

Abstract
In normal aging, a peripheral immune challenge induces a sensitized and protracted neuroinflammatory response in parallel with long-term memory (LTM) impairments. Proinflammatory mediators of neuroinflammation impair LTM, synaptic plasticity and LTP. The immediate early gene Arc is considered a critical protein regulating LTM and synaptic plasticity. The present investigation examined whether 1) a peripheral E. coli infection suppresses hippocampal Arc expression, and 2) central proinflammatory cytokines (IL-1β and IL-6) mediate the effects of peripheral E. coli infection on Arc and LTM. In 24 mo F344 × BN F1 rats, E. coli infection suppressed basal Arc gene expression as well as contextual fear conditioning-induced Arc expression. E. coli treatment failed to alter either basal or conditioning-induced c-Fos expression. At 24 h post-infection, intracisterna magna (ICM) treatment with the anti-inflammatory cytokine IL-1RA blocked the E. coli-induced suppression of hippocampal Arc and increases in IL-6 protein. At 4 d post-infection, IL-1RA blocked the E. coli-induced LTM impairments and increases in IL-6 protein. The present results suggest that central proinflammatory cytokines play a salient role in the suppression of Arc and impairments of LTM by a peripheral immune challenge in older animals.
Keywords: aging, neuroinflammation, proinflammatory cytokines, IL-1RA, memory, Arc
1. Introduction
In older animals, the CNS exhibits a sensitized neuroinflammatory response to peripheral as well as central administration of proinflammatory agents (Abraham et al., 2008; Barrientos et al., 2009; Barrientos et al., 2006; Chen et al., 2008; Godbout et al., 2005; Huang et al., 2007). Induction of neuroinflammatory processes or treatment with neuroinflammatory mediators/products has profound effects on learning and memory, in particular hippocampus-dependent memory processes (Barrientos et al., 2009; Barrientos et al., 2006; Barrientos et al., 2002; Barrientos et al., 2003; Barrientos et al., 2004; Gibertini et al., 1995; Hauss-Wegrzyniak et al., 1998; Hein et al., 2007; Oitzl et al., 1993; Pugh et al., 2000; Pugh et al., 1998; Shaw et al., 2001; Tanaka et al., 2006; Thomson and Sutherland, 2005). The time course of hippocampal-dependent memory deficits occurs in parallel with a protracted neuroinflammatory response to peripheral infection in aged animals (Barrientos et al., 2009; Barrientos et al., 2006). It is important to note that peripheral infection in young animals (3 mo) does not induce a protracted neuroinflammatory response as well as a deficit in hippocampal dependent memory (Barrientos et al., 2009). However, how the neuroinflammatory response to peripheral infection may compromise memory processes in older animals is unknown and is the focus of the present investigation.
Mediators of neuroinflammation, including proinflammatory cytokines (IL-1β, IL-6, TNFα, and IL-18), are known to modulate the putative neurobiological substrates (LTP) of memory formation (Cumiskey et al., 2007; Curran and O'Connor, 2003; Tancredi et al., 2000; Vereker et al., 2000). Aged animals show deficits in LTP, which are accompanied by increases in proinflammatory cytokines (Lynch, 1998). However, the molecular mechanism(s) mediating proinflammatory cytokine suppression of LTP has yet to be fully characterized.
Investigations into the molecular basis of LTP have yielded several genetic targets (Tzingounis and Nicoll, 2006). Of these targets, the effector immediate early gene (IEG) Arc (activity-dependent cytoskeletal-associated protein) exhibits several unique features, which underscore its importance in memory consolidation (Bramham et al., 2008). Arc mRNA is rapidly and specifically distributed throughout the dendritic arbor post-induction (Link et al., 1995; Lyford et al., 1995) and localized to regions receiving direct synaptic activation (Steward et al., 1998). Suppression of Arc impairs long -term memory (LTM) consolidation, whereas acquisition and short- term memory (STM) are unaffected (Guzowski et al., 2000). This is noted because we have previously found that a peripheral immune challenge in older animals impairs LTM, but not STM (Barrientos et al., 2006).
To understand how the neuroinflammatory sequelae of peripheral infection may impair LTM in older animals, we examined whether 1) a peripheral E. coli infection suppresses hippocampal Arc expression, and 2) central proinflammatory cytokines (IL-1β and IL-6) mediate the effects of peripheral E. coli infection on Arc as well as LTM.
2. Materials and Methods
2.1. Subjects
As in our prior investigations (Barrientos et al., 2009; Barrientos et al., 2006; Frank et al., 2006), the present set of experiments compared 3 and 24 mo old male F344 × BN F1 rats. 24 mo was selected as the older age because at this age animals do not show major age-related pathologies and in the absence of a peripheral immune challenge, STM and LTM processes do not differ between 24 mo and 3 mo old animals in this hybrid strain. Therefore, differences in basal and contextual fear conditioning-induced Arc expression should be minimized between these age groups prior to immune challenge.
Subjects were obtained from the National Institute on Aging (Bethesda, MD). Upon arrival at our facility, older rats were 24 mo old and had a mean weight of 573.7 g±S.D. 60.5. Younger rats were 3 mo old and had a mean weight of 282.5 g±S.D. 31.8. Older and younger rats were housed 2 or 4 to a cage (52 cm × 30 cm × 21 cm; L × W × H), respectively. The animal colony was maintained at 22 C° on a 12-h light/dark cycle (lights on at 07:00 h). All rats were allowed free access to food and water and were given 1 week to acclimate to colony conditions before experimentation began. All experiments were conducted in accordance with protocols approved by the University of Colorado Animal Care and Use Committee.
2.2. Experimental Designs
2.2.1. Experiment 1: Effect of peripheral Escherichia coli (E. coli) on basal hippocampal Arc gene expression
The present investigation was restricted to examining hippocampal changes in Arc because we have previously shown that 1) peripheral bacterial infection induces pro-inflammatory cytokine changes specific to the hippocampus (Barrientos et al., 2009) and 2) the impairment of LTM by peripheral infection is restricted to hippocampal dependent memory processes and do not affect hippocampal independent memory processes (Barrientos et al., 2006). 3 and 24 mo old animals were injected intraperitoneal (i.p.) with either vehicle or E. coli. All injections occurred between 08:00 and 10:00 h. Either 2h, 4h, 24h, or 96h post-infection, animals were anesthetized with pentobarbital, transcardially perfused with ice-cold saline (0.9%) and hippocampi collected.
2.2.2. Experiment 2: Effect of peripheral E. coli on conditioning-induced hippocampal Arc and c-Fos gene expression
In addition to Arc, we examined the effects of E. coli infection on the IEG c-Fos to determine whether the effect of E. coli infection on Arc generalizes to other IEGs. The regulatory transcription factor c-Fos has been implicated in memory processes (Davis et al., 2003; Morrow et al., 1999). In addition, c-Fos and Arc expression are induced to a similar degree in the hippocampus 30 min after behavioral training (Guzowski et al., 2001). 3 and 24 mo old animals were injected with either vehicle or E. coli i.p. All injections occurred between 08:00 and 10:00 h. 96h post-treatment, animals underwent contextual fear conditioning or served as home cage controls (HCC). 30 min post-conditioning, animals were briefly anesthetized using halothane, rapidly decapitated and hippocampi collected.
2.2.3. Experiment 3: Effect of exogenous IL-1 receptor antagonist (IL-1RA) on E. coli-induced suppression of hippocampal Arc 24 h post-infection
24 mo animals were briefly anesthetized under halothane and injected intracisterna magna (ICM) with either vehicle or IL-1RA. IL-1RA binds to the IL-1 type 1 receptor with an affinity equal to IL-1β without any agonist activity, thereby effectively preventing IL-1β signal transduction (Dinarello, 1997). Substances injected ICM spread throughout the CNS (Proescholdt et al., 2000). Immediately following ICM injection, animals were injected i.p. with either vehicle or E. coli. 24h post-injections, animals were perfused with saline and hippocampi collected. A single injection of IL-1RA prior to E. coli injection was employed because the signal from the periphery to the brain after i.p. E. coli is rapid (< 2 h) and persists for < 24 h. As the duration of action of IL-1RA in the CNS is at least 3 hr, a single injection should yield blockade of brain IL-1 receptors for a significant part of the duration of E. coli-induced signaling to the brain. We have previously shown that IL-1RA (100 μg) injected ICM completely blocks the stress-induced sensitization of the hippocampal IL-1β response to peripheral LPS (Johnson et al., 2004). This issue is more fully addressed in the Discussion below.
2.2.4. Experiment 4: Effect of exogenous IL-1RA on E. coli-induced memory impairments
24 mo animals were briefly anesthetized under halothane and injected ICM with either vehicle or IL-1RA. Immediately following ICM injection, animals were injected i.p. with either vehicle or E. coli. 4 d post-injections, all animals underwent contextual fear conditioning. 3 d post-conditioning, all animals were given a long-term memory test for the context. 4 d post-conditioning, animals were perfused with saline and hippocampi collected. A single administration of IL-1RA was employed for the same reasons as provided above.
2.3. Peripheral immune challenge
3 and 24 mo animals were given a single exposure to a peripheral bacterial E. coli infection at a concentration that we have previously shown to induce a protracted proinflammatory response in the hippocampus of older animals relative to younger subjects, as well as impairments in hippocampal dependent LTM only in older animals (Barrientos et al., 2006). Hippocampal IL-1β or IL-6 protein was measured as an index of E. coli-induced neuroinflammation. In all experiments, animals received an i.p. injection of either E. coli or vehicle. One day prior to experimentation, stock E. coli cultures (ATCC 15746; American Type Culture Collection, Manassas, VA) were thawed and cultured overnight (15–20 h) in 40 ml of brain-heart infusion (BHI; DIFCO Laboratories, Detroit, MI) in an incubator (37 C°, 5% CO2). The number of bacteria in cultures was quantified by extrapolating from previously determined growth curves. Cultures were then centrifuged for 15 min at 4° C, 3000 rpm, supernatants discarded, and bacteria resuspended in sterile phosphate buffered saline (PBS). Bacteria were resuspended with a volume of PBS to achieve a concentration of 1.0 × 1010 colony forming units (CFU)/ml. A volume of 250 μl was injected i.p. regardless of body weight for a final dose of 2.5 × 109 CFU. Thus, as in prior studies, the older rats received a lower dose than did the younger subjects relative to body weight, a procedure that was adopted as a conservative measure. Vehicle treated rats received an injection of sterile PBS of an equal volume (250 μl).
2.4. ICM administration of IL-1RA
Animals were briefly anesthetized with halothane. The dorsal aspect of the skull was shaved and swabbed with 70% ETOH. A 27-gauge needle attached via PE50 tubing to a 25 μl Hamilton syringe was inserted into the cisterna magna. To verify entry into the cisterna magna, ~ 2 μl of fluid was drawn. In all cases, fluid was clear of red blood cells indicating the presence of CSF and entry into the cisterna magna. IL-1RA (112 μg; Amgen, Thousand Oaks, CA) was administered in 3μl total volume.
2.5. Contextual fear conditioning (Experiment 2)
We have utilized the conditioning procedure described below to induce robust increases in both hippocampal Arc and c-Fos gene expression, which were measured 30 min post-conditioning (Huff et al., 2006). Each conditioning chamber consisted of an Igloo ice chest (54 cm × 30 cm × 27 cm; L × W × H) with a white interior. A speaker and an activated 6-W clear light bulb were mounted on the ceiling of each chest. The ice chest door was open the entire time, and the room was illuminated by two 60-W light bulbs. The conditioning chambers (26 cm × 21 cm × 24 cm; L × W × H) placed inside the chest were made of clear plastic and had window screen tops. The shock was delivered through a removable floor of stainless-steel rods (model E63-23-MOD001; Coulbourn Instruments, Allentown, PA), each of which was 0.5 cm in diameter and spaced 1.75 cm, center to center. Each rod was wired to a shock generator and scrambler (model H13-16; Coulbourn Instruments). The chamber was cleaned with water before each rat was conditioned.
Animals were taken in a black bucket to the conditioning chambers. Animals were allowed to explore the conditioning context for 5 min, at the end of which a 2 s, 1.5 mA shock was presented. After the shock, animals were immediately removed and returned to their home cage via a black bucket where they remained for 30 min until sacrifice.
2.6. Contextual fear conditioning (Experiment 4)
The conditioning context was identical to that used in experiment 2. Rats were allowed to explore the chamber for 2 min before the onset of a 15 s tone (76 dB), followed immediately by a 2 s footshock (1.5 mA). Immediately after the termination of the shock, rats were removed from the chamber and returned to their home cage. Three days later, all rats were tested for fear of the conditioning context—a hippocampal-dependent task, and then for fear of the tone—a hippocampal-independent task. That is, the hippocampus is required to form a long-term memory of fear of the context, but not for fear of the tone (Kim and Fanselow, 1992). Fear of the context was assessed by placing the rat in the conditioning context for 6 min. Fear of the tone was assessed by placing the rat in an altered context for 3 min, followed by a 3 min presentation of the tone. Scoring began approximately 10 s after the animal was placed into the chamber. Every 10 s each rat was judged as either freezing or active at the instant the sample was taken. Rats were first tested for fear of the context and then for fear to the tone about 4 h later. Behavior was scored by observers blind to experimental treatment.
2.7. Real time RT-PCR measurement of gene expression
Total RNA was isolated from whole hippocampus utilizing a standard method of phenol:chloroform extraction (Chomczynski and Sacchi, 1987). For detailed descriptions of RNA isolation, Dnase treatment, cDNA synthesis, and PCR amplification protocols refer to prior publication (Frank et al., 2006). cDNA sequences were obtained from Genbank at theNational Center for Biotechnology Information (NCBI; www.ncbi.nlm.nih.gov). Primer sequences were designed to amplify Arc (F: 5′-ACAGAGGATGAGACTGAGGCAC – 3′; R: 5′-TATTCAGGCTGGGTCCTGTCAC –3′) and c-Fos (F: 5′-CTTCCTTTGTCTTCACCTACC-3′; R: 5′- CCTTCTCTGACTGCTCACA-3′). Glyceraldehyde-6-phosphate dehydrogenase (GAPDH) (F: 5′-GTTTGTGATGGGTGTGAACC-3′; R: 5′-TCTTCTGAGTGGCAGTGATG-3′) and 18s ribosomal RNA (F: 5′-ATGGTAGTCGCCGTGCCTA-3′; R: 5′-CTGCTGCCTTCCTTGGATG-3′) served as housekeeping genes. Primer sequences were designed using the Qiagen Oligo Analysis & Plotting Tool (oligos.qiagen.com/oligos/toolkit.php?) and tested for sequence specificity using the Basic Local Alignment Search Tool at NCBI (Altschul et al., 1990). Primers were obtained from Sigma (St. Louis, MO). Primer specificity was verified by melt curve analysis (see Quantitative real-time PCR).
PCR amplification of cDNA was performed using the Quantitect SYBR Green PCR Kit (Qiagen, Valencia, CA). Formation of PCR product was monitored in real time using the MyiQ Single-Color Real-Time PCR Detection System (BioRad, Hercules, CA).
Relative gene expression was determined using the 2−ΔΔCT method (Livak and Schmittgen, 2001). Mean CT of triplicate measures was computed for each sample. Sample mean CT of GAPDH or 18s rRNA (internal controls) was subtracted from the sample mean CT of the respective gene of interest (Δ CT). The sample with the highest absolute ΔCT was selected as a calibrator and subtracted from the ΔCT of each experimental sample (ΔΔCT). 2−ΔΔCT yields fold change in gene expression of the gene of interest normalized to the internal control gene expression and relative to the calibrator sample.
2.8. Enzyme-linked immunoSorbent assay (ELISA) of IL-1β and IL-6 protein
Tissue was added to 0.5 ml of Iscove's culture medium containing 5% fetal calf serum and a cocktail enzyme inhibitor (100 mM amino-n-caproic acid, 10 mM EDTA, 5 mM benzamidine HCl, and 0.2 mM phenylmethyl sulfonyl fluoride). Total protein was mechanically dissociated from tissue using an ultrasonic cell disrupter (Fisher Scientific, Pittsburgh, PA). Sonication consisted of 10 s of cell disruption. Sonicated samples were centrifuged at 10,000 × g at 4°C for 10 min. Supernatants were removed and stored at −80°C until ELISA was performed. Bradford protein assays were also performed to determine total protein concentrations in sonication samples. Levels of IL-1β and IL-6 protein were determined using a commercially available rat IL-1β and IL-6 ELISA kit (R&D Systems, Minneapolis, MN). The assay was performed according to the manufacturer's instructions. Fifty l of tissue sonicates was used for the assay.
2.9. Statistical analysis
All data are presented as mean + SEM. Sample sizes are provided in figure captions. Statistical analyses consisted of ANOVA followed by Bonferroni post-hoc comparisons. In text and figure captions, Bonferroni corrected p values are reported. Threshold for statistical significance was set at α = .05.
3. Results
3.1 Experiment 1: Peripheral E. coli reduced basal hippocampal Arc gene expression in 24 mo animals (Fig. 1)
Fig. 1.
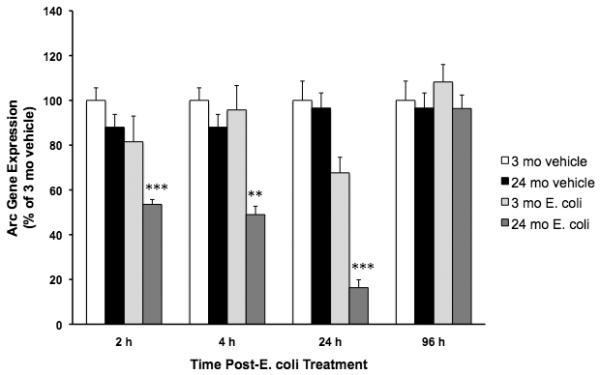
Peripheral E. Coli reduced basal hippocampal Arc gene expression in 24 mo animals. 3 mo and 24 mo animals were administered E. coli or vehicle i.p. At 2 h, 4 h, 24 h and 96 h post-treatment, hippocampal Arc expression was measured. E. Coli treatment reduced basal Arc expression 2 h, 4 h, and 24 h post-E. coli in 24 mo animals compared to all other experimental groups (**p < .01, *** p < .001). E. coli treatment did not significantly alter Arc expression in 3 mo animals nor did Arc expression differ between vehicle treated 3 and 24 mo animals. At 96 h post-infection, Arc expression was not altered by E. coli treatment in either 3 or 24 mo animals. N = 5-9 animals/group.
As an initial experiment to assess whether a peripheral immune challenge differentially alters Arc expression in 3 mo and 24 mo animals, E. coli was administered to animals at a dose that induces increased hippocampal IL-1β along with memory impairments in older animals 4 d post-infection (Barrientos et al., 2006). Here, the effects of E. coli on Arc expression were measured 2 h, 4 h, 24 h, and 96 h post-infection. In the absence of E. coli, basal Arc expression was similar in 3 mo and 24 mo animals at each time point post-treatment. E. coli treatment differentially suppressed Arc gene expression in 24 mo animals compared to E. coli treated 3 mo animals at different time points post-infection. E. coli treatment induced comparable decreases in body weight in younger and older animals 24 h post-infection indicating that all animals received an active infection (data not shown).
3.1.1. E. coli suppressed Arc 2 hours post-infection
E. Coli treatment differentially suppressed Arc expression in 24 mo compared to 3 mo old animals (F = 10.33(df=1,24), p = .0037). In 24 mo animals, E. coli treatment reduced Arc expression compared to 3 mo vehicle (4.4 fold decrease, p < .00001), 3 mo E. coli (3.9 fold decrease, p < .001) and 24 mo vehicle (3.7 fold decrease, p < .0001) groups. All other experimental groups did not differ in Arc expression.
3.1.2. E. coli suppressed Arc 4 hours post-infection
Similar to the effects observed at 2 h post-infection, E. coli treatment resulted in a robust Arc reduction only in 24 mo animals (F = 11.67(df=1,21), p = .0026). E. coli significantly suppressed Arc expression in 24 mo animals compared to vehicle treated 3 mo animals (3.7 fold decrease, p < .001), E. coli treated 3 mo animals (3.5 fold decrease, p < .01) and 24 mo vehicle animals (3.3 fold decrease, p < .001). All other experimental groups did not differ in Arc expression.
3.1.3. E. coli suppressed Arc 24 hours post-infection
E. coli treatment interacted with age to modulate Arc expression (F = 11.401(df=1,19), p = .0032). E. coli significantly suppressed Arc expression in 24 mo animals compared to vehicle treated 3 mo animals (6.1 fold decrease, p < .00001), E. coli treated 3 mo animals (4.1 fold decrease, p < .001) and vehicle treated 24 mo animals (5.9 fold decrease, p < .00001). 3 mo E. coli-treated animals did not significantly differ from vehicle treated 3 mo animals (p = .13) and vehicle treated 24 mo animals (p = .1). 3 mo and 24 mo vehicle treated animals did not significantly differ.
3.1.4. E. coli did not suppress Arc 96 hours post-infection
96 hours post-infection, E. coli treatment did not significantly alter Arc expression compared to vehicle treatment in either 3 mo or 24 mo animals.
3.2. Experiment 2: Effect of peripheral E. coli on hippocampal IL-1β protein and conditioning-induced Arc and c-Fos gene. expression 96 h post-infection
In light of the results of experiment 1 showing that a peripheral immune challenge substantially reduces basal Arc only in older animals, we investigated whether E. coli treatment would differentially suppress conditioning-induced Arc in 24 mo compared to 3 mo animals. 96 h post-infection was selected as the time point to test this hypothesis because 1) our initial experiment showed that at 96 h post-infection, basal Arc expression was similar among experimental groups, which obviates potential floor effects and 2) at this time point post-infection, we have previously observed not only LTM impairments, but also a protracted proinflammatory cytokine response in 24 mo animals (Barrientos et al., 2006).
3.2.1. Peripheral E. coli treatment induced a protracted IL-1β protein response in the hippocampus of 24 mo animals (Fig. 2A)
Fig. 2A.
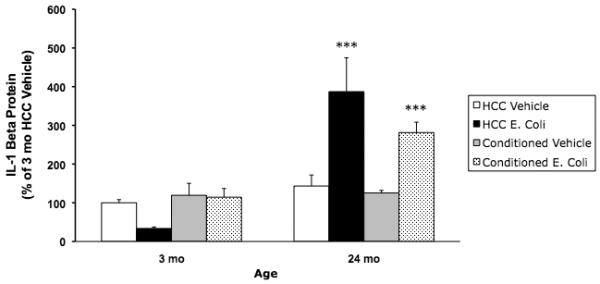
Peripheral E. coli treatment induced a protracted IL-1β protein response in the hippocampus of 24 mo animals. 3 mo and 24 mo animals were administered E. coli or vehicle i.p. 4 d post-treatment, animals underwent contextual fear conditioning or served as HCC. 30 min post-conditioning, hippocampal IL-1β was measured. Peripheral E. coli treatment increased IL-1β protein in 24 mo animals compared to all other experimental groups (*** p < .001). E. coli treatment had no effect on IL-1β in 3 mo animals. N = 4 animals/group.
Previously we have demonstrated that peripheral E. coli induces a protracted hippocampal IL-1β response only in older animals (Barrientos et al., 2006). In the present experiment, hippocampal IL-1β was measured 30 min post-conditioning, which occurred 4 d post-E. coli treatment. Consistent with our prior findings, E. coli treatment differentially increased IL-1β in 24 mo animals compared to 3 mo animals. E. coli treatment in 24 mo animals resulted in robust increases in IL-1β compared to all other experimental groups (p < .001). IL-1β levels did not significantly differ between 3 mo vehicle, 3 mo E. coli, and 24 mo vehicle groups. E. coli treatment induced comparable decreases in body weight in younger and older animals 24 h post-infection indicating that all animals received an active infection (data not shown).
3.2.2. E. coli treatment suppressed conditioning-induced Arc gene expression in 24 mo animals (Fig. 2B)
Fig. 2B.
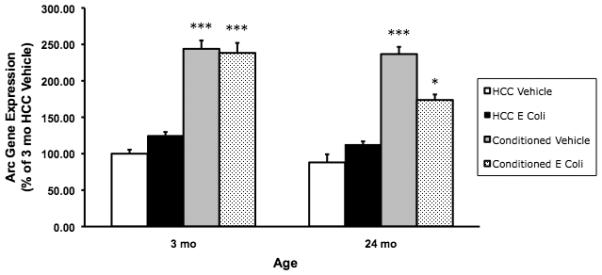
E. coli treatment suppressed conditioning-induced Arc gene expression in 24 mo animals. 3 mo and 24 mo animals were administered E. coli or vehicle i.p. 4 d post-treatment, animals underwent contextual fear conditioning or served as HCC. 30 min post-conditioning, hippocampal Arc was measured. E. coli- treatment suppressed conditioning-induced Arc (174% of control) in 24 mo animals compared to 3 mo and 24 mo vehicle conditioned animals as well as 3 mo E. coli-conditioned animals (* p < .05). In the vehicle treated animals, conditioning increased Arc gene expression to similar levels in 3 mo (244% of control) and 24 mo (237% of control) animals and significantly above 3 mo and 24 mo vehicle and E. coli HCC animals (*** p < .001). In E. coli treated 3 mo animals, conditioning-induced Arc was not altered (238% of control) compared to 3 mo and 24 mo vehicle conditioned animals. E. coli treatment in HCC animals did not significantly increase Arc expression compared to vehicle treated HCC in both 3 mo and 24 mo animals. N = 6 - 8 animals/group.
E. coli treatment, age, and conditioning interacted to alter Arc gene expression (F = 5.868(df=1,47), p = .019). In the vehicle treated animals, conditioning increased Arc gene expression to similar levels in 3 mo (244% of control) and 24 mo (237% of control) animals and significantly above 3 mo and 24 mo vehicle and E. coli HCC animals (p < .00001). In E. coli-treated 3 mo animals, conditioning-induced Arc was not altered (238% of control) compared to 3 mo and 24 mo vehicle conditioned animals. However, in 24 mo animals, E. coli treatment suppressed conditioning-induced Arc (174% of control) compared to 3 mo and 24 mo vehicle conditioned animals as well as 3 mo E. coli conditioned animals (p < .02). E. coli treatment in HCC animals did not significantly increase Arc expression compared to vehicle treated HCC in both 3 mo and 24 mo animals.
3.2.3. E. coli treatment had no effect on conditioning-induced c-Fos gene expression (Fig. 2C)
Fig. 2C.
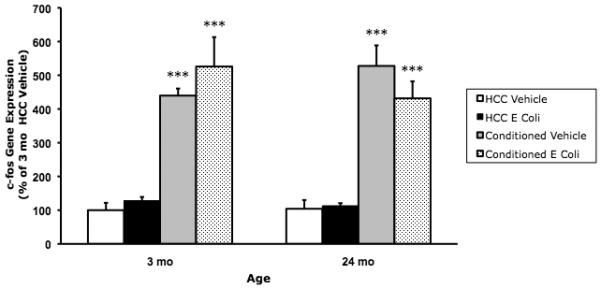
E. coli treatment did not suppress conditioning-induced c-Fos gene expression in 24 mo animals. 3 mo and 24 mo animals were administered E. coli or vehicle i.p. 4 d post-treatment, animals underwent contextual fear conditioning or served as HCC. 30 min post-conditioning, hippocampal c-Fos was measured. Conditioning induced a robust increase in c-Fos expression compared to HCC (*** p < .001). E. Coli treatment had no effect on c-Fos expression. N = 6 - 8 animals/group.
In light of the effects of E. coli treatment on conditioning-induced Arc in older animals, we examined whether this effect generalized to other IEGs, which show robust induction in response to fear conditioning (Huff et al., 2006). E. coli treatment, age, and conditioning did not interact to modulate c-Fos expression (p = .21). In addition, none of the 2-way interactions between variables had a significant effect on c-Fos expression. However, conditioning induced a robust increase in c-Fos expression in both 3 mo and 24 mo animals compared to HCC animals (p < .0001).
3.3. Experiment 3: Effect of exogenous IL-1RA on E. coli-induced suppression of hippocampal Arc 24 h post-infection
We have previously demonstrated that hippocampal IL-1β protein levels parallel the E. coli-induced memory impairments only in 24 mo animals for 8 d post-infection (Barrientos et al., 2009), however these findings were obscure as to whether IL-1β or other neuroinflammatory products mediated the memory impairments in 24 mo animals. These prior findings as well as the E. coli-induced suppression of Arc observed in experiment 1 suggest that IL-1β may play a role in the E. coli-induced memory impairments observed in 24 mo animals. Therefore, we examined whether IL-1β protein mediates the suppression of hippocampal Arc in 24 mo animals. To block the central effects of IL-1β, the anti-inflammatory cytokine IL-1RA was administered ICM concurrent with E. coli infection. 3 mo animals were not examined because E. coli failed to alter Arc expression in experiment 1 and E. coli does not impair memory in 3 mo animals at any time point post-infection (Barrientos et al., 2009).
3.3.1. Exogenous IL-1RA blocked the E. coli-induced increase in hippocampal IL-6 protein (Fig. 3A)
Fig. 3A.
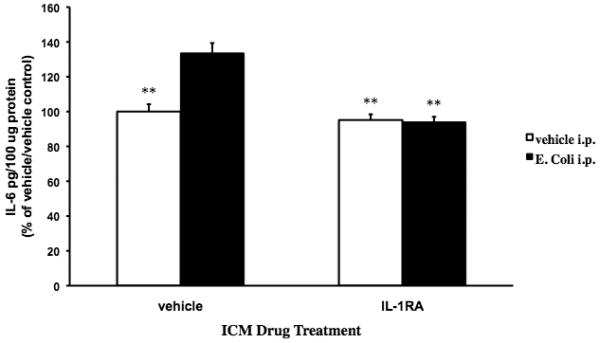
Exogenous IL-1RA blocked the E. coli-induced increase in hippocampal IL-6 protein. 24 mo animals were injected ICM with either vehicle or IL-1RA. Immediately following ICM injection, animals were administered vehicle or E. coli i.p. 24 h post-treatments, hippocampal IL-6 protein levels were measured. In ICM vehicle treated animals, E. coli i.p. treatment significantly increased IL-6 levels compared to all other experimental groups (** p < .01). IL-1RA treatment completely blocked the E. coli-induced increase in IL-6 protein resulting in IL-6 protein levels similar to the vehicle ICM/vehicle i.p. and IL-1RA ICM/vehicle i.p. groups. N = 4 animals/group.
To verify that IL-1RA blocked the neuroinflammatory effects of peripheral E. coli, hippocampal IL-6 protein levels were measured 24 h post-infection. We measured the effect of IL-1RA on IL-6 protein levels because IL-1β is a potent inducer of IL-6 in the CNS (Van Wagoner and Benveniste, 1999) and therefore E. coli-induced changes in IL-6 levels is a sensitive measure of IL-1RA anti-inflammatory effectiveness. IL-1RA treatment interacted with E. coli treatment to significantly alter IL-6 protein levels (F = 18.788(df=1,15), p = .0006). Post-hoc comparisons show that in animals receiving E. coli i.p. and vehicle ICM treatment, IL-6 levels were significantly increased above the vehicle ICM/vehicle i.p. (p < .01), IL-1RA ICM /vehicle i.p. (p < .003) and IL-1RA ICM/E. coli i.p./ (p < .002) groups. In animals that received E. coli i.p., ICM treatment with IL-1RA completely blocked the E. coli-induced IL-6 to levels that did not significantly differ from the vehicle ICM/vehicle i.p. and IL-1RA ICM/vehicle i.p. groups.
3.3.2. Exogenous IL-1RA blocked the E. coli-induced decrease in hippocampal Arc expression (Fig. 3B)
Fig. 3B.
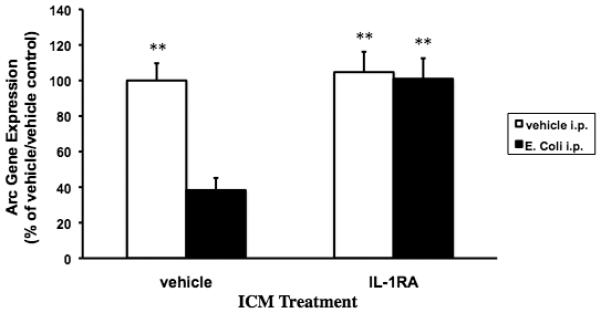
Exogenous IL-1RA blocked the E. coli-induced decrease in hippocampal Arc expression. 24 mo animals were injected ICM with either vehicle or IL-1RA. Immediately following ICM injection, animals were administered vehicle or E. coli i.p. 24 post-treatments, hippocampal Arc gene expression levels were measured. In vehicle ICM treated animals, E. coli treatment significantly decreased Arc levels compared to the vehicle ICM/vehicle i.p. (** p < .01), IL-1RA ICM/vehicle i.p. (** p < .01), and IL-1RA ICM/E. coli i.p. (** p < .01) groups. IL-1RA treatment completely blocked the E. coli-induced decrease in Arc gene expression resulting in Arc levels similar to the vehicle ICM/vehicle i.p. and IL-1RA ICM/vehicle i.p. groups. N = 5-7 animals/group.
IL-1RA ICM treatment differentially modulated hippocampal Arc expression in E. coli i.p. treated animals (F = 8.335(df=1,22), p = .0086). E. coli induced a significant decrease in Arc expression in vehicle ICM treated animals compared to vehicle ICM/vehicle i.p. (p < .002), IL-1RA ICM/vehicle i.p. (p < .002) and IL-1RA ICM/E. coli i.p. (p < .004) groups. In animals treated with IL-1RA ICM and E. coli i.p., Arc levels did not significantly differ from vehicle ICM/vehicle i.p. and IL-1RA ICM/ vehicle i.p. groups. IL-1RA and E. coli treatment did not significantly alter c-Fos expression (data not shown).
3.4. Experiment 4: Effect of exogenous IL-1RA on E. coli-induced long-term memory (LTM) impairments in 24 mo animals
Given the role of Arc in memory formation, the results of experiment 3 suggested that the E. coli-induced memory impairments observed in 24 mo animals (Barrientos et al., 2009) may be mediated by the neuroinflammatory sequelae of a peripheral infection. To investigate this possibility, we examined whether IL-1RA ICM would block the LTM impairments induced by E. coli in 24 mo animals.
3.4.1. Exogenous IL-1RA blocked the E. coli-induced LTM impairments in 24 mo animals (Fig. 4A)
Fig. 4A.
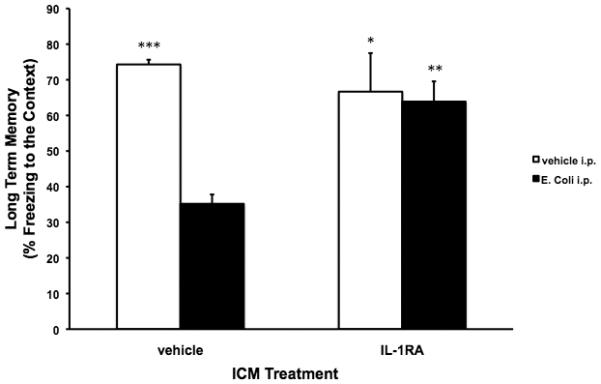
Exogenous IL-1RA blocked the E. coli-induced LTM impairments in 24 mo animals. 24 mo animals were injected ICM with either vehicle or IL-1RA. Immediately following ICM injection, animals were injected i.p. with either vehicle or E. coli. 4 d post-injections, all animals underwent contextual fear conditioning. 3 d post-conditioning, all animals were given a LTM test for the context. In vehicle ICM treated animals, E. coli treatment resulted in a significant impairment of LTM (% freezing to the context) compared to vehicle ICM/vehicle i.p. (*** p < .001), IL-1RA ICM/vehicle i.p. (* p < .05), and IL-1RA ICM/E. coli i.p. (** p < .01) groups. IL-1RA treatment completely blocked the E. coli-induced impairment in LTM resulting in LTM similar to the vehicle ICM/vehicle i.p. and IL-1RA ICM/vehicle i.p. groups. N = 4 animals/group.
IL-1RA ICM treatment interacted with E. coli i.p. treatment to significantly alter LTM (F = 8.37(df=1,12), p = .0135). Consistent with our prior studies (Barrientos et al., 2009; Barrientos et al., 2006), E. coli i.p. administration in animals treated with vehicle ICM significantly impaired hippocampus-dependent LTM (% freezing to the context) compared to the vehicle ICM/vehicle i.p. (p < .0001), IL-1RA ICM/vehicle i.p. (p < .03), and IL-1RA ICM/E. coli i.p. (p < .004) groups. In animals treated with E. coli, IL-1RA ICM treatment blocked the E. coli-induced impairment of LTM to levels similar to vehicle ICM/vehicle i.p. and IL-1RA ICM/vehicle i.p. groups. In the tone-cued fear test, all experimental groups showed robust fear of the tone, relative to the pre-tone testing session, however, there were no significant differences between the groups during the pre-tone or the tone periods (data not shown) indicating that hippocampus-independent memory processes were unaffected by E. coli treatment.
3.4.2. Exogenous IL-1RA blocked the E. coli-induced increase in hippocampal IL-6 protein 8 d post-infection (Fig. 4B)
Fig. 4B.
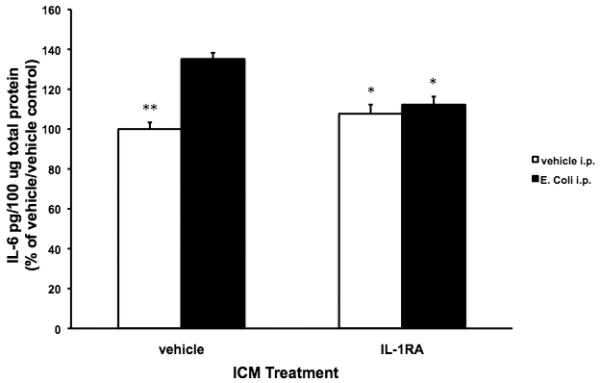
Exogenous IL-1RA blocked the E. coli-induced increase in hippocampal IL-6 protein 8 d post-infection. 24 mo animals were injected ICM with either vehicle or IL-1RA. Immediately following ICM injection, animals were injected i.p. with either vehicle or E. coli. 1 d post-test for fear of the context, hippocampal IL-6 protein was measured. In vehicle ICM treated animals, E. coli induced a significant increase in IL-6 protein compared to vehicle ICM/vehicle i.p. (** p < .01), IL-1RA ICM/vehicle i.p. (* p < .05), and IL-1RA ICM/E. coli i.p. (* p < .05) groups. IL-1RA blocked the E. coli-induced increase in hippocampal IL-6 resulting in IL-6 levels that did not significantly differ from the vehicle ICM/vehicle i.p. and IL-1RA ICM/vehicle i.p. groups. N = 4 animals/group.
To verify an E. coli-induced neuroinflammatory response as well as the anti-inflammatory effect of IL-1RA, hippocampal IL-6 protein was measured 24 h after the LTM test (i.e. 8 d post-E. coli infection). Similar to the results of experiment 3, there was a significant interaction between IL-1RA and E. coli treatment (F = 16.14(df=1,12), p = .0017). E. coli i.p. treatment significantly increased IL-6 protein in vehicle ICM treated animals compared to the vehicle ICM/vehicle i.p. (p < .002 ), IL-1RA ICM/vehicle i.p. (p < .02) and IL-1RA ICM/E. coli i.p. (p < .03) groups. IL-1RA ICM blockade of E. coli-induced IL-6 resulted in IL-6 protein levels similar to the levels observed in the vehicle ICM/vehicle i.p. and IL-1RA ICM/vehicle i.p. groups. Thus, a single administration of IL-1RA blocked the hippocampal IL-6 increase that is normally still present 8 days after E. coli infection in 24 mo animals.
4. Discussion
Prior work has shown that aging renders hippocampal-based LTM formation vulnerable to disruption by peripheral immune-challenge (Barrientos et al., 2009; Barrientos et al., 2006). At 24 mo of age, male F344 × BN F1 rats show normal memory formation in both contextual fear conditioning and spatial water maze tasks. However, for a number of days after infection with E. coli these subjects are impaired in forming LTM for both contextual fear and spatial water maze escape (hippocampal). However, LTM for conditioned fear to a tone and non-spatial water maze performance (non-hippocampal) is unaffected, as is STM for contextual fear and spatial water maze learning. Importantly, infection with E. coli has no effect on memory at all in young (3 mo) animals of the same strain (Barrientos et al., 2009; Barrientos et al., 2006). Clearly, potential mediators of aging-induced vulnerability of memory to infection have to share these properties. One such mediator is the IEG Arc (Guzowski et al., 2000). The present set of experiments demonstrates that a peripheral bacterial infection suppresses the expression of Arc in older animals, but not younger animals. In light of Arc's role in LTM formation, reductions in Arc expression may underlie the LTM impairments in older animals after exposure to a peripheral immune challenge. In the absence of infection, Arc mRNA did not differ between older and younger animals, and contextual fear conditioning induced Arc to similar levels in both vehicle treated younger and older animals. However, a peripheral immune challenge induced a protracted suppression of basal Arc expression in older animals, whereas in younger animals, E. coli infection failed to alter Arc expression. In older animals, Arc expression was suppressed 2 h, 4 h and 24 h post-infection compared to all other experimental groups. It should be noted that basal Arc expression was no longer suppressed by E. coli 96 h post-infection in 24 mo animals, however at this time point post-infection, hippocampal IL-1β remains elevated. If an ongoing neuroinflammatory process is necessary for the suppression of Arc, why does basal Arc expression normalize in the presence of a neuroinflammatory response? One explanation is that the normalization of basal Arc expression is compensatory for the prolonged suppression observed within 24 h post-infection. However, the E. coli-induced suppression of conditioning-induced Arc 96 h post-infection in 24 mo animals indicates that E. coli was still negatively regulating conditioning-induced Arc 96 h post-infection, although basal expression normalized.
Induction of Arc appears necessary for the formation of LTM, but not acquisition or STM (Guzowski et al., 2000; Plath et al., 2006). In the present study, E. coli infection partially suppressed conditioning-induced Arc 96 h post-infection only in older animals, but not younger animals. The results of experiment 1 and 2 suggest that E. coli-induced suppression of Arc could be a neural mechanism by which a peripheral immune challenge impairs LTM selectively in older animals. The present findings are consistent with studies showing that attenuating the behavioral induction of hippocampal Arc results in LTM deficits (Guzowski et al., 2000). It is important to underscore here that the present set of studies was limited to examining whole hippocampus, which precluded examining whether Arc and IL-1β co-localized within discrete hippocampal sub-regions.
Older animals show not only a cognitive vulnerability, but also a neuroinflammatory susceptibility to a peripheral immune challenge. Therefore, we explored the possibility that the neuroinflammatory response to a peripheral immune challenge impairs LTM in older animals via the suppression of Arc. Consistent with our prior findings (Barrientos et al., 2009; Barrientos et al., 2006), a single exposure to a peripheral bacterial infection in older animals resulted in a prolonged neuroinflammatory response (elevated hippocampal IL-1β and IL-6 protein), which was evident 4 d and 8 d post-infection. At 4 d post-infection, a neuroinflammatory response was not evident in young animals. We have found that peripheral infection induces a neuroinflammatory response in young animals, however the proinflammatory cytokine response resolves rapidly (within 24 h post-infection) compared to the neuroimmune response in older animals (Barrientos et al., 2009). This protracted proinflammatory response in the CNS of 24 mo animals reflects an age-related shift in the central microenvironment from anti-inflammatory to proinflammatory as characterized by microglial and astroglial activation and sensitization to proinflammatory stimuli (Frank et al., 2006; Huang et al., 2008).
To test whether proinflammatory cytokines mediate the E. coli-induced suppression of hippocampal Arc, we assessed the effects of exogenously administered IL-1RA on E. coli-induced suppression of Arc. A single ICM administration of IL-1RA completely blocked the E. coli-induced suppression of Arc 24 h post-infection in 24 mo animals and abrogated the E. coli-induced increase in the proinflammatory cytokine IL-6. The ability of IL-1RA to block E. coli-induced suppression of Arc suggested that IL-1RA had the potential to ameliorate the LTM impairments induced by E. coli in 24 mo animals. Indeed, IL-1RA did block the LTM impairments observed in 24 mo animals 3 d post-contextual fear conditioning (i.e. 7 d post-E. coli infection). Of note, hippocampal-independent memory (tone fear memory) was unaffected by E. coli treatment, which replicates our prior findings that hippocampal-dependent memory processes are particularly vulnerable to central proinflammatory insults (Barrientos et al., 2009). At 8 d post-infection, we found that IL-1RA administered immediately before E. coli blocked the E. coli-induced increase in hippocampal IL-6 in those animals that underwent contextual fear conditioning as well as a test for memory of the context.
It is important to note here that IL-1RA exhibits a terminal half-life < 2 h (Granowitz et al., 1993), however we observed that the anti-inflammatory effects of a single administration of IL-1RA persisted for 8 d post-E. coli infection. This raises an important question: what is the mechanism whereby IL-1RA, a protein with a short duration of action, blocks the prolonged proinflammatory cytokine response in the hippocampus of 24 mo animals after E. coli infection? Several prior findings (Barrientos et al., 2009) may help explain the durability of IL-1RA's anti-inflammatory effects observed here. First, E. coli infection induces a rapid hippocampal proinflammatory response (< 2 h post-E. coli) in both younger and older animals. Within the first 24 h post-infection, the hippocampal proinflammatory response is similar in magnitude in younger and older animals. By 24 h post-infection, the neuroinflammatory response resolves in younger animals, but remains elevated for 8 d post-infection only in 24 mo animals. Second, the E. coli-induced chronic neuroinflammation in 24 mo animals occurs despite the resolution of the peripheral cytokine response within ~ 1 d post-infection. Third, the peripheral immune response to E. coli does not differ in magnitude or duration between 3 and 24 mo animals. Finally, immune effector cells in the CNS of older animals, notably microglia, exhibit a shift towards a proinflammatory state compared to younger animals. These lines of evidence suggest that the hippocampal proinflammatory response is initiated soon after E. coli infection in both 3 and 24 mo animals, but chronically maintained in the hippocampus of older animals due to the presence of immunologically “primed” microglia or other immune effector cells. While in 3 mo animals, hippocampal neuroinflammation parallels the peripheral immune response to E. coli, 24 mo animals exhibit a disconnect between the central and peripheral immune response. Once set in motion, the neuroinflammatory response in older animals becomes a central self-sufficient phenomenon and independent of the peripheral immune response. Therefore, if an intervention can block the initial transduction of the peripheral immune signal to the CNS, the induction of a chronic neuroinflammatory state may be obviated in older animals. Indeed, the present results suggest that IL-1RA did serve to block the transduction of the peripheral proinflammatory signal to the CNS within the first few hours post-infection. This is because IL-1RA given at the time of E. coli blocked the E. coli-induced increase in hippocampal IL-6 protein at 24 h and 8 d post-infection, suggesting that IL-1RA prevented the initiation of a chronic neuroinflammatory cascade in 24 mo animals. These results are consistent with a recent report showing that a single administration of IL-1RA at a dose (4 ug ICV) considerably lower than the dose we utilized blocks the E. coli LPS-induced sickness and IL-1β mRNA responses in aged mice (Abraham and Johnson, 2008). However, it is important to note that in the rat, larger doses of IL-1RA (50 – 200 ug ICV), comparable to the dose utilized here (112 ug ICM), are required to attenuate the sickness response (fever) to a peripheral immune challenge (Cartmell et al., 1999; Luheshi et al., 1996; Miller et al., 1997), whereas IL-1RA (60 ug ICV) failed to attenuate the behavioral responses to peripheral LPS (Bluthe et al., 1992).
Taken together, the effects found here of the anti-inflammatory cytokine IL-1RA on E. coli suppression of Arc and LTM suggest that central proinflammatory cytokines play a salient role in the suppression of Arc and impairments of LTM by a peripheral immune challenge in older animals. The present findings are consistent with a body of evidence showing that neuroinflammation or proinflammatory cytokines elevated above basal levels impairs memory formation (Barrientos et al., 2002; Barrientos et al., 2004; Hein et al., 2007; Pugh et al., 2000; Tanaka et al., 2006) and suppresses LTP in vivo and in vitro (Cumiskey et al., 2007; Curran and O'Connor, 2003; Tancredi et al., 2000; Vereker et al., 2000). Hippocampal LTP is impaired in aged animals in parallel with increased IL-1β (Lynch, 1998) and chronic blockade of pro-IL-1β processing in the CNS alleviates age-related deficits in LTM (Gemma et al., 2005).
In older animals exposed to a peripheral immune challenge, the suppression of conditioning-induced Arc as well as the induction of a protracted neuroimmune response further illustrates the cognitive and neuroinflammatory vulnerability to proinflammatory insults that emerges with normal aging. The present findings observed in “middle-aged” animals are highly consistent with a considerable body of work showing similar neuroimmune and cognitive changes in “aged” or senescent animals (Dilger and Johnson, 2008). The present findings may be relevant to clinical observations where, upon exposure to a challenge such as surgery, stress, or severe infection (Bekker and Weeks, 2003; Holmes et al., 2003; Lupien et al., 1997), aged humans develop cognitive impairments, which otherwise were not apparent pre-challenge. The present data suggest obvious targets for intervention strategies.
Acknowledgments
The present work was supported by an NIH grant (AG028271) to M.G.F., R.M.B, and S.F.M.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abraham J, Jang S, Godbout JP, Chen J, Kelley KW, Dantzer R, Johnson RW. Aging sensitizes mice to behavioral deficits induced by central HIV-1 gp120. Neurobiol Aging. 2008;29:614–621. doi: 10.1016/j.neurobiolaging.2006.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham J, Johnson RW. Central inhibition of interleukin-1beta ameliorates sickness behavior in aged mice. Brain Behav Immun. 2008 doi: 10.1016/j.bbi.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Barrientos RM, Frank MG, Hein AM, Higgins EA, Watkins LR, Rudy JW, Maier SF. Time course of hippocampal IL-1 beta and memory consolidation impairments in aging rats following peripheral infection. Brain Behav Immun. 2009;23:46–54. doi: 10.1016/j.bbi.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos RM, Higgins EA, Biedenkapp JC, Sprunger DB, Wright-Hardesty KJ, Watkins LR, Rudy JW, Maier SF. Peripheral infection and aging interact to impair hippocampal memory consolidation. Neurobiol Aging. 2006;27:723–732. doi: 10.1016/j.neurobiolaging.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Barrientos RM, Higgins EA, Sprunger DB, Watkins LR, Rudy JW, Maier SF. Memory for context is impaired by a post context exposure injection of interleukin-1 beta into dorsal hippocampus. Behav Brain Res. 2002;134:291–298. doi: 10.1016/s0166-4328(02)00043-8. [DOI] [PubMed] [Google Scholar]
- Barrientos RM, Sprunger DB, Campeau S, Higgins EA, Watkins LR, Rudy JW, Maier SF. Brain-derived neurotrophic factor mRNA downregulation produced by social isolation is blocked by intrahippocampal interleukin-1 receptor antagonist. Neuroscience. 2003;121:847–853. doi: 10.1016/s0306-4522(03)00564-5. [DOI] [PubMed] [Google Scholar]
- Barrientos RM, Sprunger DB, Campeau S, Watkins LR, Rudy JW, Maier SF. BDNF mRNA expression in rat hippocampus following contextual learning is blocked by intrahippocampal IL-1beta administration. J Neuroimmunol. 2004;155:119–126. doi: 10.1016/j.jneuroim.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Bekker AY, Weeks EJ. Cognitive function after anaesthesia in the elderly. Best Pract Res Clin Anaesthesiol. 2003;17:259–272. doi: 10.1016/s1521-6896(03)00005-3. [DOI] [PubMed] [Google Scholar]
- Bluthe RM, Dantzer R, Kelley KW. Effects of interleukin-1 receptor antagonist on the behavioral effects of lipopolysaccharide in rat. Brain Res. 1992;573:318–320. doi: 10.1016/0006-8993(92)90779-9. [DOI] [PubMed] [Google Scholar]
- Bramham CR, Worley PF, Moore MJ, Guzowski JF. The immediate early gene arc/arg3.1: regulation, mechanisms, and function. J Neurosci. 2008;28:11760–11767. doi: 10.1523/JNEUROSCI.3864-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartmell T, Luheshi GN, Rothwell NJ. Brain sites of action of endogenous interleukin-1 in the febrile response to localized inflammation in the rat. J Physiol. 1999;518(Pt 2):585–594. doi: 10.1111/j.1469-7793.1999.0585p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Buchanan JB, Sparkman NL, Godbout JP, Freund GG, Johnson RW. Neuroinflammation and disruption in working memory in aged mice after acute stimulation of the peripheral innate immune system. Brain Behav Immun. 2008;22:301–311. doi: 10.1016/j.bbi.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Cumiskey D, Curran BP, Herron CE, O'Connor JJ. A role for inflammatory mediators in the IL-18 mediated attenuation of LTP in the rat dentate gyrus. Neuropharmacology. 2007;52:1616–1623. doi: 10.1016/j.neuropharm.2007.03.006. [DOI] [PubMed] [Google Scholar]
- Curran BP, O'Connor JJ. The inhibition of long-term potentiation in the rat dentate gyrus by pro-inflammatory cytokines is attenuated in the presence of nicotine. Neurosci Lett. 2003;344:103–106. doi: 10.1016/s0304-3940(03)00440-3. [DOI] [PubMed] [Google Scholar]
- Davis S, Bozon B, Laroche S. How necessary is the activation of the immediate early gene zif268 in synaptic plasticity and learning? Behav Brain Res. 2003;142:17–30. doi: 10.1016/s0166-4328(02)00421-7. [DOI] [PubMed] [Google Scholar]
- Dinarello CA. Interleukin-1. Cytokine Growth Factor Rev. 1997;8:253–265. doi: 10.1016/s1359-6101(97)00023-3. [DOI] [PubMed] [Google Scholar]
- Frank MG, Barrientos RM, Biedenkapp JC, Rudy JW, Watkins LR, Maier SF. mRNA up-regulation of MHC II and pivotal pro-inflammatory genes in normal brain aging. Neurobiol Aging. 2006;27:717–722. doi: 10.1016/j.neurobiolaging.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Gemma C, Fister M, Hudson C, Bickford PC. Improvement of memory for context by inhibition of caspase-1 in aged rats. Eur J Neurosci. 2005;22:1751–1756. doi: 10.1111/j.1460-9568.2005.04334.x. [DOI] [PubMed] [Google Scholar]
- Gibertini M, Newton C, Friedman H, Klein TW. Spatial learning impairment in mice infected with Legionella pneumophila or administered exogenous interleukin-1-beta. Brain Behav Immun. 1995;9:113–128. doi: 10.1006/brbi.1995.1012. [DOI] [PubMed] [Google Scholar]
- Godbout JP, Chen J, Abraham J, Richwine AF, Berg BM, Kelley KW, Johnson RW. Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. Faseb J. 2005;19:1329–1331. doi: 10.1096/fj.05-3776fje. [DOI] [PubMed] [Google Scholar]
- Granowitz EV, Porat R, Mier JW, Orencole SF, Callahan MV, Cannon JG, Lynch EA, Ye K, Poutsiaka DD, Vannier E, et al. Hematologic and immunomodulatory effects of an interleukin-1 receptor antagonist coinfusion during low-dose endotoxemia in healthy humans. Blood. 1993;82:2985–2990. [PubMed] [Google Scholar]
- Guzowski JF, Lyford GL, Stevenson GD, Houston FP, McGaugh JL, Worley PF, Barnes CA. Inhibition of activity-dependent arc protein expression in the rat hippocampus impairs the maintenance of long-term potentiation and the consolidation of long-term memory. J Neurosci. 2000;20:3993–4001. doi: 10.1523/JNEUROSCI.20-11-03993.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzowski JF, Setlow B, Wagner EK, McGaugh JL. Experience-dependent gene expression in the rat hippocampus after spatial learning: a comparison of the immediate-early genes Arc, c-fos, and zif268. J Neurosci. 2001;21:5089–5098. doi: 10.1523/JNEUROSCI.21-14-05089.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauss-Wegrzyniak B, Dobrzanski P, Stoehr JD, Wenk GL. Chronic neuroinflammation in rats reproduces components of the neurobiology of Alzheimer's disease. Brain Res. 1998;780:294–303. doi: 10.1016/s0006-8993(97)01215-8. [DOI] [PubMed] [Google Scholar]
- Hein AM, Stutzman DL, Bland ST, Barrientos RM, Watkins LR, Rudy JW, Maier SF. Prostaglandins are necessary and sufficient to induce contextual fear learning impairments after interleukin-1 beta injections into the dorsal hippocampus. Neuroscience. 2007;150:754–763. doi: 10.1016/j.neuroscience.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes C, El-Okl M, Williams AL, Cunningham C, Wilcockson D, Perry VH. Systemic infection, interleukin 1beta, and cognitive decline in Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2003;74:788–789. doi: 10.1136/jnnp.74.6.788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Henry CJ, Dantzer R, Johnson RW, Godbout JP. Exaggerated sickness behavior and brain proinflammatory cytokine expression in aged mice in response to intracerebroventricular lipopolysaccharide. Neurobiol Aging. 2007 doi: 10.1016/j.neurobiolaging.2007.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Henry CJ, Dantzer R, Johnson RW, Godbout JP. Exaggerated sickness behavior and brain proinflammatory cytokine expression in aged mice in response to intracerebroventricular lipopolysaccharide. Neurobiol Aging. 2008;29:1744–1753. doi: 10.1016/j.neurobiolaging.2007.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huff NC, Frank M, Wright-Hardesty K, Sprunger D, Matus-Amat P, Higgins E, Rudy JW. Amygdala regulation of immediate-early gene expression in the hippocampus induced by contextual fear conditioning. J Neurosci. 2006;26:1616–1623. doi: 10.1523/JNEUROSCI.4964-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JD, O'Connor KA, Watkins LR, Maier SF. The role of IL-1beta in stress-induced sensitization of proinflammatory cytokine and corticosterone responses. Neuroscience. 2004;127:569–577. doi: 10.1016/j.neuroscience.2004.05.046. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Fanselow MS. Modality-specific retrograde amnesia of fear. Science. 1992;256:675–677. doi: 10.1126/science.1585183. [DOI] [PubMed] [Google Scholar]
- Link W, Konietzko U, Kauselmann G, Krug M, Schwanke B, Frey U, Kuhl D. Somatodendritic expression of an immediate early gene is regulated by synaptic activity. Proc Natl Acad Sci U S A. 1995;92:5734–5738. doi: 10.1073/pnas.92.12.5734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Luheshi G, Miller AJ, Brouwer S, Dascombe MJ, Rothwell NJ, Hopkins SJ. Interleukin-1 receptor antagonist inhibits endotoxin fever and systemic interleukin-6 induction in the rat. Am J Physiol. 1996;270:E91–95. doi: 10.1152/ajpendo.1996.270.1.E91. [DOI] [PubMed] [Google Scholar]
- Lupien SJ, Gaudreau S, Tchiteya BM, Maheu F, Sharma S, Nair NP, Hauger RL, McEwen BS, Meaney MJ. Stress-induced declarative memory impairment in healthy elderly subjects: relationship to cortisol reactivity. J Clin Endocrinol Metab. 1997;82:2070–2075. doi: 10.1210/jcem.82.7.4075. [DOI] [PubMed] [Google Scholar]
- Lyford GL, Yamagata K, Kaufmann WE, Barnes CA, Sanders LK, Copeland NG, Gilbert DJ, Jenkins NA, Lanahan AA, Worley PF. Arc, a growth factor and activity-regulated gene, encodes a novel cytoskeleton-associated protein that is enriched in neuronal dendrites. Neuron. 1995;14:433–445. doi: 10.1016/0896-6273(95)90299-6. [DOI] [PubMed] [Google Scholar]
- Lynch MA. Age-related impairment in long-term potentiation in hippocampus: a role for the cytokine, interleukin-1 beta? Prog Neurobiol. 1998;56:571–589. doi: 10.1016/s0301-0082(98)00054-9. [DOI] [PubMed] [Google Scholar]
- Miller AJ, Hopkins SJ, Luheshi GN. Sites of action of IL-1 in the development of fever and cytokine responses to tissue inflammation in the rat. Br J Pharmacol. 1997;120:1274–1279. doi: 10.1038/sj.bjp.0701049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow BA, Elsworth JD, Inglis FM, Roth RH. An antisense oligonucleotide reverses the footshock-induced expression of fos in the rat medial prefrontal cortex and the subsequent expression of conditioned fear-induced immobility. J Neurosci. 1999;19:5666–5673. doi: 10.1523/JNEUROSCI.19-13-05666.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oitzl MS, van Oers H, Schobitz B, de Kloet ER. Interleukin-1 beta, but not interleukin-6, impairs spatial navigation learning. Brain Res. 1993;613:160–163. doi: 10.1016/0006-8993(93)90468-3. [DOI] [PubMed] [Google Scholar]
- Plath N, Ohana O, Dammermann B, Errington ML, Schmitz D, Gross C, Mao X, Engelsberg A, Mahlke C, Welzl H, Kobalz U, Stawrakakis A, Fernandez E, Waltereit R, Bick-Sander A, Therstappen E, Cooke SF, Blanquet V, Wurst W, Salmen B, Bosl MR, Lipp HP, Grant SG, Bliss TV, Wolfer DP, Kuhl D. Arc/Arg3.1 is essential for the consolidation of synaptic plasticity and memories. Neuron. 2006;52:437–444. doi: 10.1016/j.neuron.2006.08.024. [DOI] [PubMed] [Google Scholar]
- Proescholdt MG, Hutto B, Brady LS, Herkenham M. Studies of cerebrospinal fluid flow and penetration into brain following lateral ventricle and cisterna magna injections of the tracer [14C]inulin in rat. Neuroscience. 2000;95:577–592. doi: 10.1016/s0306-4522(99)00417-0. [DOI] [PubMed] [Google Scholar]
- Pugh CR, Johnson JD, Martin D, Rudy JW, Maier SF, Watkins LR. Human immunodeficiency virus-1 coat protein gp120 impairs contextual fear conditioning: a potential role in AIDS related learning and memory impairments. Brain Res. 2000;861:8–15. doi: 10.1016/s0006-8993(99)02445-2. [DOI] [PubMed] [Google Scholar]
- Pugh CR, Kumagawa K, Fleshner M, Watkins LR, Maier SF, Rudy JW. Selective effects of peripheral lipopolysaccharide administration on contextual and auditory-cue fear conditioning. Brain Behav Immun. 1998;12:212–229. doi: 10.1006/brbi.1998.0524. [DOI] [PubMed] [Google Scholar]
- Shaw KN, Commins S, O'Mara SM. Lipopolysaccharide causes deficits in spatial learning in the watermaze but not in BDNF expression in the rat dentate gyrus. Behav Brain Res. 2001;124:47–54. doi: 10.1016/s0166-4328(01)00232-7. [DOI] [PubMed] [Google Scholar]
- Steward O, Wallace CS, Lyford GL, Worley PF. Synaptic activation causes the mRNA for the IEG Arc to localize selectively near activated postsynaptic sites on dendrites. Neuron. 1998;21:741–751. doi: 10.1016/s0896-6273(00)80591-7. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Ide M, Shibutani T, Ohtaki H, Numazawa S, Shioda S, Yoshida T. Lipopolysaccharide-induced microglial activation induces learning and memory deficits without neuronal cell death in rats. J Neurosci Res. 2006;83:557–566. doi: 10.1002/jnr.20752. [DOI] [PubMed] [Google Scholar]
- Tancredi V, D'Antuono M, Cafe C, Giovedi S, Bue MC, D'Arcangelo G, Onofri F, Benfenati F. The inhibitory effects of interleukin-6 on synaptic plasticity in the rat hippocampus are associated with an inhibition of mitogen-activated protein kinase ERK. J Neurochem. 2000;75:634–643. doi: 10.1046/j.1471-4159.2000.0750634.x. [DOI] [PubMed] [Google Scholar]
- Thomson LM, Sutherland RJ. Systemic administration of lipopolysaccharide and interleukin-1beta have different effects on memory consolidation. Brain Res Bull. 2005;67:24–29. doi: 10.1016/j.brainresbull.2005.05.024. [DOI] [PubMed] [Google Scholar]
- Tzingounis AV, Nicoll RA. Arc/Arg3.1: linking gene expression to synaptic plasticity and memory. Neuron. 2006;52:403–407. doi: 10.1016/j.neuron.2006.10.016. [DOI] [PubMed] [Google Scholar]
- Van Wagoner NJ, Benveniste EN. Interleukin-6 expression and regulation in astrocytes. J Neuroimmunol. 1999;100:124–139. doi: 10.1016/s0165-5728(99)00187-3. [DOI] [PubMed] [Google Scholar]
- Vereker E, O'Donnell E, Lynch MA. The inhibitory effect of interleukin-1beta on long-term potentiation is coupled with increased activity of stress-activated protein kinases. J Neurosci. 2000;20:6811–6819. doi: 10.1523/JNEUROSCI.20-18-06811.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]