

Abstract
The mechanism of status epilepticus-induced neuronal death in the immature brain is not fully understood. In the present study, we examined the contribution of caspases in our lithium-pilocarpine model of status epilepticus in 14 days old rat pups. In CA1, upregulation of caspase-8, but not caspase-9, preceded caspase-3 activation in morphologically necrotic cells. Pretreatment with a pan-caspase inhibitor provided neuroprotection, showing that caspase activation was not an epiphenomenon but contributed to neuronal necrosis. By contrast, upregulation of active caspase-9 and caspase-3, but not caspase-8, was detected in apoptotic dentate gyrus neurons, which were immunoreactive for doublecortin and calbindin-negative, two features of immature neurons. These results suggest that, in cells which are aligned in series as parts of the same excitatory hippocampal circuit, the same seizures induce neuronal death through different mechanisms. The regional level of neuronal maturity may be a determining factor in the execution of a specific death program.
Keywords: Necrosis, apoptosis, caspase, doublecortin, hippocampus, status epilepticus, pilocarpine, development
Introduction
The immature brain is thought to be more susceptible but less vulnerable to status epilepticus (SE) than the adult brain. Epidemiologic data indicate that the immature brain is highly susceptible to developing epileptic seizures (Hauser, 1994), and that SE occurs more frequently in children than in adults (DeLorenzo et al., 1995, 1996). Experimental SE induces neuronal death in the developing brain (Thompson and Wasterlain, 1997, 1998; Sankar et al., 1998; Kubova et al., 2001; Silva et al., 2005; Nairismagi et al., 2006), but its mechanisms have not been fully characterized.
In our experimental model of SE induced by lithium-pilocarpine in fourteen days old (P14) rat pups (Sankar et al., 1998, Suchomelova et al 2006), neuronal damage predominates in CA1 pyramids and is milder in dentate gyrus (DG). One to three days following SE, many CA1 neurons have ultrastructural features of necrosis characterized by severe cytoplasmic swelling, pyknosis of the nucleus and tigroid fragmentation of the chromatin (Niquet et al., 2007). Interestingly, DNA fragmentation, detected by TUNEL assay, and caspase-3 activation were found in many CA1 eosinophilic neurons displaying pyknotic nuclei (Tan et al., 2002; Niquet et al., 2007). These findings raise the possibility that SE induces neuronal necrosis in the developing brain through a caspase-dependent program, but light microscopy could not unequivocally show whether necrosis and caspase activation occurred in the same cells. We have previously reported the contribution of a mitochondrial mechanism involving caspase activation to neuronal necrosis following hypoxia-ischemia or glutamate-induced excitotoxicity (Niquet et al., 2003, 2006; Seo et al., 2009). The present study used postembedding EM immunohistochemistry to examine these mechanisms in seizure-induced neuronal injury.
Traditional mechanisms for caspase-3 activation involve the intrinsic and extrinsic cell death pathways, through caspase-9 and caspase-8 activation, respectively (Kroemer et al., 2007). In adult rats, seizures can activate either the intrinsic or the extrinsic pathway of cell death (Henshall et al., 2001a, b). This study carried out a semi-quantitative analysis to determine the hippocampal distribution and the time course of caspase-3, −9 and −8 activation and/or expression following SE in the immature brain. We found that injured CA1 and DG neurons both expressed the active form of caspase-3, but showed upregulation of different initiator caspases: caspase-8 was upregulated in CA1, while active caspase-9 was upregulated in DG, demonstrating that the same seizures can lead to region-specific cell death pathways. Furthermore, the highly differentiated CA1 neurons displayed the morphological features of necrosis, the most common mode of seizure-induced death in adult neurons. By contrast, injured DG neurons were morphologically apoptotic and located in the inner, most recently formed layers of granule cells. They expressed doublecortin, a marker of immaturity, and were devoid of calbindin, a marker of maturity. These results suggest that the level of an individual neuron’s maturation may be more important than the level of maturation of the whole organism in determining neuronal vulnerability to seizures.
Experimental procedures
Animals
P14 Wistar rat pups of either sex (Simonsen Lab, Gilroy, CA) were used. The day of birth was considered as day 0. Pups were housed with their dams in a temperature- and humidity- controlled room with 12 h light-dark cycles (7 am–7 pm) and had free access to food. All experiments were conducted with the approval and in accordance with the regulations of the Institutional Animal Care and Use Committee of West Los Angeles VA Medical Center.
Induction of SE
13 days old (P13) rat pups were administered lithium chloride (3 mEq/kg; #L-0505 Sigma, St. Louis MO, USA) intraperitoneally (i.p.) and, 20 h later, SE was induced with subcutaneous pilocarpine hydrochloride (60 mg/kg; #P6503 Sigma) as described previously (Sankar et al., 1998). Control rats were given an equal volume of saline subcutaneously. Behavioral seizures were scored using a modified Racine (1972) scale: (1) behavioral arrest, (2) rhythmic head nodding, (3) forelimb clonus with hyperextension of the tail, (4) forelimb clonus with rearing, (5) Bilateral clonus with loss of postural control and hyperextension of the tail. Only animals reaching SE (defined as near-continuous seizure activity lasting over 10 min) were included in the study. After SE, pups received 5% of their body weight of isotonic 5% dextrose in water subcutaneously to avoid dehydration without stressing the cardiovascular system. Temperature and time of separation from the mother were strictly controlled, since separation alone can trigger neuronal apoptosis in rat pups (Lee et al., 2001).
Preparation of tissue for immunohistochemistry and detection of neuronal damage
Rats were anesthetized with an overdose of pentobarbital (100 mg/kg i.p.) 7 (n=10), 24 (n=13), 48 (n=16), or 72 h (n=16) after induction of SE. Then, pups underwent transcardiac perfusion with 4% (caspase-3 and −9) or 2% (caspase-8) phosphate-buffered paraformaldehyde (#P-6148 Sigma; vol. 100 ml at 7 ml/min). Control animals (n=12) where subjected to the same procedure. Brains were kept in situ at 4 °C overnight, after which they were removed and postfixed in the same perfusate for 2–3 h. Subsequently, brains were dehydrated, embedded in paraffin and cut into 10-μm-thick coronal sections at the level of dorsal hippocampus at 2.0 to 1.8 mm anterior to the horizontal zero plane that passes through the interaural line (A 2.0 to 1.8 mm; Sherwood and Timiras, 1970). Brain sections were deparaffinized, rehydrated in a graded series of ethanol and dH2O with a final wash in 0.1 M phosphate buffer (PB) before any procedure. TUNEL staining and neuronal damage were assessed in either 4% or 2% paraformaldehyde fixed brain sections.
Immunohistochemical studies
Briefly, coronal brain sections were subjected to an antigen retrieval procedure (#HK080-9K; BioGenex Laboratories), in which the slides were put in a slide tray with 10 mM citrate solution (pH 6.0) in a microwave oven, heated at high power for 2 min 30 s, at low power for 10 min, after which they were cooled to room temperature. Slides were then rinsed in distilled water, washed in 0.1 M PB for 10 min and incubated at room temperature for 1 h in a blocking buffer: 2% gelatin (#G-2500, Sigma) in 0.1 M PB for active caspase-3 (caspase-3a) or active caspase-9 (caspase-9a) immunodetection, or 5% donkey serum (#530; Chemicon) in 0.1 M PB, for caspase-8 immunolabeling. They were then incubated with primary antibody [rabbit polyclonal anti-caspase-3a (#9661; Cell Signaling Technology), anti-caspase-9a (#9507; Cell Signaling Technology) and monoclonal mouse anti-NeuN (#MAB377 Chemicon)], diluted 1:100 with blocking buffer, two nights at 4 °C in a humidified chamber. For caspase-8 immunolabeling, sections were incubated with mouse monoclonal anti-total caspase-8 antibody (1:100; #sc-5263; Santa Cruz Biotechnology) overnight at 4°C in a humidified chamber. For doublecortin immunodetection, sections were incubated for two days with goat polyclonal anti-doublecortin antibody (1/1000; #sc-8066; Santa Cruz Biotechnology). For calbindin imunolabelling, sections were incubated with mouse monoclonal anti-calbindin-D-28K antibody (C9848; Sigma-Aldrich) diluted 1/300 in the blocking buffer containing 0.3 % Triton. Then, sections were washed in PB three times and were exposed to corresponding donkey secondary antibodies linked to fluorochrome Alexa Fluor (Molecular probes) diluted 1:100 with the blocking solution for 2 h at room temperature. Slides were then washed three times in PB for 10 min. Sections were counterstained with the chromatin dye Hoechst 33342 (#H-1399 Molecular Probes) and mounted with buffer or aqueous mounting medium (#MØ2 Biomeda) to perform cell counts. As a negative control, sections were incubated without primary antibodies or non-immune rabbit, mouse or goat immunoglobulins.
Determination of caspase-3 activity on unfixed frozen sections
In situ labeling of caspase-3 activity was performed using a detection kit (APO LOGIX FAM; Cell Technology Inc) following the manufacturer’s instructions. Briefly, frozen, unfixed brains from control and experimental pups (n=6) were cut coronally on a cryostat. Sections (15 μM) were labeled for 1h at 37°C with FAM-DEVD-fmk, a cell-permeant, irreversible inhibitor of caspase-3. After 3 washes, the sections were fixed with a paraformaldehyde solution and counterstained with Hoechst 33342.
Systemic administration of a pan-caspase inhibitor
Quinolyl-valyl-O-methylaspartyl-[−2, 6-difluorophenoxy]-methyl ketone (Q-VD-OPh; #OPH001; R&D systems) is a cell permeable, irreversible, third-generation broad-spectrum caspase inhibitor (Caserta et al., 2003; Yang et al., 2004; Chauvier et al., 2007; Renolleau et al., 2007). A 10 mM stock solution was prepared in dimethylsulfoxide (DMSO; #D2650 Sigma). Male rats were injected i.p. with vehicle (10% DMSO in 0.9% saline; n=9) or Q-VD-OPh (n=9) at a dose of 1 mg/kg in 10% DMSO 5 min before pilocarpine administration similarly to described by Renolleau et al. (2007). Behavioral seizures were measured as described above. Twenty-four hours after SE induction, animals were anesthetized with an overdose of pentobarbital and perfused transcardially with 4% phosphate buffered paraformaldehyde. Brains were kept in situ at 4 °C overnight and processed as described above.
Histological evaluation of the extent of neuronal damage
Neuronal damage after SE induction was assessed with a modified hematoxylin & eosin staining (H&E; Sankar et al., 1998), in sections that previously were used for immunohistochemistry as described by Niquet et al. (2007). Injured neurons were identified, under light and fluorescent microscopy, by their eosinophilic (acidophilic) cytoplasm and pyknotic or fragmented nuclei. Neuronal injury in rats treated with QVD-OPh was addressed with Fluoro-Jade B (#AG310, Chemicon). Sections for Fluoro-Jade B staining were incubated with 0.06% KMnO4 (#P-9810, Sigma) followed by 0.001% Fluoro-Jade B and evaluated under fluorescent light (Schmued et al., 1997).
Cell counting
For immunohistochemical studies, count of caspase-3a, caspase-9a or caspase-8-positive neurons was performed bilaterally in the CA1-subiculum and DG granule cell layer. Three sections per animal were randomly selected from the dorsal hippocampus (A 2.0 to 1.8 mm; Sherwood and Timiras, 1970).
For the caspase inhibition experiment, a count of Fluoro-Jade B-positive neurons was performed by unbiased stereological methods using the physical disector. Digital images were taken with an Olympus 20X objective with sufficient depth of field to identify cells at all depths in the histological section in a single image. Fluorescence images of CA1-subiculum were taken from contiguous sections and were aligned using the registration function in Image-Pro (Media Cybernetics, Silver Spring, MD) to superimpose 4 fiduciary points (usually small blood vessels passing through both sections at near 90 degrees to the plane of sectioning) by rotation and translation of one image. By overlaying the registered images and turning one layer on and off rapidly, the relation of Fluoro-Jade B-positive cells in the two sections could be observed. Only positive cells in CA1-subiculum that occurred in one image (reference section) and not the other (lookup section) were counted. The section pair was selected in the dorsal hippocampus (A 1.8 mm; Sherwood and Timiras, 1970). Both left and right sides were counted. Counting was performed along the whole length of CA1-subiculum in each section. To compensate for anatomical variations, the length of the CA1-subiculum was measured and the number of positive cells was expressed per mm of CA1-subiculum.
Ultrastructural studies
Morphologic changes were observed by routine electron microscopy (EM) procedures. Twenty-four h after lithium-pilocarpine SE (n=6), anesthetized animals were perfused with 2.5 % glutaraldehyde and 1.6% paraformaldehyde in 0.1 M cacodylate buffer (CB). Brains were kept in situ at 4°C overnight, after which they were removed and postfixed in the same fixative. Hippocampi were dissected out and then subsectioned into approximately 2–4 mm blocks. Blocks were postfixed with 1% osmium tetroxide in CB, rinsed with distilled water, dehydrated with graded ethanols and embedded in Spurr Epon. The thin sections, stained with uranyl acetate and lead citrate, were viewed using a Philips 201C TEM. Necrosis was defined as severe cytoplasmic swelling (often with plasma membrane rupture) and a tigroid nucleus. Apoptosis was defined by fragmentation of the nucleus into large, round clumps of chromatin.
Electron microscopic immunohistochemical studies
Mode of death, i.e. necrosis or apoptosis, of caspase-3a-immunoreactive (IR) cells were evaluated by post-embedding immunogold method. Twenty-four h after lithium-pilocarpine SE, anesthetized animals (n=6) were perfused as described above with 4% phosphate-buffered paraformaldehyde. Brains were then removed and kept in the same fixative for 24 h at 4°C. Hippocampi were dissected out, subsectioned into approximately 2–4 mm blocks and washed in 0.1 M PB. Blocks were dehydrated and embedded in London Resin white. Thin sections were picked up on nickel grids, blocked with normal goat serum, fish gelatin and skimmed milk, and incubated overnight at 4°C with rabbit antibody against the active form of caspase-3 (CM1; #55150; Pharmigen). After washes in PBS, the grids were incubated in anti-rabbit immunoglobulin conjugated to 10 nm gold particles (1:60 in cold fish gelatin), followed by a wash in PBS containing 0.075% cold fish gelatin and 2.5 M NaCl and several washes in water. The 10 nm gold was intensified by silver enhancement (Ted Pella, Inc.). Grids, stained with uranyl acetate and lead citrate, were viewed on a Philips 201C TEM. As a negative control, sections were incubated without the primary antibody or with normal rabbit serum instead of primary antibody. The mode of death (necrotic or apoptotic) of 50 randomly chosen caspase-3a-IR cells was determined in CA1 from the dorsal hippocampus.
In situ detection of DNA fragmentation by TUNEL staining
DNA fragmentation was detected by the terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL), by using the ApopTag Red In situ Apoptosis Detection Kit (#S7165; Chemicon). Staining was performed according with the manufacturer instructions. Briefly, sections were incubated with proteinase K (20 μg/mL; #P6556; Sigma) followed by terminal deoxynucleotidyl transferase and unlabeled and digoxigenin-nucleotides. Then, staining was developed with a rhodamine-conjugated anti-dioxigenin antibody and counterstained with Hoechst 33342. Stain was visualized with fluorescent light. Count of TUNEL-positive cells in the CA1-subiculum and DG granule cell layer was performed on three adjacent sections from 5–8 animals perfused 7 or 24 h following SE onset and control rats.
Statistical analysis
Experimental groups that showed different variances were analyzed with non parametric statistical methods: Kruskal Wallis one way ANOVA followed by Dunn’s or Mann Whitney test versus the control (Fig 1B-C, 2A, 3A, 4 A-B). Those groups that followed a normal distribution were analyzed with parametric statistical methods: one way ANOVA followed by Bonferroni or Student t-test versus the control (graphs in Fig. 2D, 4C). In Fig. 2D (table), seizure severity was analyzed by chi-square test (Sigma stat, version 3.5). Statistical significances were considered when p< 0.05. In all graphs, data are presented as the mean ± S.E.M.
Figure 1. SE induces necrosis in CA1 and apoptosis in DG.
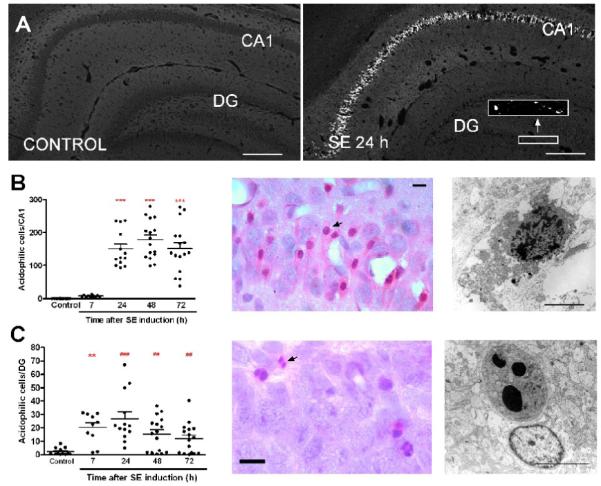
(A) Images of Fluoro-jade B staining in hippocampus from control animal and 24 h following SE. (B-C) Time course of neuronal injury in CA1 (B) and DG (C) following SE. Left panels: The graphs represent the number of eosinophilic cells detected with H&E staining from CA1 (B) and DG (C) 7–72 h after SE. Analyzed by Dunn’s test (***p<0.001, **p<0.01 vs control) or Mann Whitney test (###p<0.001, ##p<0.01 vs control). Middle panels: High magnification images of eosinophilic cells with pyknotic nuclei from CA1 (B) and fragmented nuclei from DG (C) obtained 24 h after SE. Arrows point to injured neurons. Right panels: Electron microscopy micrographs obtained 24 h after SE, showing a CA1 necrotic cell, characterized by swollen organelles, ruptured plasma membrane and tigroid fragmentation of the chromatin (B) and a DG cell with a late stage of apoptosis, characterized by large round chromatin clumps and mild organelle swelling (C). Scale bars in A = 100 μm; B-C = 25 μm (light microscopy) and 3 μm (electron microscopy).
Figure 2. SE activates caspase-3 in morphologically necrotic CA1 neurons.
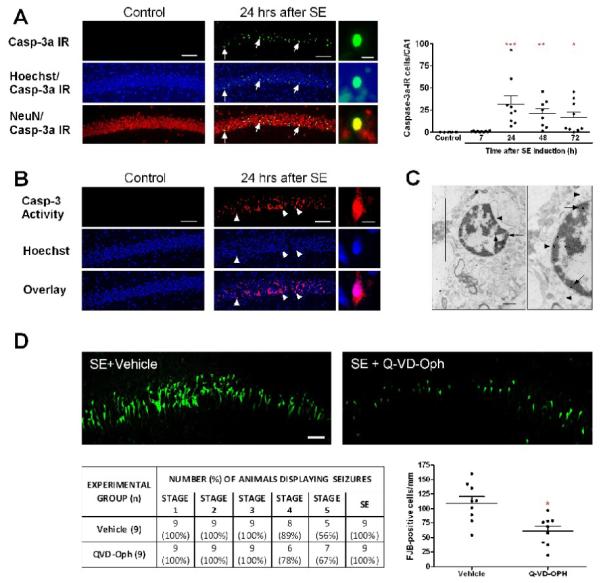
(A) Images of caspase-3a (green), NeuN (neuronal marker, red) IR and Hoechst staining (chromatin dye, blue). Arrows point to caspase-3a-IR neurons in CA1 24 h following SE. High magnification images show that caspase-3a IR is found in pyknotic nuclei. The graph represents the number of caspase-3a-IR neurons from CA1 7–72 h after SE (***p<0.001, **p<0.01, *p<0.05 vs control). (B) Images of caspase-3 activity (red) on unfixed frozen sections from control and experimental animal (24 h following SE). Arrowheads show that caspase-3 activity is mainly seen in pyknotic nuclei. (C) Example of a caspase-3a-IR neuron with necrotic morphology 24 h following SE. Gold particles, revealing caspase-3 localization (arrows), are mainly visible in the nucleus. Arrowheads show mitochondrial swelling. (D) The images show fluoro-Jade B staining in CA1 from a rat pretreated with vehicle and a rat pretreated with Q-VD-OPh 24 h following SE. The table shows that Q-VD-Oph did not modify the seizure severity (Chi-square= 0.619, df= 5, p = 0.987). The graph shows a reduction of the number of Fluoro-Jade B-positive cells (*p=0.003, Student’s t test vs vehicle). Scale bars in A-B = 100 μm (low magnification images) and 25 μm (high magnification images). Scale bars in D = 25 μm and C = 1 μm.
Figure 3. SE activates the extrinsic pathway in CA1 neurons.
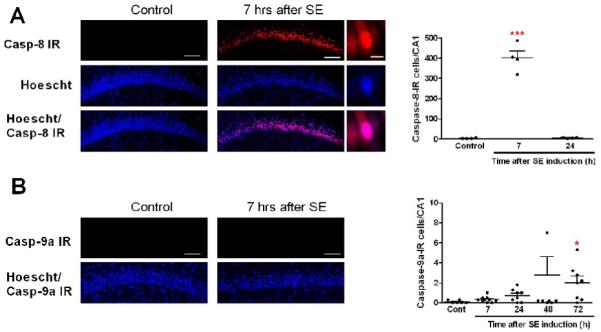
(A) Images of caspase-8 IR (red) show that many CA1 cells are IR 7 h following SE. High magnification images show that caspase-8 IR is mainly seen in the nuclei. The graph represents the number of caspase-8-IR cells from CA1 7–24 h after SE (***p<0.001 vs control). (B) Images showing the absence of caspase-9a IR in CA1. The graph represents the number of caspase-9a-IR cells from CA1 7–72 h after SE (*p<0.05 vs control). Scale bars in A-B = 100 μm (low magnification images) and 25 μm (high magnification images).
Figure 4. SE activates the intrinsic pathway in morphologically apoptotic DG neurons.
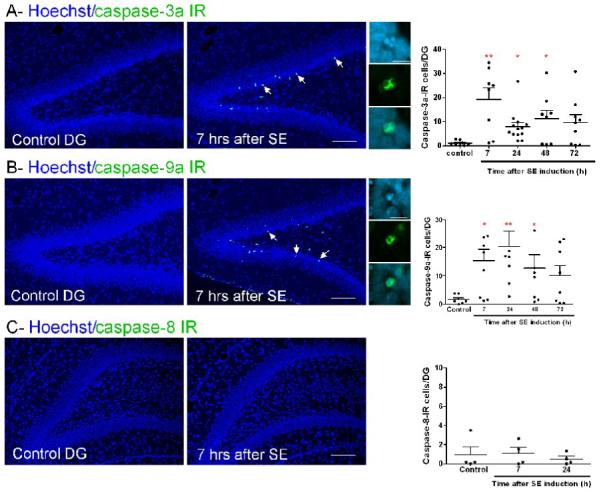
(A-B) Images of caspase-3a or −9a IR (green) and Hoechst staining (chromatin dye, blue). Arrows point to IR cells in DG 7 h following SE. High magnification images show that IR is observed mainly in the cytosol of cells with fragmented nuclei. The graphs represent the number of IR cells in DG 7–72 h after SE (**p<0.001, *p<0.05 vs control). (C) Images showing the absence of caspase-8 IR in DG. The graph represents the number of caspase-8-IR cells in DG 7–24 h after SE. Scale bars = 100 μm (low magnification images) and 25 μm (high magnification images).
Results
Neuronal damage induced by SE in CA1 and DG
By H&E staining, no sign of neuronal injury, indicated for the presence of acidophilic neurons with a pyknotic nucleus, was detected in CA1 of control animals, but rare isolated eosinophilic cells were observed in control DG. Neuronal damage in hippocampus was examined at 7, 24, 48 and 72 h after SE induction (Fig. 1). In CA1, eosinophilic neurons were detected 24 to 72 h following SE (Fig. 1B); whereas in DG, eosinophilic cells were first observable 7 h after SE (Fig. 1C). CA1 injured neurons were characterized by eosinophilic cytosol and pyknotic nucleus and by EM displayed a necrotic morphology with cytoplasmic and organelle swelling, loss of plasma membrane and a tigroid nucleus (Fig. 1B). In DG, most of the neurons displayed fragmented nuclei, and by EM displayed an apoptotic morphology with large rounded chromatin clumps, loss of nuclear membrane and preservation of the plasma membrane (Fig 1C). It is worth mentioning that 7 h following SE, some CA1 pyramidal neurons displayed abnormal morphology, but were not eosinophilic. At that time, our previous ultrastructural studies of CA1 pyramidal neurons revealed organelle swelling whereas the nucleus was almost intact, which is compatible with an early stage of necrosis (Niquet et al., 2007).
SE activates caspase-3 in CA1
Activation of caspase-3 may lead to DNA fragmentation and/or cell death. The contribution of caspase-3 to SE-induced neuronal death was assessed by light immunohistochemistry using an antibody which recognizes caspase-3a, the active form of that enzyme. Sparse, scattered staining was detected in CA1 and DG from control rats. The number of caspase-3a-IR cells was significantly increased in CA1 neurons 24 and 48 h following SE, and this was still evident at 72 h (Fig. 2A). It is noteworthy that this time course is identical to the H&E one (Fig. 1A). Cell counting showed that 24 h following SE, 19.3 ± 7.9% of eosinophilic neurons in CA1 were caspase-3a-IR, whereas this number decreased to 10.3 ± 3.7% and 9.6 ± 3.4% 48 and 72 h after SE, respectively. A caspase activation assay confirms the activation of caspase-3 in CA1 (Fig. 2B). Using a TUNEL assay, we found a good correlation between caspase-3a IR and DNA fragmentation time courses. When compared to control animals (1.2 ± 0.3 cells/CA1), a clear increase in the number of TUNEL-positive cells was detected in CA1 24 h after SE (187 ± 61 cells/CA1; p<0.01, ANOVA followed by Bonferroni test), but not at 7 h (1.1 ± 0.5 cells/CA1; p>0.05, ANOVA followed by Bonferroni test).
In CA1, caspase-3a-IR neurons have morphological features of necrosis
At all times studied or methods used, caspase-3 activation was mainly found in the nucleus (Fig. 2 A-C). By double-labeling with the chromatin dye Hoechst 33342, most caspase-3a-IR neurons had pyknotic nuclei (98.2 % at 24 h; 71.6% at 48 h and 91.9% at 72 h), a feature suggestive of necrosis. H&E staining from the same sections confirmed that all caspase-3a-IR cells from CA1 had an acidophilic cytoplasm and pyknotic nuclei. We also studied by EM the type of death (necrotic or apoptotic) of 50 caspase-3a-IR CA1 pyramidal neurons 24 h following SE. Forty-eight out of 50 IR neurons had a necrotic morphology characterized by cytoplasmic and mitochondrial swelling, and pyknosis of the nucleus (Fig. 2C).
Effect of a caspase inhibitor on SE-induced neuronal necrosis
We then determined whether caspase activation contributes to SE-induced necrosis by injecting rat pups intraperitoneally with the pan-caspase inhibitor QVD-Oph before SE induction. According with the behavioral scoring system used, seizure severity was similar in animals from control (vehicle-treated) and QVD-Oph-treated groups (Fig. 2D). In addition, 100% of the animals in both groups had SE of similar duration (318.3 ± 9.5 min and 332.6 ± 13.3 min, respectively; p=1.0, Mann Whitney test). As measured by Fluoro-Jade B staining, QVD-Oph reduced by 44% the number of CA1 injured neurons detected 24 h following SE (Fig. 2D). This result suggests that caspase activation is not an epiphenomenon but does contribute to neuronal necrosis in CA1.
Caspase-8, but not caspase-9, is upregulated in CA1
Caspase-8 and/or caspase-9 may activate caspase-3, and their participation in CA1 neuronal damage was evaluated by immunohistochemistry. Using an antibody that recognizes both the precursor and active form of caspase-8, staining was absent in control animals and strong and mostly nuclear 7 h following SE (Fig. 3A). This increase was transient, since it was not observed 24 h after SE (Fig. 3A). By contrast, using an antibody that recognizes the active form of caspase-9, we found a few caspase-9a-IR neurons in control CA1. No significant change was detected from 7 to 24 h after SE. A non-statistically significant trend toward an increase was found at 48 h, and a slight but significant increase was observed at 72 h (Fig. 3B), when 2 ± 0.7% of CA1 eosinophilic neurons were caspase-9a-IR, suggesting that caspase-9 contributes modestly to a delayed activation of caspase-3. These results show that caspase-8 upregulation precedes activation of caspase-3 in CA1, suggesting that the extrinsic pathway (caspase-8 and −3 activation) contributes to SE-induced CA1 neuronal injury.
In DG, caspase-3a-IR cells have morphological features of apoptosis
In DG, a significant increase of caspase-3a-IR neurons was already detectable 7 h following SE onset, and was still visible at 24 and 48 h. A non-significant increase of caspase-3a-IR neurons was found 72 h following SE (Fig. 4A). In agreement with these results, increased DNA fragmentation in DG was already observable 7 h after SE (14.9 ± 4.5 cells/DG; p<0.05, Student’s t test), and peaked at 24 h (77.1 ± 10.7 cells/DG; p<0.01, ANOVA followed by Bonferroni test) when compared to control rats (2.8 ± 0.2 cells/DG). Caspase-3a-IR neurons were mostly seen in the DG inner granule cell layer (Fig. 4A). Staining was mainly cytosolic and many caspase-3a-IR cells had large round clumps of chromatin (42.5% at 7 h and 45% at 24 h after SE) or displayed chromatin margination (17.9% at 7 h and 39% at 24 h after SE); both features are suggestive of apoptosis (Fig. 4A). H&E staining from the same sections confirmed that most caspase-3a-IR cells are eosinophilic and showed nuclear fragmentation. Cell counts showed that 79.9 ± 35.7 % of eosinophilic neurons in DG were caspase-3a-IR 7 h following SE and this proportion was reduced to 31.2 ± 12.7% at 24 h.
Caspase-9a, but not caspase-8, is upregulated in DG
By contrast of what observed in CA1, in the DG inner granule cell layer, the number of caspase-9a-IR neurons was significantly increased 7, 24 and 48 h after SE (Fig. 3B), and there was no increase of caspase-8-IR cells (Fig. 4C). Hoechst counterstaining showed that many caspase-9a-IR cells from DG had large round chromatin clumps (50.1% at 7 h; 48% at 24 h and 49.7% at 48 h following SE) or displayed chromatin margination (21.6 % at 7 h; 27% at 24 h and 18.3% at 48 h following SE); both features are suggestive of apoptotic neuronal death (Fig. 4B). Cell counts showed that 72 ± 32.2%, 70.9 ± 26.8% and 73.2 ± 25.9% of eosinophilic neurons in DG 7, 24 and 48 h following SE, respectively, were caspase-9a-IR. Altogether, these results suggest that in DG SE activates the intrinsic pathway (caspase-9 and −3 activation) leading to apoptosis.
Apoptotic DG cells have features of immature neurons
Altogether, our results suggest that SE activates distinct cell death programs in two different neuronal populations. The reason for this difference is unknown. In several models of acute brain injuries, apoptosis is often found in the developing brain and progressively disappears during brain maturation. Interestingly, the inner granule cell layer is an area of neuronal maturation. DG cells are generated throughout life and their inner layer may not yet express the calcium buffer calbindin at that age (Altman and Bayer, 1990; Goodman et al., 1993), but express doublecortin, a marker of immature neurons (Francis et al., 1999). Concordantly, caspase-3a-IR cells in DG were doublecortin-positive and calbindin-negative, confirming that immature neurons are more likely to die by apoptosis than necrosis (Fig. 5).
Figure 5. SE-injured DG cells have features of immature neurons.
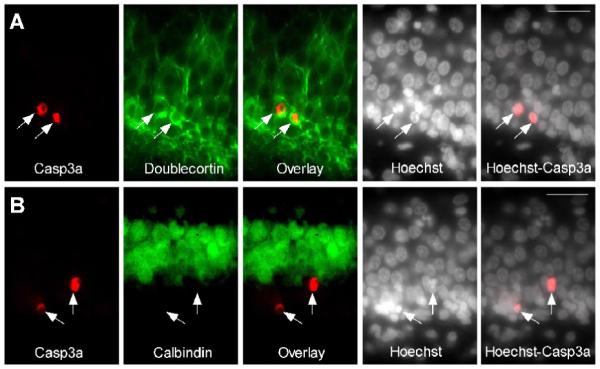
Caspase-3a-IR DG neurons (arrows) are calbindin-negative and doublecortin-positive. (A) Images of active caspase-3-IR (red), doublecortin-IR (immature neuron marker, green) and Hoechst staining (chromatin dye, white) in DG 24 h following SE. (B) Images of active caspase-3-IR (red), calbindin-IR (mature granule cell marker, green) and Hoechst staining (white) in DG 24 h following SE. Scale bars = 100 μm.
Discussion
The current study examined the mechanism of neuronal injury induced by SE in the immature hippocampus. The results showed that the same seizures induced morphologically and biochemically different forms of neuronal death in different parts of the same seizure circuit, the hippocampal formation. Since hippocampal principal cells are connected in series, excitation moves from DG granule cells to CA3 pyramids and from them to CA1 pyramids, and one would expect these neuronal populations to be exposed to roughly similar degrees of firing during seizures, yet their outcome differed markedly. One possible reason is that maturational levels are not uniform through the hippocampus. CA1 matures, physiologically and anatomically, much earlier than the DG cells (Harris and Teyler, 1984; Bekenstein et al., 1991a,b). In the highly differentiated CA1 neurons, caspase-8 expression was seen 7 h after SE, and was followed by caspase-3a expression and necrosis, the overwhelming form of seizure-induced death in adult neurons (Fujikawa et al., 2000). In DG, the mature (calbindin-expressing) granule cells exhibited little damage, as expected, while the immature (doublecortin-expressing) inner DG cells displayed apoptosis, the preferred form of seizure-induced death of immature neurons (Sankar et al, 1998). In these apoptotic DG cells, caspase-3 activation was accompanied by caspase-9 activation, without any involvement of caspase-8, suggesting that the mitochondrial “intrinsic pathway” had been activated. This suggests that it is the level of maturation of individual cells or cell groups, rather than the level of maturation of the organism, which determines the mode and mechanism of neuronal injury. It also implies that, in regards to neuronal vulnerability, the concept of the “immature brain” may not be useful, and that closer attention should be paid to regional levels of maturation.
This study demonstrated SE-induced caspase-8 upregulation in CA1 neurons from developing rats, but did not show how this caspase may be activated by SE. Seizures induce the brain expression of inflammatory cytokines including Interleukin-1β, Interleukin-6, and Tumor Necrosis Factor-α (Vezzani et al., 1999; Vezzani and Granata, 2005; Ravizza et al., 2005; De Simoni et al., 2000) and it has been reported that caspase-8 activation after seizures can involve recruitment to the TNFR1 signaling complex (Henshall et al., 2003; Shinoda et al., 2003; Yamamoto et al., 2006). Although the relationship between neuronal damage induced by seizures and cytokines is complex, we cannot rule out the possibility that inflammatory mediators play a role in activation of caspase-8 in CA1, which then cleaves caspase-3 to induce necrotic death. Alternatively, necrotic neuronal death in CA1 per se may also contribute to a sustained inflammatory reaction as observed after perinatal ischemia (Benjelloun et al., 1999).
Our results show that caspase activation plays an active role in necrotic cell death. The conventional view that necrotic cell death is a passive process has been called into question by accumulating evidence that necrotic cell death may involve caspase-dependent, programmed mechanisms. In primary neuronal cultures, neuronal necrosis induced by chemical hypoxia and/or glutamate excitotoxicity depends on an orderly cell death program leading to caspase-3 activation, which has been described as “active necrosis” (Niquet et al., 2003; 2005; 2006; Seo et al, 2009). The present study shows that a caspase cascade (caspase-8 preceding caspase-3 activation) may contribute to SE-induced neuronal necrosis. To our knowledge, this is the first in vivo demonstration that an active form of necrosis contributes to injury induced by SE in the immature brain. Indirect observations suggest that “active necrosis” is a common form of neuronal death following SE or hypoxia-ischemia. A recent paper (Tokuhara et al., 2007) reported that kainic acid-induced SE in adult rats caused two types of pyramidal cell death: early necrosis (1 day after SE) and delayed TUNEL-positive and caspase-3-dependent programmed cell death (3–7 days following SE). Interestingly, apoptotic morphology could not be found at 1, 3 and 7 days following seizures, despite evidence of caspase-3 activation, suggesting that necrosis was the main form of CA1 neuronal death (Tokuhara et al., 2007). Another study showed that 24 h after hypoxia in neonatal rats, many hippocampal and cortical propidium iodide-positive neurons displayed caspase-3 immunoreactivity (Carloni et al., 2007). Propidium iodide is a fluorescent dye used as an indicator of disrupted plasma membrane integrity, which is an early feature of necrosis. Those findings were interpreted as a “secondary necrosis”, i.e., apoptosis that progressed toward a necrotic type of cell death. In our model, caspase-3 activation is probably neither the result of “failed apoptosis” nor an epiphenomenon, since caspase inhibition blocked necrosis, and early activation of a specific caspase pathway (caspase-8 but not caspase-9) was linked to necrosis but not apoptosis. However, our studies do not exclude the involvement of other cell death factors, such as calpain or mitochondrial death factors, in neuronal necrosis. The multiplicity of cell death mechanisms raises formidable barriers to therapeutic applications in seizures and other brain diseases, but the involvement of caspase cascades in both necrotic and apoptotic forms of neuronal death in this study raises the hope that global, cell-permeant caspase inhibitors may have therapeutic potential that extends beyond classical apoptosis.
Acknowledgements
Supported by Epilepsy Foundation (JN), VHA Research Service (CW), NINDS (grant RO1 NS13515; CW), and UC MEXUS-CONACyT (CW and LR). MLLM received a UC MEXUS-CONACYT postdoctoral fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altman J, Bayer SA. Migration and distribution of two populations of hippocampal granule cell precursors during the perinatal and postnatal periods. J. Comp. Neurol. 1990;30:365–81. doi: 10.1002/cne.903010304. [DOI] [PubMed] [Google Scholar]
- Bekenstein JW, Lothman EW. An in vivo study of the ontogeny of long-term potentiation (LTP) in the CA1 region and in the dentate gyrus of the rat hippocampal formation. Brain Res. Dev. 1991a;63:245–51. doi: 10.1016/0165-3806(91)90084-v. [DOI] [PubMed] [Google Scholar]
- Bekenstein JW, Lothman EW. A comparison of the ontogeny of excitatory and inhibitory neurotransmission in the CA1 region and dentate gyrus of the rat hippocampal formation. Dev. Brain Res. 1991b;63:237–43. doi: 10.1016/0165-3806(91)90083-u. [DOI] [PubMed] [Google Scholar]
- Benjelloun N, Renolleau S, Represa A, Ben-Ari Y, Charriaut-Marlangue C. Inflammatory responses in the cerebral cortex after ischemia in the P7 neonatal Rat. Stroke. 1999;30:1916–23. doi: 10.1161/01.str.30.9.1916. [DOI] [PubMed] [Google Scholar]
- Carloni S, Carnevali A, Cimino M, Balduini W. Extended role of necrotic cell death after hypoxia-ischemia-induced neurodegeneration in the neonatal rat. Neurobiol Dis. 2007;27:354–61. doi: 10.1016/j.nbd.2007.06.009. [DOI] [PubMed] [Google Scholar]
- Caserta TM, Smith AN, Gultice AD, Reedy MA, Brown TL. Q-VD-OPh, a broad spectrum caspase inhibitor with potent antiapoptotic properties. Apoptosis. 2003;8:345–52. doi: 10.1023/a:1024116916932. [DOI] [PubMed] [Google Scholar]
- Chauvier D, Ankri S, Charriaut-Marlangue C, Casimir R, Jacotot E. Broad-spectrum caspase inhibitors: from myth to reality? Cell Death Differ. 2007;14:387–91. doi: 10.1038/sj.cdd.4402044. [DOI] [PubMed] [Google Scholar]
- De Simoni MG, Perego C, Ravizza T, Moneta D, Conti M, Marchesi F, De Luigi A, Garattini S, Vezzani A. Inflammatory cytokines and related genes are induced in the rat hippocampus by limbic status epilepticus. Eur. J. Neurosci. 2000;12:2623–2633. doi: 10.1046/j.1460-9568.2000.00140.x. [DOI] [PubMed] [Google Scholar]
- DeLorenzo RJ, Pellock JM, Towne AR, Boggs JG. Epidemiology of status epilepticus. J. Clin. Neurophysiol. 1995;12:316–325. [PubMed] [Google Scholar]
- DeLorenzo RJ, Hauser WA, Towne AR, Boggs JG, Pellock JM, Penberthy L, Garnett L, Fortner CA, Ko D. A prospective, population-based epidemiologic study of status epilepticus in Richmond, Virginia. Neurology. 1996;46:1029–1035. doi: 10.1212/wnl.46.4.1029. [DOI] [PubMed] [Google Scholar]
- Francis F, Koulakoff A, Boucher D, Chafey P, Schaar B, Vinet MC, Friocourt G, McDonnell N, Reiner O, Kahn A, McConnell SK, Berwald-Netter Y, Denoulet P, Chelly J. Doublecortin is a developmentally regulated, microtubule-associated protein expressed in migrating and differentiating neurons. Neuron. 1999;23:247–56. doi: 10.1016/s0896-6273(00)80777-1. [DOI] [PubMed] [Google Scholar]
- Fujikawa DG, Shinmei SS, Cai B. Kainic acid-induced seizures produce necrotic, not apoptotic, neurons with internucleosomal DNA cleavage: implications for programmed cell death mechanisms. Neuroscience. 2000;98:41–53. doi: 10.1016/s0306-4522(00)00085-3. [DOI] [PubMed] [Google Scholar]
- Goodman JH, Wasterlain CG, Massarweh WF, Dean E, Sollas AL, Sloviter RS. Calbindin-D28k immunoreactivity and selective vulnerability to ischemia in the dentate gyrus of the developing rat. Brain Res. 1993;606:309–14. doi: 10.1016/0006-8993(93)90999-4. [DOI] [PubMed] [Google Scholar]
- Harris KM, Teyler TJ. Developmental onset of long-term potentiation in area CA1 of the rat hippocampus. J. Physiol. 1984;346:27–48. doi: 10.1113/jphysiol.1984.sp015005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser W. The prevalence and incidence of convulsive disorders in children. Epilepsia. 1994;35:1–6. doi: 10.1111/j.1528-1157.1994.tb05932.x. [DOI] [PubMed] [Google Scholar]
- Henshall DC, Bonislawski DP, Skradski SL, Araki T, Lan JQ, Schindler CK, Meller R, Simon RP. Formation of the Apaf-1/cytochrome c complex precedes activation of caspase-9 during seizure-induced neuronal death. Cell Death Differ. 2001a;8:1169–81. doi: 10.1038/sj.cdd.4400921. [DOI] [PubMed] [Google Scholar]
- Henshall DC, Bonislawski DP, Skradski SL, Lan JQ, Meller R, Simon RP. Cleavage of bid may amplify caspase-8-induced neuronal death following focally evoked limbic seizures. Neurobiol Dis. 2001b;8:568–80. doi: 10.1006/nbdi.2001.0415. [DOI] [PubMed] [Google Scholar]
- Henshall DC, Arakmi T, Schindler CK, Shinoda S, Lan JQ, Simon RP. Expression of death-associated protein kinase and recruitment to the tumor necrosis factor signaling pathway following brief seizures. J. Neurochem. 2003;86:1260–70. doi: 10.1046/j.1471-4159.2003.01934.x. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Galluzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- Kubova H, Druga R, Lukasiuk K, Suchomelova L, Haugvicova R, Jirmanova I, Pitkanen A. Status epilepticus causes necrotic damage in the mediodorsal nucleus of the thalamus in immature rats. J. Neurosci. 2001;21:3593–3599. doi: 10.1523/JNEUROSCI.21-10-03593.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Kim JW, Yim SV, Kim MJ, Kim SA, Kim YJ, Kim CJ, Chung JH. Fluoxetine enhances cell proliferation and prevents apoptosis in dentate gyrus of maternally separated rats. Mol. Psychiatry. 2001;6:725–728. doi: 10.1038/sj.mp.4000954. [DOI] [PubMed] [Google Scholar]
- Nairismagi J, Pitkanen A, Kettunen MI, Kauppinen RA, Kubova H. Status epilepticus in 12-day-old rats leads to temporal lobe neurodegeneration and volume reduction: a histologic and MRI study. Epilepsia. 2006;47:479–488. doi: 10.1111/j.1528-1167.2006.00455.x. [DOI] [PubMed] [Google Scholar]
- Niquet J, Baldwin RA, Allen SG, Fujikawa DG, Wasterlain CG. Hypoxic neuronal necrosis: protein synthesis-independent activation of a cell death program. Proc. Natl. Acad. Sci. USA. 2003;100:2825–2830. doi: 10.1073/pnas.0530113100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niquet J, Liu H, Wasterlain CG. Programmed neuronal necrosis and status epilepticus. Epilepsia. 2005;46(Suppl 5):43–48. doi: 10.1111/j.1528-1167.2005.01025.x. [DOI] [PubMed] [Google Scholar]
- Niquet J, Seo DW, Allen SG, Wasterlain CG. Hypoxia in presence of blockers of excitotoxicity induces a caspase-dependent neuronal necrosis. Neuroscience. 2006;141:77–86. doi: 10.1016/j.neuroscience.2006.03.073. [DOI] [PubMed] [Google Scholar]
- Niquet J, Auvin S, Archie M, Seo DW, Allen S, Sankar R, Wasterlain CG. Status Epilepticus Triggers Caspase-3 Activation and Necrosis in the Immature Rat Brain. Epilepsia. 2007;48:1203–1206. doi: 10.1111/j.1528-1167.2007.01102.x. [DOI] [PubMed] [Google Scholar]
- Racine R. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Ravizza T, Rizzi M, Perego C, Richichi C, Velísková J, Moshé SL, De Simoni MG, Vezzani A. Inflammatory response and glia activation in developing rat hippocampus after status epilepticus. Epilepsia. 2005;46(Suppl 5):113–117. doi: 10.1111/j.1528-1167.2005.01006.x. [DOI] [PubMed] [Google Scholar]
- Renolleau S, Fau S, Goyenvalle C, Joly LM, Chauvier D, Jacotot E, Mariani J, Charriaut-Marlangue C. Specific caspase inhibitor Q-VD-OPh prevents neonatal stroke in P7 rat: a role for gender. J. Neurochem. 2007;100:1062–1071. doi: 10.1111/j.1471-4159.2006.04269.x. [DOI] [PubMed] [Google Scholar]
- Sankar R, Shin DH, Liu H, Mazarati A, Pereira de Vasconcelos A, Wasterlain CG. Patterns of status epilepticus-induced neuronal injury during development and long-term consequences. J. Neurosci. 1998;18:8382–8393. doi: 10.1523/JNEUROSCI.18-20-08382.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo DW, Lopez-Meraz ML, Allen S, Wasterlain CG, Niquet J. Contribution of a mitochondrial pathway to excitotoxic neuronal necrosis. J. Neurosci. Res. 2009;87:2087–94. doi: 10.1002/jnr.22035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmued LCY, Albertson C, Slikker W., Jr Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res. 1997;751:37–46. doi: 10.1016/s0006-8993(96)01387-x. [DOI] [PubMed] [Google Scholar]
- Shinoda S, Skradski SL, Araki T, Schindler CK, Meller R, Lan JQ, Taki W, Simon RP, Henshall DC. Formation of a tumour necrosis factor receptor 1 molecular scaffolding complex and activation of apoptosis signal-regulating kinase 1 during seizure-induced neuronal death. Eur. J. Neurosc.i. 2003;17:2065–2076. doi: 10.1046/j.1460-9568.2003.02655.x. [DOI] [PubMed] [Google Scholar]
- Sherwood NM, Timiras PS. A stereotaxic atlas of the developing rat brain. University of California Press; USA: 1970. [Google Scholar]
- Silva AV, Regondi MC, Cipelletti B, Frassoni C, Cavalheiro EA, Spreafico R. Neocortical and hippocampal changes after multiple pilocarpine-induced status epilepticus in rats. Epilepsia. 2005;46:636–642. doi: 10.1111/j.1528-1167.2005.31604.x. [DOI] [PubMed] [Google Scholar]
- Suchomelova L, Baldwin RA, Kubova H, Thompson KW, Sankar R, Wasterlain CG. Treatment of experimental status epilepticus in immature rats: dissociation between anticonvulsant and antiepileptogenic effects. Pediatr. Res. 2006;59:237–243. doi: 10.1203/01.pdr.0000196333.16608.30. [DOI] [PubMed] [Google Scholar]
- Tan Z, Sankar R, Shin D, Sun N, Liu H, Wasterlain CG, Schreiber SS. Differential induction of p53 in immature and adult rat brain following lithium-pilocarpine status epilepticus. Brain Res. 2002;928:187–193. doi: 10.1016/s0006-8993(01)03359-5. [DOI] [PubMed] [Google Scholar]
- Thompson K, Wasterlain C. Lithium-pilocarpine status epilepticus in the immature rabbit. Dev. Brain Res. 1997;100:1–4. doi: 10.1016/s0165-3806(96)00209-x. [DOI] [PubMed] [Google Scholar]
- Thompson K, Holm AM, Schousboe A, Popper P, Micevych P, Wasterlain CW. Hippocampal stimulation produces neuronal death in the immature brain. Neuroscience. 1998;82:337–348. doi: 10.1016/s0306-4522(97)00195-4. [DOI] [PubMed] [Google Scholar]
- Tokuhara D, Sakuma S, Hattori H, Matsuoka O, Yamano T. Kainic acid dose affects delayed cell death mechanism after status epilepticus. Brain Dev. 2007;29:2–8. doi: 10.1016/j.braindev.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Vezzani A, Conti M, De Luigi A, Ravizza T, Moneta D, Marchesi F, De Simoni MG. Interleukin-1beta immunoreactivity and microglia are enhanced in the rat hippocampus by focal kainate application: functional evidence for enhancement of electrographic seizures. J. Neurosci. 1999;19:5054–5065. doi: 10.1523/JNEUROSCI.19-12-05054.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, Granata T. Brain inflammation in epilepsy: experimental and clinical evidence. Epilepsia. 2005;46:1724–1743. doi: 10.1111/j.1528-1167.2005.00298.x. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Schindler CK, Murphy BM, Bellver-Estelles C, So NK, Taki W, Meller R, Simon RP, Henshall DC. Evidence of tumor necrosis factor receptor 1 signaling in human temporal lobe epilepsy. Exp. Neurol. 2006;202:410–420. doi: 10.1016/j.expneurol.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Yang L, Sugama S, Mischak RP, Kiaei M, Bizat N, Brouillet E, Joh TH, Beal MF. A novel systemically active caspase inhibitor attenuates the toxicities of MPTP, malonate, and 3NP in vivo. Neurobiol. Dis. 2004;17:250–259. doi: 10.1016/j.nbd.2004.07.021. [DOI] [PubMed] [Google Scholar]