition is vital for long-term therapeutic applications of HDACi. We report in the present study specific cap group substitution patterns and spacer-group chain lengths that enhance the antimalarial and antileishmanial activity of aryltriazolylhydroxamates-based HDACi. We identified many compounds that are several folds selectively cytotoxic to the plasmodium parasites compared to standard HDACi. Also, a few of these compounds have antileishmanial activity that rivals that of miltefosine, the only currently available oral agent against visceral leishmaniasis. The anti-parasite properties of several of these compounds tracked well with their anti-HDAC activities. The results presented here provide further evidence on the suitability of HDAC inhibition as a viable therapeutic option to curb infections caused by apicomplexan protozoans and trypanosomatids.
Introduction
Inhibition of histone deacetylase (HDAC) activity has been largely validated as a viable therapeutic strategy for cancer treatment.1 Of the several mechanisms proposed to explain the molecular basis of the anti-proliferative activity of HDAC inhibitors (HDACi), perturbation of chromatin remodeling and acetylation states of key non-histone proteins have been widely accepted.2 Consequently, intense research activities are ongoing on the application of HDACi to other diseases where chromatin remodeling and protein acetylation states may play significant roles.3
Human pathogenic apicomplexan protozoans and trypanosomatids, such as the causative agents of malaria and leishmaniasis respectively, have been shown to be responsive to HDACi.4 Malaria is a serious infectious disease that is prevalent in sub- Saharan Africa and Asia.5 It is caused by members of the genus Plasmodium, with an estimated 500 million infections and 2 million fatalities annually.6 Similarly, leishmaniasis is a tropical/sub-tropical parasitic disease, caused by the members of the genus Leishmania, which infects over 2 million people annually.7 These two diseases constitute an emerging serious threat to public health due to the emergence of multi-drug resistant strains and the occurrence of leishmania as opportunistic infective agents in human immunodeficiency virus-infected patients.8–10 Therefore, there is an urgent need for affordable alternative agents to curtail these diseases.
Plasmodium falciparum, the principal malarial protozoan parasite in humans, has at least five putative HDAC enzymes.11 Genes encoding two of these putative HDAC - PfHDAC-1 and PfSir2 - have been partially characterized.11–13 Because of its homology with the class I family of HDACs from human, chicken, frog and Saccharomyces cerevisiae, PfHDAC-1 may be one of the intracellular targets whose interaction with HDACi elicits the observed antimalarial activity of HDACi.4c, f, g, 11 Similarly, Leishmania parasite’s genome contains multiple genes encoding different HDACs isozymes some of which have been shown to be essential for the survival and proliferation of Leishmania parasites.14–16 However, the structure–activity relationship (SAR) of the antimalarial and antileishmanial activities of HDACi is not entirely clear4g.
Previously, we and others have reported the synthesis and SAR for aryltriazolyl-hydroxamates, HDACi incorporating 1,2,3-triazole into the cap group-linking and surface recognition moieties.17 We showed that these compounds displayed cap group- and spacer-length dependent anti-HDAC and whole cell antiproliferative activities that tracked with the three-motif pharmacophoric model of all HDACi (Fig. 1).17a In this contribution, we sought to clarify the specific structural attributes that confer antimalarial and antileishmanial activities to aryltriazolylhydroxamates HDACi. We found that for a given cap group, HDACi spacer-group length is a major structural determinant of antimalarial and antileishmanial activities. Specifically, the antimalarial and antileishmenial activities of these aryltriazolylhydroxamates generally peak in analogs having 5 or 6 methylene spacer groups separating the active site zinc binding hydroxamate moiety and the aryltriazolyl group.
Figure 1.

(a) A three-motif pharmacophoric model of HDAC inhibitors. (b) Selected examples of small molecule HDAC inhibitors.
Chemistry
Our previous protocol for the synthesis of aryltriazolylhydroxamates required the unmasking of the hydroxamic acid moiety of the desired product from a penultimate ester intermediate by treatment with aqueous hydroxylamine in the presence of a catalytic amount of KCN.17a This reaction sometime gives the carboxylic acid derivative of the desired product as a contaminant, thereby complicating compound isolation. We describe here an alternative route that allows for a facile synthesis of the aryltriazolyl-hydroxamates of interest while avoiding the formation of carboxylic acid contaminants. Reaction of azido acid 1a–c with O-tritylhydroxylamine under standard activation condition gave O-tritylated hydroxamates 2a–c in good to excellent yields. Subsequent Cu (I) catalyzed cycloaddition reaction between 2a–c and terminal alkynes 3a–g resulted in O-trityl protected aryltriazolylhydroxamates 4–14.17a The unmasking of the O-trityl protection group was facilitated by BF3.OEt2 or TFA treatment to furnish the requisite compounds, 19, 20, 23, 24, 26, 27, 34, 42, 43, 47 and 50, all new aryltriazolyl-hydroxamates having longer methylene spacer-groups (n = 6 – 9) and varied HDAC surface recognition cap groups (Scheme 1).
Scheme 1.

Synthesis of aryltriazolylhydroxamates for SAR studies. Conditions: (a) NH2-O-Trityl, IBCF, THF, −15°C, 2h, (b) 3a–g, CuI, Hunig’s base, THF, rt, (c) BF3.OEt2, THF, rt, 20 min or CH2Cl2, TFA/Thioanisole (1:1), 0°C, 3h. Abbreviation: 3-Bp, 3-biphenyl; DMA, p-N,N-dimethylanilyl; Nap, 6-methoxynapthyl; Ph, phenyl; 3-Py, 3-pyridyl; PyP, 4-pyridylphenyl; 2-Tol, 2-tolyl.
Results and Discussion
In vitro assays
Our goal in the present study is to elucidate the structural determinants that confer antimalarial and antileishmanial activities to aryltriazolylhydroxamates HDACi. To complement our previous studies on the anti-HDAC activities of this class of compounds and also establish a SAR of their anti-protozoan activities, we synthesized additional aryltriazolylhydroxamates having longer methylene spacer-groups (n = 6 – 9) and varied HDAC surface recognition cap groups. We first evaluated the anti-HDAC activity of the new compounds using the Fluor de Lys assay18 as described previously.17a, 19a
All aryltriazolylhydroxamates were tested for their ability to induce in vitro inhibition of the proliferation of chloroquine-sensitive (D6, Sierra Leone) and chloroquine-resistant (W2, Indochina) strains of P. falciparum. The in vitro antileishmanial activities of compounds were tested against the promastigote stage of L. donovani, the causative agent of visceral leishmaniasis. Plasmodium growth inhibition was determined by a parasite lactate dehydrogenase assay using Malstat reagent,20 while inhibition of viability of the promastigote stage of L. donovani was determined using standard Alamar blue assay, modified to a fluorometric assay.21 Amphotericin B and pentamidine, standard antileishmanial agents; chloroquine and artemisinin, standard antimalarials; and suberoylanilide hydroxamic acid (SAHA) and trichostatin A (TSA), stardard HDACi were all used as positive controls. To determine selective toxicity index, all compounds were simultaneously tested against a non-transformed mammalian cell line namely, monkey kidney epithilial (Vero) using Neutral Red assay.22
Structure–Activity Relationship
The HDAC inhibition profiles of the newly synthesized aryltriazolylhydroxamates showed a trend that paralleled those of the previously reported compounds.17a In general; anti-HDAC activities are dependent on two key features of these molecules, namely - the length of the methylene spacer-group (n) and the identity of the cap group. Again, for a given cap group, the optimum n is either 5 or 6 (Table 1).
Table 1.
In Vitro HDAC Inhibition (nM), Antilieshmanial (μg/mL) and Antimalarial (ng/mL) Activities of Aryltriazolylhydroxamates HDACi.
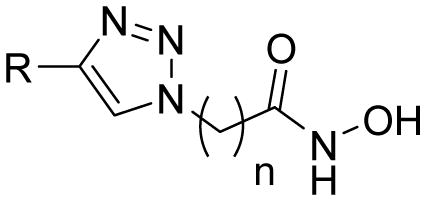 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compd | R | n | HDAC Inhibition IC50 (nM) | Antileshmanial Activity c | Antimalarial Activity c | Cytotoxicity (VERO) IC50 (ng/ml) | S. I. D6 (W2) | ||
| IC50 (μg/ml) ±SD | IC90 (μg/ml) ±SD | Plasmodium falciparum (D6 clone) IC50 (ng/ml) | Plasmodium falciparum (W2 clone) IC50 (ng/ml) | ||||||
| 15 | 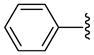 |
3 | N.D. a | NA | NA | 2850 ±50 | 3500 ±800 | NC | >1.7 (>1.4) |
| 16 | 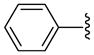 |
4 | 110.0 a | NA | NA | 125 ±5 | 120 ±10 | NC | >38.08 (>39.6) |
| 17 | 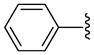 |
5 | 14.2 a | 17.5 ±0.71 | >40 | 88 ±2 | 130 ±30 | 3950 ±550 | 44.9 (30.4) |
| 18 | 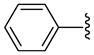 |
6 | 9.6 a | 4.55 ±0.35 | 40.0 ±0.0 | 38 ±5 | 24.5 ±1.5 | 430 ±20 | 11.3 (17.6) |
| 19 | 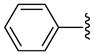 |
7 | 363.6 | 17.75 ±0.35 | >40 | 645 ±55 | 695 ±115 | NC | >7.4 (>6.8) |
| 20 | 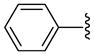 |
8 | 55.4 | 18.5 ±0.71 | >40 | 2650 ±450 | 2750 ±50 | 3350 ±550 | 1.9 (1.2) |
| 21 | 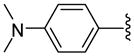 |
5 | 4.3 a | 15.5 ±0.71 | >40 | 68 ±8 | 38.5 ±1.5 | 3400 ±200 | 50.0 (88.3) |
| 22 | 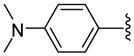 |
6 | 106.1 a | 5.35 ±0.35 | 36.0 ±1.41 | 24.5 ±5.5 | 32 ±13 | 765 ±35 | 31.2 (23.9) |
| 23 | 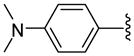 |
7 | 15.9 | 24.0 ±2.83 | >40 | 175 ±105 | 185 ±15 | 970 ±130 | 3.8 (3.6) |
| 24 | 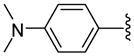 |
8 | 596.6 | 20.75 ±0.35 | >40 | 1800 ±500 | 1650 ±50 | NC | >2.6 (>2.9) |
| 25 | 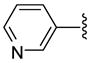 |
5 | 287.2 a | 11.45 ±3.61 | >40 | 175 ±45 | 205 ±55 | NC | >27.2 (>23.2) |
| 26 | 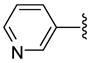 |
6 | 365.8 | 15.5 ±0.71 | 40 | 75.5 ±21.5 | 81 ±49 | 3780 ±980 | 50.1 (46.7) |
| 27 | 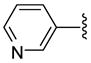 |
7 | 425.8 | 19.75 ±0.35 | >40 | 1495 ± 253 | 1687± 321 | NC | >3.2 (>2.8) |
| 28 | 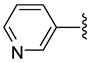 |
5 | 112.5 a | 17.75 ±0.35 | >40 | 195 ±65 | 132.5 ±57.5 | NC | >24.4 (>35.9) |
| 29 | 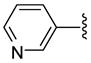 |
5 | 67.6 a | 20.5 ±0.71 | >40 | 985 ±15 | 625 ±95 | 2900 ±300 | 2.9 (4.6) |
| 30 | 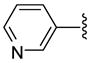 |
6 | 23.9 a | 5.75 ±0.35 | 40.0 ±0.0 | 69 ±1 | 40 ±5 | 950 ±150 | 13.8 (23.8) |
| 31 | 5 | 43.4 a | 21.5 ±2.12 | >40 | 130 ±40 | 140 ±40 | 3850 ±650 | 29.6 (27.5) | |
| 32 | 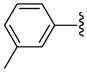 |
5 | 31.9 a | 17.0 ±1.41 | >40 | 79.5 ±0.5 | 68 ±8 | 2700 ±300 | 34.0 (39.7) |
| 33 | 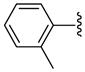 |
5 | 17.4 a | 23.0 ±1.41 | >40 | 1250 ±50 | 1950 ±750 | 3300 ±700 | 2.6 (1.7) |
| 34 | 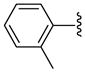 |
6 | 2.8 | 26.0 ±0.0 | 40 | 120 ±20 | 140 ±20 | 710 ±210 | 5.9 (5.1) |
| 35 | 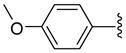 |
5 | 2.1 a | 18.5 ±0.71 | >40 | 130 ±6 | 90 ±10 | 2350 ±850 | 18.1 (26.1) |
| 36 | 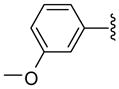 |
5 | 13.9 a | NA | NA | NA | NA | NC | - |
| 37 | 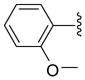 |
5 | 76.0 a | 18.75 ±0.35 | >40 | 150 ±10 | 97.5 ±2.5 | 2300 ±500 | 15.3 (23.6) |
| 38 | 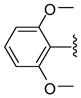 |
5 | 315.9 a | 40 | >40 | 2000 ±300 | 2600 ±200 | NC | >2.4 (>1.8) |
| 39 | 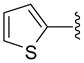 |
5 | 31.7 a | 11.5 ±0.71 | >40 | 46 ±6 | 36.5 ±13.5 | NC | >103 (>130) |
| 40 | 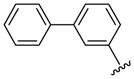 |
5 | 1.9 a | 11.25 ±0.35 | 32.5 ±0.71 | 37.5 ±17.5 | 26 ±4 | NC | >127 (>183.1) |
| 41 | 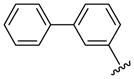 |
6 | 5.4 a | 20.5 ±0.71 | >40 | 25 ±5 | 25 ±5 | NC | >190.4 (>190.4) |
| 42 | 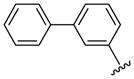 |
7 | 108.9 | 20.25 ±0.35 | 40 | 840 ±40 | 650 ±50 | NC | >5.6 (>7.3) |
| 43 | 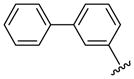 |
8 | 147.7 | 20.0 ±0.0 | 40.0 ±0.0 | 3350 ±150 | 3200 ±200 | NC | >1.4 (>1.5) |
| 44 | 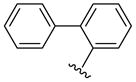 |
5 | 162.6 a | 13.0 ±2.83 | 38.0 ±0.0 | 2100 ±800 | 2600 ±100 | 2700 ±200 | 1.3 (1.0) |
| 45 | 5 | 2.3 a | 28.0 ±2.83 | >40 | 45.5 ±9.5 | 47.5 ±22.5 | NC | >104.6 (>100.2) | |
| 46 | 6 | 16.6 a | NA | NA | 1150 ±150 | 1250 ±50 | NC | >4.1 (>3.8) | |
| 47 | 7 | 212.9 | NA | NA | 88.5 ±6.5 | 127.5 ±2.5 | 3850 ±650 | 43.5 (30.2) | |
| 48 | 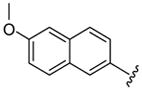 |
5 | 1.8 a | 18.0 ±2.83 | >40 | 35 ±5 | 31 ±11 | 2850 ±50 | >81.4 (>91.9) |
| 49 | 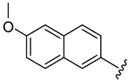 |
6 | 15.3 a | 18.5 ±2.12 | >40 | 47.5 ±4.5 | 53 ±13 | 2650 ±350 | 55.8 (50.0) |
| 50 |  |
7 | 226.1 | NA | NA | 112 ±13 | 165 ±35 | 650 ±250 | 5.8 (3.9) |
| 51 | 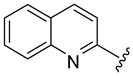 |
5 | 2.1 a | 9.5 ±0.71 | 39.0 ±1.41 | 33 ±3 | 34 ±16 | 2400 ±400 | 72.7 (70.6) |
| 52 | 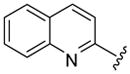 |
5 | 151.5 a | 18.5 ±0.71 | >40 | 52.5 ±12.5 | 33 ±3 | NC | >90.7 (>144.2) |
| Chloroquine | - | NT | NT | NT | 17 | 125 | NT | NT | |
| Artemisinin | - | NT | NT | NT | 4 | 6 | NT | NT | |
| Pentamidine | - | NT | 0.90 ±0.9 | 1.80 ±0.0 | NT | NT | NT | NT | |
| Amphotericine B | - | NT | 0.14 ±0.01 | 0.31 ±0.01 | NT | NT | NT | NT | |
| SAHA | - | 65 | 29.7 ±5.4 | 57.9± 6.8 | 261.6 ± 39.5 | 478.9± 33.4 | 1200 | 4.8 (2.5) | |
| TSA | - | 5 | 1.37± 0.16 | 20.5 ±0.71 | 41.2 ± 6.1 | 49.5 ± 7.4 | 95 | 2.6 (2.2) | |
Each value is obtained from three independent experiments
Cited from ref. 17
Values are mean ± SD of triplicates NA = Not active up to the highest concentration tested NC = No cytotoxic up to highest concentration tested NT = Not tested
The antiprotozoan activities of several of these aryltriazolylhydroxamates are also dependent on the two structural features described above. For example, homologous compounds 15–20, derived from unsubstituted phenyl ring, have spacer length-dependent antimalarial activities that peaked when n = 6 (i.e. 6 methylene spacers separating the triazole ring from the zinc binding hydroxamic acid group) (Table 1). This antimalarial trend is in close agreement with the anti-HDAC activities of these compounds against HDACs 1 and 2 from HeLa cell nuclear fraction.17a Additionally, a similar spacer-length restriction in antimalarial activity has been recently observed for acyl arylhydrazone based compounds.4f Introduction of a N,N-dimethylamino moiety to the para position of the cap group, similar to the substitution pattern on TSA, had no effect on the methylene spacer length dependence of the antimalarial activities. However, these N,N-dimethylamino compounds have a relatively relaxed preference for either 5 or 6 methylene spacers (Table 1, comparing compounds 21 and 22). Incorporation of nitrogen into the phenyl ring did not improve the potency of the simple phenyl-substituted compounds. Nevertheless, the resulting pyridine derivatives 25–30 possessed antimalarial activity profile that was dependent on the ring location of the nitrogen atom. For compounds with 5 methylene spacers, the antimalarial activity was intolerant of nitrogen substitution at the ortho position. This observation is contrary to the trend of the anti-HDAC activities of these pyridyl compounds. However, the ortho N-substitution intolerance was relieved with one additional methylene group (Table 1, comparing compounds 29 and 30). For compounds with the same pyridyl group, we observed spacer length-dependent antimalarial activities in similar manner to those of the unsubstituted phenyl analogs.
Other aryl group substitutions resulted in distinct antimalarial activities that may give further indications of the electronic and steric environment of the intracellular drug target(s) within the parasite. Methyl substituted compounds 31–33 (n = 5) showed a ring meta-position preference despite anti-HDAC activity that favored a ring ortho-position substitution. Extension of the linker length by one methylene group drastically relieved the ortho-substitution intolerance of these series of methyl substituted compounds (Table 1, comparing compounds 33 and 34). Conversely, meta-substitution with a stronger electron donating methoxy moiety (relative to the methyl group) eliminated anti-parasite activity while there was no clear preference between ortho- and para-substitution (Table 1, comparing compounds 35–37). This lack of preference is surprising considering the fact that these compounds had anti-HDAC activity that was about forty-fold in favor of the para-methoxy substitution (Table 1, comparing compounds 35 with 37). However, bisortho methoxy-substitution, incorporated into compound 38, led to a reduction of the antimalarial activity, an observation that may suggest that the parasite’s intracellular target(s) may have similar steric constraints as the mammalian HDAC1.
Larger cap groups such as biphenyls, naphthalenes and quinolines also furnished compounds with potent antimalarial activities. We observed that this class of compounds displayed spacer length-dependent antimalarial activities in similar manner to their simple aryl counterparts. Moreover, compounds’ antimalarial potency was dependent on the substitution pattern on these larger cap group moieties. Similar to their anti-HDAC activities, the biphenyl compounds 40–47 displayed varying antimalarial activities that were dependent on the relative position of the triazole ring.17 Specifically, the meta-placement of the triazole ring was about 65–100 fold preferred over an ortho-substitution by either strains of P. falciparum (Table 1, comparing compounds 40 and 44). While there was no strong preference for either 5 or 6 methylene spacer group among the meta-biphenyl compounds 40 and 41, 5 methylene-linked 4(4-pyridyl) compound 45 was surprisingly 25-times more potent compared to the 6 methylene congener 46. The antimalarial activities of the 5 and 6 methylene spacer, six-six fused ring naphthalenes 48–49 and quinolines 51–52 were virtually indistinguishable. These compounds showed potent antimalarial activities with IC50 values that were comparable to those of TSA and 5- to 15-fold lower than those of SAHA against either strain of P. falciparum (Table 1).
We also investigated the effects of these aryltriazolylhydroxamates on the viability of the promastigote stage of L. donovani and found that they have modest to moderate in vitro antileishmanial activities. Similar to their antimalarial effects, the antileishmanial activities of these compounds were generally maximal in analogs with 5 or 6 methylene spacer groups. Worthy of specific note are compounds 18, 22, 30, 39, 40 and 51 which inhibited the proliferation of the promastigote stage of L. donovani with IC50 values that were 2- to 4-fold higher than that of SAHA and comparable to that of miltefosine, the only clinically approved oral drug against visceral leishmaniasis.23, 24 It is imperative to point out that the antileishmanial activities of some of these compounds may be primarily due to their general cytotoxicity at the measured IC50 values, in similar manner to that of SAHA.
Finally, several of the compounds that potently inhibited parasite proliferation were found to be less cytotoxic to normal mammalian Vero cells. For example, eighteen compounds – 16, 17, 21, 22, 25, 26, 28, 31, 32, 39–41, 45, 47–49, 51 and 52 - were several folds selectively toxic to plasmodium parasite compared to the two standard HDAC inhibitors - SAHA and TSA (Table 1).
Conclusion
We have identified in the present study specific cap group substitution patterns and spacer-group chain lengths that enhanced the antimalarial and antileishmanial activities to aryltriazolylhydroxamates HDACi. The anti-parasite properties of several of these compounds tracked well with their anti-HDAC activities. Many of the compounds herein disclosed are several folds selectively cytotoxic to the Plasmodium parasite compared to standard HDACi. Also, a few of these compounds have antileishmanial activities that rivaled the only currently available oral agent against visceral leishmaniasis. Our results provide additional evidence on the suitability of HDAC inhibition as a viable therapeutic approach to curtail infections caused by apicomplexan protozoans and trypanosomatids. The remarkable and selective anti-parasitic properties of several of these aryltriazolylhydroxamates warrant further investigation. A few lead compounds are being advanced further for in vivo evaluation in murine malarial models.
Experimental
General
ω-Bromoalkanoic acids and 7-bromoheptane nitrile were purchased from Sigma–Aldrich. Anhydrous solvents and other reagents were purchased and used without further purification. Analtech silica gel plates (60 F254) were used for analytical TLC, and Analtech preparative TLC plates (UV 254, 2000 μm) were used for purification. UV light was used to examine the spots. Silica gel (200–400 Mesh) was used in column chromatography. NMR spectra were recorded on a Varian-Gemini 400 magnetic resonance spectrometer. 1H NMR spectra were recorded in parts per million (ppm) relative to the peak of CDCl3, (7.24 ppm), CD3OD (3.31 ppm), or DMSO-d6 (2.49 ppm). 13C spectra were recorded relative to the central peak of the CDCl3 triplet (77.0 ppm), CD3OD (49.0 ppm), or the DMSO-d6 septet (39.7 ppm), and were recorded with complete heterodecoupling. Multiplicities are described using the abbreviation s, singlet; d, doublet, t, triplet; q, quartet; m, multiplet; and app, apparent. High-resolution mass spectra were recorded at the Georgia Institute of Technology mass spectrometry facility in Atlanta. Melting points (uncorrected) were recorded on a Mel-Temp II apparatus. 7-Azidoheptanoic acid 1a was synthesized starting from 7-bromoheptane nitrile while 8-azidooctanoic acid 1b and 9-azidononanoic acid 1c were synthesized from the corresponding w-bromo acids as described previously.19a–c
Analogue synthesis
Representative Procedure for Conversion of ω-Azidoalkanoic acid to O-Trityl protected Hydroxamates. 7-Azido-O-tritylheptahydroxamate (2a)
7-azidoheptanoic acid 1a (434 mg, 2.54 mmol) was dissolved in anhydrous THF. N-methylmorpholine (257 mg, 2.54 mmol) was added to the solution. The reaction mixture was then cooled down to −15 °C and stirred for 5 min. Isobutylchloroformate (346 mg, 2.55 mmol) was added and the mixture was stirred for 10 min at −15 °C. O-tritylhydroxylamine (700 mg, 2.55 mmol) was added followed by 2 more equivalents of N-methylmorpholine. Stirring continued for 15 min at −15 °C and 2 h at room temperature. Afterwards the mixture was poured into 2M HCl and extracted 3 times in each case with water, sodium bicarbonate solution (5 %) and water. After washing with brine and drying over Na2SO4, solvent was evaporated in vacuo. Column chromatography using 25 % EtoAc in Hexanes as the eluent system gave compound 2 837 mg (77%) as a white solid. 1H NMR (CDCl3, 400 MHz) δ 0.91–0.99 (2H, m), 1.11–1.21 (4H, m), 1.44 (2H, p, J = 7.6 Hz), 1.77 (2H, t, J = 7.2 Hz), 3.24 (2H, t, J = 6.8 Hz), 7.29–7.38 (15H, m); 13C NMR (CDCl3, 100 MHz) δ 23.0, 26.1, 28.3, 30.8, 51.0, 93.0, 127.3, 127.9, 128.8, 140.9, 176.9.
8-Azido-O-trityloctahydroxamate (2b)
Reaction of 8-azidooctanoic acid 1b (1.71 g, 9.21 mmol) and O-tritylhydroxylamine (2.55 g, 9.27 mmol) as described for the synthesis of 2a, followed by flash chromatography (eluent 2:1 hexanes/EtOAc) gave 2.59 g (88%) of 2b as a white solid. 1H NMR (CDCl3, 400MHz) δ 0.88–1.39 (8H, m), 1.39–1.54 (4H, m), 3.12 (2H, t, J = 6.9 Hz), 7.10–7.49 (15H, m), 7.67 (1H, s); 13C NMR (CDCl3, 100 MHz) δ 23.2, 24.9, 26.4, 28.7, 31.1, 33.2, 51.3, 93.1, 127.1, 128.0, 128.9, 141.0, 141.8, 146.8, 177.1.
9-Azido-O-tritylnonahydroxamate (2c)
Reaction of 9-azidononanoic acid 1c (724 mg, 3.63 mmol) and O-tritylhydroxylamine (1.00 g, 3.63 mmol) overnight as described for the synthesis of 2a, followed by flash chromatography (eluent 2:1 hexanes/EtOAc) gave 940 mg (56%) of 2c as a sticky white solid. 1H NMR (CDCl3, 400MHz) δ 1.26 (10H, m), 1.57 (4H, m), 3.24 (2H, t, J = 6.8 Hz), 7.34 (15H, m), 7.74 (1H, s).
Representative Procedure for Cu(I)-catalyzed Cycloaddition Reaction. O-Trityl-3-pyridyltriazolylheptahydroxamate (8)
7-Azido-O-tritylheptahydroxamate 2a (207 mg, 0.48 mmol) and 3-ethynylpyridine (50 mg, 0.48 mmol) were dissolved in anhydrous THF (10 mL) and stirred under argon at room temperature. Copper (I) iodide (9 mg, 0.05 mmol) and Hunig’s base (0.1 mL) were then added to the reaction mixture, and stirring continued for 24 h. The reaction mixture was diluted with CH2Cl2 (30 mL) and washed with 1:4 NH4OH/saturated NH4Cl (3 × 30 mL) and saturated NH4Cl (30 mL). The organic layer was dried over Na2SO4 and concentrated in vacuo. The crude product was purified by flash chromatography (gradient 3:1 CH2Cl2:Acetone, then CH2Cl2:MeOH (5%)) to give 206 mg (81 %) of 8 as a white solid. 1H NMR (DMSO-d6, 400 MHz) δ 0.94–1.01 (2H, m), 1.06–1.19 (4H, m), 1.71–1.78 (4H, m), 3.59 (1H, s), 4.34 (2H, t, J = 6.8 Hz), 7.25–7.36 (16H, m), 7.44–7.47 (1H, m), 8.07 (1H, s), 8.17–8.20 (1H, m), 8.51–8.52 (1H, m), 8.68 (1H, s), 9.03–9.04 (1H, m), 10.16 (1H, s); 13C NMR (CDCl3, 100 MHz) δ 25.8, 29.7, 30.7, 46.9, 50.1, 53.8, 119.8, 123.5, 126.6, 127.7, 128.4, 128.8, 132.7, 144.0, 144.4, 146.7, 148.9, 176.8.
O-Tritylphenyltriazolyloctahydroxamate (4)
Reaction of 8-azido-O-trityloctahydroxamate 2b (150 mg, 0.34 mmol) and phenylacetylene (35.9 mg, 0.35 mmol) as described for the synthesis of 8, followed by purification using preperative TLC (eluent 1:1 hexanes/EtOAc) gave 90 mg (48.6%) of 4 as a colorless solid. 1H NMR (CDCl3, 400 MHz) δ 0.94–1.12 (2H, m), 1.12–1.46 (6H, m), 1.49–1.65 (2H, m), 1.75–1.97 (2H, m), 4.33 (2H, t, J = 7.3 Hz), 7.17–7.64 (18H, m), 7.72 (1H, s), 7.75 (1H, s), 7.81 (2H, d, J = 7.5); 13C NMR (CDCl3, 100 MHz) δ 23.2, 24.7, 26.1, 28.7, 31.0, 33.1, 50.3, 93.0, 119.3, 125.7, 128.0, 128.9, 130.6, 141.0, 141.7, 147.7, 178.2.
O-Tritylphenyltriazolylnonahydroxamate (5)
Reaction of 9-azido-O-tritylnonahydroxamate 2c (80.4 mg, 0.18 mmol) and phenylacetylene (23.5 mg, 0.23 mmol) as described for the synthesis of 8, followed by purification using preperative TLC (eluent 11:10 hexanes/EtOAc) gave 72.0 mg (73.2%) of 5 as a sticky white solid. 1H NMR (CDCl3, 400MHz) δ 0.88–1.44 (12H, m), 1.89 (2H, m), 4.34 (2H, t, J = 7.1 Hz), 7.15–7.6 (18H, m), 7.72 (1H, s), 7.79–7.81 (2H, d, J = 8 Hz); 13C NMR (CDCl3, 100MHz) δ 23.6, 26.6, 28.9, 29.2, 30.5, 31.2, 31.4, 50.6, 119.7, 125.9, 128.3, 129.0, 129.3, 141.3, 147.9, 177.5.
O-Trityl-p-N,N-dimethylanilyltriazolyloctahydroxamate (6)
Reaction of 8-azido-O-trityloctahydroxamate 2b (514 mg, 1.16 mmol) and 4-ethynyl-N,N-dimethylanaline (169 mg, 1.16 mmol) as described for the synthesis of 8, followed by purification using preperative TLC (eluent 8:1 CH2Cl2/Acetone) gave 560 mg (82.1%) of 6 as a white solid. 1H NMR (CD3OD, 400 MHz) δ 0.93–1.06 (2H, m), 1.11–1.36 (6H, m), 1.76–1.92 (4H, m), 2.92 (6H, s), 4.31 (2H, t, J = 7.0 Hz), 6.76 (2H, d, J = 9.0 Hz), 7.18–7.43 (15H, m), 7.62 (2H, d, J = 9.0 Hz), 8.05 (1H, s); 13C NMR (CD3OD, 100MHz) δ 24.5, 26.2, 27.3, 29.6, 31.1, 33.4, 40.8, 51.3, 94.3, 113.8, 119.8, 120.5, 127.6, 128.6, 130.4, 143.6, 149.4, 152.1, 173.2.
O-Trityl-p-N,N-dimethylanilyltriazolylnonahydroxamate (7)
Reaction of 9-azido-O-tritylnonahydroxamate 2c (100 mg, 0.22 mmol) and 4-ethynyl-N,N-dimethylanaline (49 mg, 0.34 mmol) as described for the synthesis of 8, followed by purification using preperative TLC (eluent 40:1:0.1 CH2Cl2:MeOH:NH4OH) gave 72.5 mg (55%) of 7 as a white solid. 1H NMR (CDCl3, 400MHz) δ 1.04–1.60 (12H, m), 1.88 (2H, m), 4.32 (2H, t, J = 7.2 Hz), 6.74–6.76 (2H, d, J = 8.8 Hz), 7.25–7.53 (16H, m), 7.58 (1H, s), 7.66–7.69 (2H, d, J = 8.8 Hz); 13C NMR (CDCl3, 100MHz) δ 23.6, 26.6, 29.0, 29.2, 29.9, 30.5, 31.4, 40.7, 50.5, 112.7, 118.2, 119.2, 126.9, 127.0, 128.3, 129.3, 141.3, 142.1, 148.4, 149.5, 150.6, 177.5
O-Trityl-3-pyridyltriazolyloctahydroxamate (9)
Reaction of 8-azido-O-trityloctahydroxamate 2b (165 mg, 0.37 mmol) and 3-ethynylpyridine (38.3 mg, 0.37 mmol) as described for the synthesis of 8, followed by purification using preperative TLC (eluent 5:3 CH2Cl2:Acetone) gave 170 mg (83.3%) of 9 as a white solid. 1H NMR (CD3OD, 400 MHz) δ 0.90–1.06 (2H, m), 1.09–1.38 (6H, m), 1.77–2.00 (4H, m), 4.38 (2H, t, J = 7.3 Hz), 7.10–7.59 (15H, m), 8.22 (1H, d, J = 8.3 Hz), 8.44 (1H, s), 8.46 (1H, d, J = 4.8 Hz), 8.99 (1H, d, J = 2.3 Hz); 13C NMR (CD3OD, 100 MHz) δ 24.5, 26.2, 27.2, 29.6, 31.1, 33.4, 51.6, 94.2, 123.0, 125.6, 128.7, 130.4, 134.8, 143.5, 145.2, 147.2, 149.5, 173.0.
O-Trityl-2-tolyltriazolylheptahydroxamate (10)
Reaction of 7-azido-O-tritylheptahydroxamate 2a (147 mg, 0.34 mmol) and 2-ethynyltoluene (40 mg, 0.34 mmol) as described for synthesis of 8, followed by flash chromatography (gradient CH2Cl2:Acetone 7:1, 6:1, 5:1) gave 160 mg (85 %) of compound 10 as white solid.. 1H NMR (DMSO-d6, 400 MHz) δ 0.94–1.01 (2H, m), 1.07–1.19 (4H, m), 1.72–1.79 (4H, m), 2.40 (3H, s), 4.33 (2H, t, J = 7.2 Hz), 7.22–7.30 (17H, m), 7.70–7.74 (1H, m), 8.35 (1H, s), 10.16 (1H, s); 13C NMR (DMSO-d6, 100 MHz) δ 21.1, 25.5, 27.6, 29.5, 31.8, 49.3, 54.8, 91.6, 123.0, 125.9, 127.3, 127.4, 127.6, 128.1, 128.9, 130.1, 130.8, 134.8, 142.4, 145.4, 170.2.
O-Trityl-3-biphenyltriazolyloctahydroxamate (11)
Reaction of 8-azido-O-trityloctahydroxamate 2b (137 mg, 0.31 mmol) and 1,1′-biphenyl-3-ethynyl (60 mg, 0.34 mmol) as described for the synthesis of 8, followed by purification using preperative TLC (eluent 18:1 CH2Cl2/Acetone) gave 170 mg (88.5%) of 11 as a white solid. 1H NMR (CDCl3, 400 MHz) δ 0.92–1.12 (2H, m), 1.12–1.47 (6H, m), 1.75–1.97 (4H, m), 4.34 (2H, t, J = 7.1 Hz), 7.20–7.39 (13H, m), 7.40–7.52 (5H, m), 7.54 (2H, d, J = 7.7 Hz), 7.64 (2H, d, J = 7.4 Hz), 7.77 (1H, s), 7.78 (2H, m), 8.06 (1H, s); 13C NMR (CDCl3, 100MHz) δ 23.2, 24.9, 26.1, 28.5, 30.9, 33.0, 50.4, 93.0, 119.6, 124.5, 126.8, 127.1, 127.4, 128.1, 128.7, 129.0, 129.2, 131.1, 140.7, 141.0, 141.7, 147.6, 177.4.
O-Trityl-3-biphenyltriazolylnonahydroxamate (12)
Reaction of 9-azido-O-tritylnonahydroxamate 2c (130 mg, 0.29 mmol) and 1,1′-biphenyl-3-ethynyl (65 mg, 0.37 mmol) as described for the synthesis of 8, followed by purification using preperative TLC (eluent 30:1 CH2Cl2/MeOH) gave 144.2 mg (81.5%) of 11 as a red-brown solid. 1H NMR (CDCl3, 400MHz) δ 0.9–1.2 (6H, m), 1.21–1.42 (6H, m), 1.86 (2H, m), 4.31 (2H, m), 7.16–7.47 (19H, m), 7.51 (1H, d, J = 8.0 Hz), 7.61 (2H, d, J = 7.2 Hz), 7.76 (3H, m), 8.05 (1H, s); 13C NMR (CDCl3, 100MHz) δ 23.6, 26.6, 29.0, 29.2, 30.0, 30.5, 31.4, 50.6, 119.9, 124.7, 124.8, 127.1, 127.4, 128.1, 128.2, 128.4, 129.0, 129.3, 129.5, 131.5, 141.0, 142.0, 147.8, 177.6.
O-Trityl-4-pyridylphenyltriazolyloctahydroxamate (13)
Reaction of 8-azido-O-trityloctahydroxamate 2b (150 mg, 0.34 mmol) and 4-(4-ethynylphenyl)-pyridine (61 mg, 0.34 mmol) as described for synthesis of 8, followed by purification using preparative TLC (eluent 10:1 Et2O:EtOH) gave 133 mg (63 %) of compound 13 as white solid. 1H NMR (DMSO-d6, 400 MHz) δ 0.86–0.92 (2H, m), 1.10–1.20 (6H, m), 1.73–1.81 (4H, m), 4.37 (2H, t, J = 6.8 Hz), 7.26–7.34 (15H, m), 7.74 (2H, d, J = 6.0 Hz), 7.89 (2H, d, J = 8.4 Hz), 7.97 (2H, d, J = 8.4 Hz), 8.62–8.64 (2H, m), 8.67 (1H, s), 10.1 (1H, s); 13C NMR (CDCl3, 100 MHz) δ 23.1, 26.1, 28.5, 28.7, 29.6, 30.1, 50.3, 119.7, 121.2, 123.6, 126.2, 127.3, 127.8, 128.5, 128.9, 131.4, 137.3, 140.9, 144.1, 146.7, 147.5, 150.1, 177.1.
O-Trityl-6-methoxynapthyltriazolyloctahydroxamate (14)
Reaction of 8-azido-O-trityloctahydroxamate 2b (135 mg, 0.30 mmol) and 2-ethynyl-6-methoxynapthalene (56 mg, 0.30 mmol) as described for synthesis of 8, followed by purification using preparative TLC (eluent 25:1 Et2O:EtOH) gave 142 mg (76 %) of compound 14 as white solid. 1H NMR (DMSO-d6, 400 MHz) δ 0.86–0.95 (2H, m), 1.10–1.21 (3H, m), 1.73–1.81 (2H, m), 3.86 (3H, s), 4.37 (2H, t, J = 6.8 Hz), 7.15–7.32 (17H, m), 7.83–7.92 (3H, m), 8.29 (1H, m), 8.61–8.62 (1H, m), 10.12 (1H, s); 13C NMR (CDCl3, 100 MHz) δ 23.1, 26.1, 28.5, 28.7, 29.6, 30.1, 50.3, 55.2, 93.6, 105.7, 119.1, 119.3, 124.1, 124.3, 125.8, 127.2, 127.8, 127.8, 128.0, 128.9, 128.9, 129.6, 134.2, 140.9 147.8, 157.8, 177.1.
Representative procedure for conversion of O-tritylhydroxamate to hydroxamic acid via TFA deprotection. 7-(3-Pyridyl)triazolylheptahydroxamic acid (26)
To a solution of O-trityl-3-pyridyltriazolylheptahydroxamate 8 (90 mg, 0.16 mmol) in CH2Cl2 (7 ml) was added trifluoroacetic acid (TFA) (0.2 mL) and thioanisole (0.2 mL) dropwise at 0 °C. Reaction mixture stirred for next 3 h at 0 °C. Solvent evaporated off in vacuo. The crude mixture was loaded on preparative TLC, eluted with 12:1:0.2 CH2Cl2:MeOH:NH4OH to give 39 mg (84%) of 26 as a white solid; mp 146–148 °C. 1H NMR (DMSO-d6, 400 MHz) δ 1.21–1.29 (4H, m), 1.46 (2H, m), 1.84 (2H, m), 1.91 (2H, m), 4.39 (2H, t, J = 7.2 Hz), 7.45–7.48 (1H, m), 8.18 (1H, m), 8.52 (1H, s), 8.65 (1H, s), 8.70 (1H, s), 9.03 (1H, s), 10.31 (1H, s). 13C NMR (DMSO-d6, 100 MHz) δ 24.9, 25.5, 27.9, 29.4, 32.1, 49.5, 121.9, 124.0, 132.4, 143.4, 146.2, 148.7, 169.0. HRMS (FAB, thioglycerol) calcd for [C14H19N5O2 + H]+ 290.1617, found 290.1615.
8-(Phenyl)triazolyloctahydroxamic acid (19)
Reaction of O-trityl-phenyltriazolyloctahydroxamate 4 (70 mg, 0.13 mmol) with TFA (0.1 ml) and thioanisole (0.1 ml) in CH2Cl2 (10 ml) at 0 °C within 3 h as described for the synthesis of 26, followed by purification using preparative TLC (eluent 8:1 CH2Cl2:MeOH) gave 20 mg (51 %) of 19 as a white solid; mp 118–121 °C. 1H NMR (CD3OD, 400 MHz) δ 1.22–1.48 (6H, m), 1.52–1.70 (2H, m), 1.87–2.01 (2H, m), 2.02–2.18 (2H, m), 4.44 (2H, t, J = 7.1 Hz), 7.34 (1H, t, J = 7.2 Hz), 7.43 (2H, t, J = 7.9 Hz), 7.81 (2H, d, J = 7.2 Hz), 8.32 (1H, s); 13C NMR (DMSO-d6, 100 MHz) δ 26.5, 27.2, 29.6, 29.8, 31.1, 33.6, 51.4, 122.2, 126.6, 129.3, 130.0, 131.7, 148.8, 169.1. HRMS (FAB, thioglycerol) calcd for [C16H22N4O2 + H]+ 303.1821, found 303.1806
9-(Phenyl)triazolylnonahydroxamic acid (20)
Reaction of O-trityl-phenyltriazolylnonahydroxamate 5 (57 mg, 0.10 mmol) with TFA (0.1 ml) and thioanisole (0.1 ml) in CH2Cl2 (10 ml) at 0 °C within 3 h as described for the synthesis of 26, followed by purification using preparative TLC (eluent 18:1:0.1 CH2Cl2:MeOH:NH4OH) gave 9.9 mg (27.5 %) of 20 as a white solid; mp 135–137 °C. 1H NMR (DMSO-d6, 400 MHz) δ 1.12–1.3 (8H, m), 1.42 (2H, m), 1.81 (2H, m), 1.87 (2H, t, J = 7.2 Hz), 4.34 (2H, t, J = 7.0 Hz), 7.28 (1H, t, J = 7.4 Hz), 7.40 (2H, t, J = 7.4 Hz), 7.80 (2H, d, J = 8.2 Hz), 8.54 (1H, s), 8.61 (1H, s), 10.28 (1H, s); 13C NMR (DMSO-d6, 100 MHz) δ 25.7, 26.5, 29.0, 29.15, 29.22, 30.3, 32.9, 50.2, 121.9, 125.8, 128.4, 129.6, 131.5, 146.9, 169.8. HRMS (FAB, thioglycerol) calcd for [C17H24N4O2 + H]+ 317.1933, found 317.1984.
9-(p-N,N-Dimethylanilyl)triazolylnonahydroxamic acid (24)
Reaction of O-trityl-(p-N,N-dimethylanilyl)triazolylnonahydroxamate 7 (65.3 mg, 0.11 mmol) with TFA (0.1 ml) and thioanisole (0.1 ml) in CH2Cl2 (10 ml) at 0 °C within 3 h as described for the synthesis of 26, followed by purification using preparative TLC (eluent 40:1 CH2Cl2:MeOH) gave 28.1 mg (72 %) of 24 as a white solid; mp 106–108 °C. 1H NMR (DMSO-d6, 400 MHz) δ 1.21 (8H, m), 1.44 (2H, m), 1.81 (2H, m), 1.90 (2H, t, J = 7.2 Hz), 4.31 (2H, t, J = 7.2 Hz), 6.75 (2H, d, J = 8.8 Hz), 7.62 (2H, d, J = 8.8 Hz), 8.33 (1H, s), 8.64 (1H, s), 10.31 (1H, s); 13C NMR (DMSO-d6, 100 MHz) δ 25.7, 26.6, 29.0, 29.16, 29.24, 30.3, 32.9, 40.7, 50.0, 113.0, 119.5, 120.0, 126.7, 147.5, 150.6, 169.8. HRMS (FAB, thioglycerol) calcd for [C19H29N5O2 + H]+ 360.2400, found 360.2402.
7-(2-Tolyl)triazolylheptahydroxamamic acid (34)
Reaction of O-trityl-2-tolyltriazolylheptahydroxamate 10 (90 mg, 0.16 mmol) with TFA (0.2 ml) and thioanisole (0.2 ml) in CH2Cl2 (7 ml) at 0 °C within 3 h as described for the synthesis of 26, followed by purification using preparative TLC (eluent 12:1:0.2 CH2Cl2:MeOH:NH4OH) gave 46 mg (93 %) of 34 as an off-white semi-solid. 1H NMR (CDCl3, 400 MHz) δ 1.26–1.34 (4H, m), 1.53–1.61 (2H, m), 1.84–1.91 (2H, m), 2.05–2.14 (2H, m), 2.40 (3H, s), 4.34 (2H, t, J = 6.8 Hz), 7.20–7.22 (3H, m), 7.64 (1H, s), 7.67–7.70 (1H, m), 9.72 (1H, s); 13C NMR (DMSO-d6, 100 MHz) δ 21.3, 24.9, 25.6, 27.8, 29.6, 29.8, 50.0, 121.9, 126.0, 128.1, 128.7, 129.7, 130.8, 135.4, 146.9, 171.3. HRMS (FAB, thioglycerol) calcd for [C16H22N4O2 + H]+ 303.1821, found 303.1810.
8-(3-Biphenyl)triazolyloctahydroxamic acid (42)
Reaction of O-trityl-3-biphenyltriazolyloctahydroxamate 11 (170 mg, 0.23 mmol) with TFA (0.1 ml) and thioanisole (0.1 ml) in CH2Cl2 (10 ml) at 0 °C within 3 h as described for the synthesis of 26, followed by purification using preparative TLC (eluent 12:1 CH2Cl2:MeOH) gave 80 mg (77.2 %) of 42 as a white solid; mp 110–112 °C. 1H NMR (CD3OD, 400 MHz) δ 1.14–1.44 (6H, m), 1.45–1.66 (2H, m), 1.78–1.96 (2H, m), 1.96-2.13 (2H, m), 4.35 (2H, t, J = 6.9 Hz), 7.31 (1H, t, J = 6.7 Hz), 7.41 (2H, t, J = 7.6 Hz), 7.46 (1H, d, J = 7.3 Hz), 7.55 (1H, d, J = 7.3 Hz), 7.63 (2H, d, J = 7.3 Hz), 7.75 (1H, d, J = 7.3 Hz), 8.07 (1H, s), 8.33 (1H, s) 13C NMR (CD3OD, 100 MHz) δ 26.5, 27.3, 29.6, 29.8, 31.1, 33.7, 51.4, 122.4, 125.1, 125.5, 127.8, 128.0, 128.6, 129.9, 130.5, 132.2, 141.8, 143.1, 148.6, 172.9. HRMS (FAB, thioglycerol) calcd for [C22H26N4O2 + H]+ 379.2134, found 379.2139.
9-(3-Biphenyl)triazolylnonahydroxamic acid (43)
Reaction of O-trityl-3-biphenyltriazolylnonahydroxamate 12 (114 mg, 0.18 mmol) with TFA (0.25 ml) and thioanisole (0.25 ml) in CH2Cl2 (10 ml) at 0 °C within 3 h as described for the synthesis of 26, followed by purification using preparative TLC (eluent 20:1 CH2Cl2:MeOH) gave 63.4 mg (91 %) of 43 as a white solid; mp 122–125 °C. 1H NMR (DMSO-d6, 400 MHz) δ 1.23 (8H, m), 1.42 (2H, m), 1.88 (4H, m), 4.36 (2H, m), 7.36 (1H, t, J = 7.6 Hz), 7.42–7.54 (3H, m), 7.59 (1H, d, J = 7.2 Hz), 7.69 (2H, d, J = 7.6 Hz), 7.82 (1H, d, J = 7.6 Hz), 8.08 (1H, s), 8.62 (1H, s), 8.68 (1H, s), 10.29 (1H, s); 13C NMR (DMSO-d6, 100 MHz) δ 25.8, 26.5, 29.0, 29.1, 29.2, 30.3, 50.2, 122.3, 124.0, 124.7, 126.8, 127.4, 128.3, 129.6, 130.2, 132.1, 140.5, 141.4, 146.9, 167.9. HRMS (FAB, thioglycerol) calcd for [C23H28N4O2 + H]+ 393.2246, found 393.2238.
8-(4-Pyridylphenyl)triazolyloctahydroxamamic acid (47)
Reaction of O-trityl-4-pyridylphenyltriazolyloctahydroxamate 13 (80 mg, 0.12 mmol) with TFA (0.2 ml) and thioanisole (0.2 ml) in CH2Cl2 (7 ml) at 0 °C within 3 h as described for the synthesis of 26, followed by purification using preparative TLC (eluent 12:1:0.2 CH2Cl2:MeOH:NH4OH) gave 29 mg (61 %) of 47 as a white solid; mp 152–154 °C. 1H NMR (DMSO-d6, 400 MHz) δ 1.21–1.26 (6H, m), 1.45 (2H, p, J = 7.2 Hz), 1.83–1.93 (4H, m), 3.86 (3H, s), 4.39 (2H, t, J = 7.2 Hz), 7.75–7.81 (2H, m), 7.91–7.93 (2H, m), 7.98–8.00 (2H, m), 8.62–8.70 (3H, s), 10.34 (1H, s); 13C NMR (DMSO-d6, 100 MHz) δ 25.0, 25.7, 28.0, 28.3, 29.5, 32.2, 49.5, 121.1, 121.8, 125.7, 127.4, 131.8, 136.1, 145.6, 146.6, 150.0, 169.1. HRMS (FAB, thioglycerol) calcd for [C21H26N5O2 + H]+ 380.2086, found 380.2102.
8-(6-Methoxynapthyl)triazolylheptaoctahydroxamamic acid (50)
Reaction of 6-methoxynapthyltriazolyloctahydroxamate 14 (90 mg, 0.14 mmol) with TFA (0.2 ml) and thioanisole (0.2 ml) in CH2Cl2 (7 ml) at 0 °C within 3 h as described for the synthesis of 26, followed by purification using preparative TLC (eluent 12:1:0.2 CH2Cl2:MeOH:NH4OH) gave 34 mg (64 %) of 50 as a white solid; mp 166–168 °C. 1H NMR (DMSO-d6, 400 MHz) δ 1.20–1.26 (6H, m), 1.45 (2H, p, J = 7.2 Hz), 1.83–1.92 (4H, m), 3.86 (3H, s), 4.38 (2H, t, J = 7.2 Hz), 7.16 (1H, dd, J = 2.3 Hz, 9.0 Hz), 7.31 (1H, d, J = 2.3 Hz), 7.84–7.92 (2H, m), 8.29 (1H, s), 8.62 (1H, s), 8.64 (1H, s), 10.31 (1H, s). 13C NMR (DMSO-d6, 100 MHz) δ 24.9, 25.7, 28.0, 28.3, 29.6, 32.1, 49.4, 55.2, 105.9, 119.1, 121.1, 123.3, 124.1, 126.0, 127.3, 133.8, 146.4, 157.3, 169.0. HRMS (FAB, thioglycerol) calcd for [C21H26N4O3 + H]+ 383.2083, found 383.2104.
Representative procedure for conversion of O-tritylhydroxamate to hydroxamic acid via BF3•OEt2 deprotection. 8-(3-Pyridyl)triazolyloctahydroxamic acid (27)
To a solution of O-trityl-3-pyridyltriazolyloctahydroxamate 9 (121 mg, 0.22 mmol) in just enough THF to dissolve (ca. 1 ml) was added thioanisole (0.31 mL) followed by BF3•OEt2 (0.34 mL) dropwise at r.t. The reaction mixture was stirred for 20 min during which a quantitative deprotection was observed bt TLC. The mixture was poured into 20 % MeOH in EtOAc (40 mL) and washed with water (10 mL). The pH of the aqueous layer was adjusted to ~7 and extracted with 20 % MeOH in EtOAc: (3 × 50mL). The combined organic layer was dried over Na2SO4 and concentrated in vacuo. Purification of the crude product using preparative TLC (eluent 10:1 CH2Cl2:MeOH) gave 62.1 mg (92.3%) of 27 as a white solid; mp 110–112 °C. 1H NMR (DMSO-d6, 400 MHz) δ 0.98–1.36 (6H, m), 1.38–1.62 (2H, m), 1.72–2.06 (4H, m), 4.39 (2H, t, J = 6.8 Hz), 7.46 (1H, t, J = 6.3 Hz), 8.19 (1H, d, J = 8.0 Hz), 8.52 (1H, d, J = 4.6 Hz), 8.64 (1H, s), 8.70 (1H, s), 9.02 (1H, s), 10.31 (1H, s); 13C NMR (DMSO-d6, 100 MHz) δ 25.0, 25.7, 28.1, 28.4, 29.6, 32.2, 49.8, 121.9, 124.0, 126.8, 132.2, 143.5, 146.3, 148.8, 169.1. HRMS (FAB, thioglycerol) calcd for [C15H21N5O2 + H]+ 304.1773, found 304.1765.
8-(p-N,N-Dimethylanilyl)triazolyloctahydroxamic acid (23)
Deprotection of O-trityl-p-N,N-dimethylanilyltriazolyloctahydroxamate 6 (186 mg, 0.32 mmol) under unoptimized conditions25 used BF3•OEt2 (0.08 ml) without thioanisole in 4:1 CHCl3:MeOH (15 ml) at r.t. for 3 h. The reaction was poured into 1:1 H2O:CHCl3 (100 mL) and the aqueous layer extracted with CHCl3 (50 mL). The combined organic layer was washed with brine (50 mL), then dried over Na2SO4 and concentrated in vacuo. Purification using preparative TLC (eluent 12:1 CH2Cl2:MeOH) gave 43 mg (40 %) of 23 as a white solid; mp 108–110 °C. 1H NMR (CD3OD, 400 MHz) δ 1.24–1.45 (6H, m), 1.49–1.68 (2H, m), 1.84–2.00 (2H, m), 2.00–2.18 (2H, m), 2.97 (6H, s), 4.39 (2H, t, J = 7.1 Hz), 6.83 (2H, d, J = 8.1 Hz), 7.65 (2H, d, J =8.7 Hz), 8.12 (1H, s); 13C NMR (DMSO-d6, 100 MHz) δ 25.1, 25.8, 28.1, 28.4, 29.7, 32.2, 40.0, 49.4, 112.3, 118.8, 119.3, 126.0, 146.8, 149.9, 169.1. HRMS (FAB, thioglycerol) calcd for [C18H27N5O2 + H]+ 346.2243, found 346.2242.
HDAC Activity Assay
In vitro HDAC inhibition was assayed using the HDAC Fluorimetric Assay/Drug Discovery Kit as previously described.17–19 Briefly, 15 μL of HeLa nuclear extract was mixed with 5 μL of 10× compound and 5 μL of assay buffer. Fluorogenic substrate (25 μL) was added, and reaction was allowed to proceed for 15 min at room temperature and then stopped by addition of a developer containing TSA. Fluorescence was monitored after 15 min at excitation and emission wavelengths of 360 and 460 nm, respectively. IC50 values were determined using logit plots.
In vitro antimalarial and antileishmanial assays
Antimalarial activity of the compounds was determined in vitro on chloroquine sensitive (D6, Sierra Leone) and resistant (W2, IndoChina) strains of Plasmodium falciparum. The 96 well microplate assay is based on evaluation of the effect of the compounds on growth of asynchronous cultures of P. falciparum, determined by the assay of parasite lactate dehydrogenase (pLDH) activity.20 The appropriate dilutions of the compounds were prepared in DMSO or RPMI-1640 medium and added to the cultures of P. falciparum (2% hematocrit, 2% parasitemia) set up in clear flat bottomed 96 well plates. The plates were placed into the humidified chamber and flushed with a gas mixture of 90% N2, 5% CO2 & 5% O2. The cultures were incubated at 37°C for 72 hours. Growth of the parasite in each well was determined by pLDH assay using Malstat® reagent.20 The medium and RBC controls were also set-up in each plates. The standard antimalarial agents, chloroquine and artemisinin, were used as the positive controls while DMSO was tested as the negative control. Antileishmanial activity of the compounds was tested in vitro on a culture of Leishmania donovani promastigotes. In a 96 well microplate assay compounds with appropriate dilution were added to the leishmania promastigotes culture (2× 106 cell/mL). The plates were incubated at 26°C for 72 hours and growth of leishmania promastigotes was determined by Alamar blue assay.21 Pentamidine and Amphotericin B were tested as standard antileishmanial agents. All the compounds were simultaneously tested for cytotoxicty on VERO (monkey kidney epithilial) cells by Neutral Red assay.22 IC50 value for each compound was computed from the growth inhibition curve.
Supplementary Material
Acknowledgments
We thank John Trott and Rajnish Sahu for their assistance with the in vitro antimalarial and antileishmanial assays respectively. This work was financially supported by Georgia Institute of Technology, by the Blanchard fellowship and by NIH Grant R01CA131217 (A.K.O.). NCNPR is partially supported by USDA-ARS scientific cooperative agreement no 58-6408-2-0009. P.C.C., W.G. and B. G. are recipients of the GAANN predoctoral fellowship from the Georgia Tech Center for Drug Design, Development and Delivery.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Marks PA. Oncogene. 2007;26:1351–1356. doi: 10.1038/sj.onc.1210204. [DOI] [PubMed] [Google Scholar]
- 2.(a) Wade PA. Hum Mol Genet. 2001;10:693–698. doi: 10.1093/hmg/10.7.693. [DOI] [PubMed] [Google Scholar]; (b) Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP. Nature. 2002;417:455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]; (c) Zhang Y, Li N, Caron C, Matthias G, Hess D, Khochbin S, Matthias P. Embo J. 2003;22:1168–1179. doi: 10.1093/emboj/cdg115. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Proc Natl Acad Sci USA. 2003;100:4389–4394. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Arts J, de Schepper S, Van Emelen K. Curr Med Chem. 2003;10:2343–2350. doi: 10.2174/0929867033456657. [DOI] [PubMed] [Google Scholar]; (f) Bali P, Pranpat M, Bradner J, Balasis M, Fiskus W, Guo F, Rocha K, Kumaraswamy S, Boyapalle S, Atadja P, Seto E, Bhalla K. J Biol Chem. 2005;280:26729–26734. doi: 10.1074/jbc.C500186200. [DOI] [PubMed] [Google Scholar]
- 3.(a) Marks PA, Dokmanovic M. Expert Opin Investig Drugs. 2005;14:1497–1511. doi: 10.1517/13543784.14.12.1497. [DOI] [PubMed] [Google Scholar]; (b) Bolden JE, Peart MJ, Johnstone RW. Nat Rev Drug Discovery. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]; (c) Butler KV, Kozikowski AP. Curr Pharm Des. 2008;14:505–528. doi: 10.2174/138161208783885353. [DOI] [PubMed] [Google Scholar]; (d) Paris M, Porcelloni M, Binaschi M, Fattori D. J Med Chem. 2008;51:1505–1528. doi: 10.1021/jm7011408. [DOI] [PubMed] [Google Scholar]; (e) Rotili D, Simonetti G, Savarino A, Palamara AT, Migliaccio AR, Mai A. Curr Top Med Chem. 2009;9:272–291. doi: 10.2174/156802609788085296. [DOI] [PubMed] [Google Scholar]
- 4.(a) Darkin-Rattray SJ, Gurnett AM, Myers RW, Dulski PM, Crumley TM, Allocco JJ, Cannova C, Meinke PT, Colletti SL, Bednarek MA, Singh SB, Goetz MA, Dombrowski AW, Polishook JD, Schmatz DM. Proc Natl Acad Sci USA. 1996;93:13143–13147. doi: 10.1073/pnas.93.23.13143. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Meinke PT, Colletti SL, Doss G, Myers RW, Gurnett AM, Dulski PM, Darkin-Rattray SJ, Allocco JJ, Galuska S, Schmatz DM, Wyvratt MJ, Fisher MH. J Med Chem. 2000;43:4919–4922. doi: 10.1021/jm0001976. [DOI] [PubMed] [Google Scholar]; (c) Mukherjee P, Pradhan A, Shah F, Tekwani BL, Avery MA. Bioorg Med Chem. 2008;16:5254–5265. doi: 10.1016/j.bmc.2008.03.005. [DOI] [PubMed] [Google Scholar]; (d) Chen Y, Lopez-Sanchez M, Savoy DN, Billadeau DD, Dow GS, Kozikowski AP. J Med Chem. 2008;51:3437–3448. doi: 10.1021/jm701606b. [DOI] [PubMed] [Google Scholar]; (e) Dow GS, Chen Y, Andrews KT, Caridha D, Gerena L, Gettayacamin M, Johnson J, Li Q, Melendez V, Obaldia N, III, Tran TN, Kozikowski AP. Antimicrob Agents Chemother. 2008;52:3467–3477. doi: 10.1128/AAC.00439-08. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Andrews KT, Tran TN, Lucke AJ, Kahnberg P, Le GT, Boyle GM, Gardiner DL, Skinner-Adams TS, Fairlie DP. Antimicrob Agents Chemother. 2008;52:1454–1461. doi: 10.1128/AAC.00757-07. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Patel V, Mazitschek R, Coleman B, Nguyen C, Urgaonkar S, Cortese J, Barker RH, Jr, Greenberg E, Tang W, Bradner JE, Schreiber SL, Duraisingh MT, Wirth DF, Clardy J. J Med Chem. 2009;52:2185–2187. doi: 10.1021/jm801654y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Mai A, Cerbara I, Valente S, Massa S, Walker LA, Tekwani BL. Antimicrob Agents Chemother. 2004;48:1435–1436. doi: 10.1128/AAC.48.4.1435-1436.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Snow RW, Guerra CA, Noor AM, Myint HY, Hay SI. Nature. 2005;434:214–217. doi: 10.1038/nature03342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wirth DF. Annu Rep Med Chem. 1999;34:349–358. [Google Scholar]
- 7.Croft SL, Yardley V. Curr Pharm Des. 2002;8:319–342. doi: 10.2174/1381612023396258. [DOI] [PubMed] [Google Scholar]
- 8.Musset L, Bouchaud O, Matheron S, Massias L, Le Bras J. Microbes Infect. 2006;8:2599–2604. doi: 10.1016/j.micinf.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 9.Ndiaye D, Daily JP, Sarr O, Ndir O, Gaye O, Mboup S, Wirth DF. Trop Med Int Health. 2005;10:1176–1179. doi: 10.1111/j.1365-3156.2005.01506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Calza L, Marinacci G, Manfredi R, Colangeli V, Fortunato L, Chiodo FJ. Chemother. 2001;13:653–657. doi: 10.1179/joc.2001.13.6.653. [DOI] [PubMed] [Google Scholar]; (b) Croft SL, Sundar S, Fairlamb AH. Drug resistance in leishmaniasis. Clin Microbiol Rev. 2006;19:111–126. doi: 10.1128/CMR.19.1.111-126.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ouellette M, Drummelsmith J, Papadopoulou B. Drug Resist Updates. 2004;7:257–266. doi: 10.1016/j.drup.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 11.Joshi MB, Lin DT, Chiang PH, Goldman ND, Fujioka H, Aikawa M, Syin C. Mol Biochem Parasitol. 1999;99:11–19. doi: 10.1016/s0166-6851(98)00177-7. [DOI] [PubMed] [Google Scholar]
- 12.Duraisingh MT, Voss TS, Marty AJ, Duffy MF, Good RT, Thompson JK, Freitas-Junior LH, Scherf A, Crabb BS, Cowman AF. Cell. 2005;121:13–24. doi: 10.1016/j.cell.2005.01.036. [DOI] [PubMed] [Google Scholar]
- 13.Freitas-Junior LH, Hernandez-Rivas R, Ralph SA, Montiel-Condado D, Ruvalcaba-Salazar OK, Rojas-Meza AP, Mancio-Silva L, Leal-Silvestre RJ, Gontijo AM, Shorte S, Scherf A. Cell. 2005;121:25–36. doi: 10.1016/j.cell.2005.01.037. [DOI] [PubMed] [Google Scholar]
- 14.Vergnes B, Sereno D, Madjidian-Sereno N, Lemesre JL, Ouaissi A. Gene. 2002;296:139–150. doi: 10.1016/s0378-1119(02)00842-9. [DOI] [PubMed] [Google Scholar]
- 15.Vergnes B, Sereno D, Tavares J, Cordeiro-da-Silva A, Vanhille L, Madjidian-Sereno N, Depoix D, Monte-Alegre A, Ouaissi A. Gene. 2005;363:85–96. doi: 10.1016/j.gene.2005.06.047. [DOI] [PubMed] [Google Scholar]
- 16.(a) Ouaissi1 M, Ouaissi A. J Biomed Biotech. 2006:1–10. doi: 10.1155/JBB/2006/13474. ID 13474. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ivens AC, Peacock CS, Worthey EA, Murphy L, Aggarwal G. Science. 2005;309:436–442. doi: 10.1126/science.1112680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Chen PC, Patil V, Guerrant W, Green P, Oyelere AK. Bioorg Med Chem. 2008;16:4839–4853. doi: 10.1016/j.bmc.2008.03.050. [DOI] [PubMed] [Google Scholar]; (b) Chen Y, Lopez-Sanchez M, Savoy DN, Billadeau DD, Dow GS, Kozikowski AP. J Med Chem. 2008;51:3437–3448. doi: 10.1021/jm701606b. [DOI] [PubMed] [Google Scholar]
- 18.HDAC Fluorimetric Assay/Drug Discovery Kit—AK-500 Manual. Fluorescent Assay System (BIOMOL_ International, Plymouth Meeting, PA), 2005.
- 19.(a) Oyelere AK, Chen PC, Guerrant W, Mwakwari SC, Hood R, Zhang Y, Fan Y. J Med Chem. 2009;52:456–468. doi: 10.1021/jm801128g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Weller RL, Rajski SR. Org Lett. 2005;7:2141–2144. doi: 10.1021/ol0504749. [DOI] [PubMed] [Google Scholar]; (c) Heine HW, Becker EB, Lane JF. J Am Chem Soc. 1953;75:4514–4515. [Google Scholar]
- 20.Makler MT, Ries JM, Williams JA, Bancroft JE, Piper RC, Gibbins BL, Hinriches DJ. Am J Trop Med Hyg. 1993;48:739–741. doi: 10.4269/ajtmh.1993.48.739. [DOI] [PubMed] [Google Scholar]
- 21.Mikus J, Steverding D. Parasitol Int. 2000;48:265–269. doi: 10.1016/s1383-5769(99)00020-3. [DOI] [PubMed] [Google Scholar]
- 22.Babich H, Borenfreund E. App Envt Microbiol. 1991;57:2101–2103. doi: 10.1128/aem.57.7.2101-2103.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saugar JM, Delgado J, Hornillos V, Luque-Ortega JR, Amat-Guerri F, Acuña AU, Rivas L. J Med Chem. 2007;50:5994–6003. doi: 10.1021/jm070595+. [DOI] [PubMed] [Google Scholar]
- 24.Sundar S, Jha TK, Thakur CP, Engel J, Sindermann H, Fischer C, Junge K, Bryceson A, Berman J. The New Engl J of Med. 2002;347:1739–1746. doi: 10.1056/NEJMoa021556. [DOI] [PubMed] [Google Scholar]
- 25.Yang SM, Lagu B, Wilson LJ. J Org Chem. 2007;72:8123–8126. doi: 10.1021/jo701411d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.