

Abstract
Polymers in the nylon-3 family contain subunits derived from β-amino acids, which are linked to one another via amide bonds. Thus, the nylon-3 backbone is homologous to the α-amino acid-based backbone of proteins. This molecular-level homology suggests that nylon-3 materials might be intrinsically protein-mimetic. The experiments described here explore this prospect in the context of cell adhesion, with tissue engineering as a long-range goal. We have evaluated a small library of sequence-random nylon-3 copolymers for the ability to render surfaces attractive to NIH 3T3 fibroblast adhesion and spreading. Library screening was accomplished in a high-throughput, parallel mode via attachment of the copolymers in a two-dimensional array to a modified glass surface. Significant variations in fibroblast adhesion and spreading were observed as a function of nylon-3 subunit identity and proportion. Several of the nylon-3 copolymers supported cell adhesion and morphology that was comparable, or even superior, to that achieved on positive control substrates such as tissue culture polystyrene and collagen-coated glass. Moreover, studies conducted under serum-free conditions demonstrated that specific nylon-3 derivatives supported cell adhesion independently of serum protein adsorption. Although cell adhesion was diminished in the absence of serum, particular copolymers demonstrated an ability to support substantially greater cell adhesion than any of the other conditions, including the positive controls. The nylon-3 copolymers that were most effective at promoting adhesion to a modified glass surface proved also to be effective at promoting adhesion when attached to a PEG-based hydrogel, demonstrating the potential for these copolymers to be used in tissue engineering applications.
INTRODUCTION
Considerable effort has been devoted to the development of synthetic substrates that can provide biomimetic signals to cells, encouraging them to attach and behave as they would in their natural environment1–5. Efforts of this type could ultimately enable valuable medical technologies, such as the ex vivo growth of tissues that can be used for transplantation. In the near-term, such research provides insight on the ways in which cells receive signals from their surroundings, and on strategies for rational control of this signaling.
Substrates for cell attachment, growth and differentiation may be generated in the laboratory by employing naturally-derived materials, such as collagen components, or non-native materials, such as synthetic polymers, or natural-synthetic combinations5. Natural components are attractive because they may contain powerful signaling elements. Disadvantages of using natural components include the possibility of inadvertently introducing viruses or other infectious agents, and the inability to control composition or nanoscale structure6. Synthetic components for cell growth can be generated free of infectious agents, and the chemist’s ability to vary the molecular and nanoscale structure of such materials should, in principle, enable control over the signals conveyed to cells (either directly or via adsorbed proteins). In practice, however, limitations in preparative capabilities and an incomplete understanding of signaling between cells and their natural substrates prevent realization of the full promise of synthetic materials at present. Evaluation of new materials as components of substrates for cell growth remains an important research goal.
Some synthetic materials employed as substrates for cell growth are generated from a single type of molecule, such as the peptide-amphiphiles introduced by Stupp et al. 7, 8, the peptides developed by Zhang et al. 9, 10 and the hairpin peptides of Schneider, Pochan et al. 11. In these cases the molecules self-assemble to create macroscopic structures that serve as cell attachment substrates. The use of perfectly monodisperse synthetic oligomers to create such substrates is attractive because, in principle, this approach allows careful tailoring of the functionality displayed; however, the preparation of these molecules requires many chemical steps, which makes these materials costly. Alternatively, cell-attachment substrates may be generated from self-assembling polymers, which are necessarily polydisperse. These polymeric materials are often relatively inexpensive to prepare, but the heterogeneity displayed by synthetic copolymers in terms of length, subunit composition and subunit sequence may limit the extent to which such materials can promote cellular attachment and growth. Hybrid approaches are possible as well, in which the self-assembly function is achieved with a readily prepared polymer that has no intrinsic signaling capability (e.g., polyethylene glycol (PEG)), and the signaling function is achieved by appending a discrete peptide, e.g., a sequence containing the RGD motif, to the polymer 12.
Here we describe an evaluation of nylon-3 polymers as components for artificial tissue engineering substrates. Very little is known about the behavior of nylon-3 materials in biological systems because until recently only limited functional variation could be achieved within this polymer class. Our long-term goal is to develop nylon-3 copolymers containing subunits that direct self-assembly linked to subunits that convey information leading to cellular attachment. The initial studies described below, however, focus exclusively on the latter goal.
Several features of the nylon-3 family render these materials attractive for biomaterials applications. First, the backbone is inherently protein-mimetic, since the repeat unit differs from that in proteins simply by the presence of an additional carbon (Figure 1; proteins are composed of α-amino acid residues, while nylon-3 polymers are comprised of β-amino acid residues). Second, nylon-3 materials are readily prepared via ring-opening polymerization (ROP) of β-lactams13. This controlled polymerization provides access to block copolymers as well as homopolymers and random copolymers. Third, recent developments in β-lactam synthesis enable considerable variation in the side chain functionality that is presented by the nylon-3 chain. In particular, we have generated β-lactams that allow incorporation of cationic subunits or polar but uncharged subunits into the polymer chain14, 15. The limited exploration to date of biological applications of nylon-3 polymers presumably reflects the fact that most previous examples contained exclusively hydrophobic subunits and displayed poor solubility.
Figure 1.

Comparison of an α-amino acid residue (left) with a β-amino acid residue (right).
This report documents a two-stage evaluation of nylon-3 materials as components of cell growth substrates. First, we use two-dimensional polymer arrays immobilized on functionalized glass to assess interactions between a small library of cationic nylon-3 copolymers and NIH 3T3 fibroblasts. This approach enabled us to screen rapidly through a variety of copolymer compositions in order to identify the most promising systems. Second, hydrogels formed from PEG bearing some of the most favorable nylon-3 derivatives are examined as substrates for cell growth. The performance of these functionalized hydrogels suggests that they represent promising candidates for tissue engineering applications.
MATERIALS AND METHODS
Materials
Chemicals, polyethylene glycol (Mn 3400, 202444) and chambered coverslips (C7735) were obtained from Sigma-Aldrich, amine functionalized glass (SMM2) from ArrayIt, collagen (PureCol™) from Inamed Biomaterials, Irgacure 2959 from Ciba Specialty Chemicals, Dulbecco’s Modified Eagle’s Medium (DMEM), cell culture supplements, LIVE/DEAD viability/cytotoxicity kit (L3224), Quant-iT™ picogreen dsDNA Assay kit (P7589) and NanoOrange Protein Quantitation Kit (N6666) from Invitrogen, syringe filter with 0.2 μm cellulose acetate membrane (192–2520) from Nalgene, round cover glass (12-545-80) and M-PER lysis buffer (78501) from Thermo Fisher Scientific, and NIH 3T3 fibroblast cells from the American Type Tissue Collection (ATCC). NIH 3T3 cells were stained using a LIVE/DEAD kit and scanned using a GeneTAC UC 4×4 scanner (Genomic Solution). All scanned data were analyzed using the GeneTAC quantitation program or GenePix Pro 6.0 demo version.
Synthetic procedures for monomers and PEG-diacrylate
β-Lactams, shown in Figure 2, were synthesized via previously reported methods 15–18.
Figure 2.

(A) Chemical structures of the β-lactams and co-initiators used in this work. (B) Example of the polymerization/deprotection process.
(±)-7-(2-Tritylthioacetyl)-7-azabicyhclo[4,2,0]octan-8-one (I)
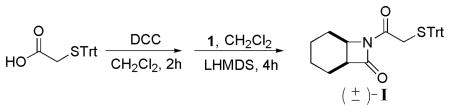
To a stirred solution of tritylthioacetic acid (8.9 g, 26.8 mmol) in dry CH2Cl2 (190 mL) was added dicyclohexylcarbodiimide (DCC) (2.8 g, 13.4 mmol). After 2 h, the reaction mixture was filtered through celite to remove dicyclohexylurea, and the celite pad was rinsed with diethyl ether. The combined filtrate was concentrated in vacuo. To a stirred solution of the concentrated residue and β-lactam 1 (1.5 g, 12.1 mmol) in dry CH2Cl2 (40 mL) was added 1 M lithium bis-(trimethylsilyl)amide (LHMDS) in THF (12 mL). After 4 h, the reaction mixture was diluted with EtOAc and washed with 1 N HCl, sat. aq. NaHCO3 and then brine. The organic layer was dried (MgSO4) and concentrated in vacuo. The crude product was purified by silica column chromatography (6:1 to 3:1 hexane/EtOAc containing 1% Et3N) to give I in 34% yield as a solid: mp 156–159 °C; 1H NMR (300 MHz, CD3OD) δ 7.48–7.38 (m, 6 H), 7.34–7.16 (m, 9 H), 4.09–4.01 (m, 1 H), 3.49–3.36 (m, 2 H), 3.24–3.14 (m, 1 H), 2.06–1.93 (m, 1 H), 1.91–1.62 (m, 3 H), 1.59–1.38 (m, 4 H); 13C NMR (75 MHz, CD3OD) δ 168.7, 166.6, 144.2, 129.7, 128.0, 126.9, 67.2, 49.8, 46.5, 36.8, 23.1, 19.4, 18.9, 16.9; HRMS (m/z, ESI) calcd for C14H19NO4 (M+Na)+ 464.1655, found 464.1676.
PEG-diacrylate
To a stirred solution of polyethylene glycol (2 g, 0.59 mmol) in dry CH2Cl2 was added triethylamine (0.18 g, 1.77 mmol) and acryloyl chloride (0.16 g, 1.77 mmol) at 0°C. After 24 h at room temperature, the reaction mixture was poured into diethyl ether (400 mL). The precipitated solid was isolated by filtration and dried. After drying, the crude product was dissolved in deionized water (20 mL) and dialyzed against 1 L deionized water (2 × 8 h) using MWCO 100 dialysis tubing to remove Et3N•HCl salt and low molecular weight impurities. After dialysis, the solution was lyophilized to give PEG-diacrylate in 85% yield as a solid: 1H NMR (300 MHz, CDCl3) δ 6.42 (dd, 1 H, J = 17.4, 1.5 Hz), 6.15 (dd, 1 H, J = 17.4, 10.5 Hz), 5.83 (dd, 1 H, J = 10.8, 1.8 Hz), 4.34–4.27 (m, 2 H), 3.91–3.37 (m, 219 H).
Polymer synthesis
Nylon-3 polymers were prepared using reported procedures 19. Polymers with Boc-protected side chain amino groups were analyzed by gel-permeation chromatography (GPC) using an instrument equipped with a Wyatt multiangle light scattering detector and a refractive index detector, and the molecular weight was calculated using ASTRA 5.3.2.15 software. In order to calculate the number-averaged molecular weight (Mn) and polydispersity index (PDI), a dn/dc value of 0.1 mL/g was used for each polymer.
Preparation of HTS glass
Aminated glass slides were placed in a staining jar. A 3% solution of succinic anhydride and iPr2EtN in DMF (30 mL) was poured into the staining jar and then shaken gently for 3 h. After thorough rinsing of the jar with DMF, a 3% solution of diisopropylcarbodiimide (DIC) and N-hydroxysuccinimide (NHS) in DMF (30 mL) was poured into the staining jar. After gentle shaking for 3 h, the slides were washed with DMF and then dried with N2 gas. The dried NHS-activated glass slide was covered with a coverslip containing 50 wells. An aliquot (8 μL) of control solution C1 = 5% (v/v) aminoethanol in 100 mM NaHCO3 containing 15% glycerol, or C2 = 5% (v/v) ethylene diamine in 100 mM NaHCO3 containing 15% glycerol, or C3 = 100 mM NaHCO3 containing 15% glycerol, or C4 = 5 %(w/v) NH2-PEG-OH (Mn = 3000) in 100 mM NaHCO3 containing 15% glycerol, or C5 = 5 %(w/v) NH2-PEG-NH2 (Mn = 3000) in 100 mM NaHCO3 containing 15% glycerol) or of a solution containing a specific polymer (1 mM in 100 mM NaHCO3 containing 15% glycerol) was added into each well. The glass slide was placed in a water-loaded petri dish as a humidified chamber. After 12 h at room temperature, the glass slides were thoroughly rinsed with deionized water and then dried with N2 gas.
Preparation of collagen-coated wells
Untreated, aminated glass slides were covered with a glass coverslip, placed in a petri dish, and 8 μL of type I collagen solution (1.75 μg/mL in bicarbonate coating buffer) was added to each well. The petri dish and enclosed slide were incubated for 20 h at 4°C. After incubation, the slide was rinsed twice with deionized water (10 μL/well) and then dried under N2 gas.
Screening of NIH 3T3 cell adhesion to polymer-functionalized substrates
NIH 3T3 fibroblasts were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, 100 μg/mL streptomycin, and 2 mM L-glutamine at 37°C in a 5% CO2 environment. A cell suspension was prepared for seeding on HTS glass surfaces following detachment of the cells via trypsinization (0.05% trypsin + 0.02% EDTA), centrifugation, and resuspension of the cell pellet in culture medium to a final concentration of 0.125 × 106 cells/mL. Cells were seeded into wells of the glass coverslip via addition of 8 μL of cell suspension to each well. After incubation for 2 h, the petri dish containing the slide was filled with 25–30 mL culture medium. Serum-free experiments were performed using the aforementioned medium formulation in the absence of FBS. At one day post-seeding, cell viability was assessed using a Live/Dead fluorescent staining assay. In this assay, calcein AM (2 μM) is cleaved into a fluorescent green product by intracellular esterases found only in viable cells, while the red fluorescent ethidium homodimer-1 (4 μM) penetrates only dead cells. Twenty min following the addition of the viability staining solution, the number of adherent cells in each well was quantified using a GeneTAC UC 4×4 scanner, and fluorescent photomicrographs of the slides were captured using a fluorescence microscope.
Protein adsorption to polymer-functionalized substrates
Polymer-functionalized and corresponding control substrates were placed in a humidified petri dish environment, and 8 μL of complete growth medium (containing 10% FBS) was deposited in each well of the slide. At time points of 30 min and 2 h following the addition of solution to the wells, each well was washed twice with deionized water and then dried with N2 gas. The amount of protein adsorbed onto the substrates was measured via the NanoOrange protein assay, in which 10 μL 1X NanoOrange working solution was added to each well and incubated for 1 h at room temperature. The slide was then scanned using a GeneTAC UC 4×4 scanner, and protein concentration was determined using a standard curve generated from known concentrations of bovine serum albumin (BSA).
Modification of PEG hydrogels for cell adhesion
A solution of 20% (w/v) PEG-diacrylate was prepared in phosphate buffered saline solution (PBS, pH 7.4) and sterilized via syringe filtration. The sterilized PEG-diacrylate solution was mixed with a photoinitiator (0.05% Irgacure-2959) and then combined with 1 mM CGRGDS, or 1 mM cysteine-terminated nylon-3 derivative (Pol-5, Pol-9, or Pol-15), or left unmodified to yield a final PEG-diacrylate concentration of 15 wt%. The cysteine-terminated sequences were covalently coupled to the PEG-diacrylate polymer chains via a thiol-ene reaction as previously described 20, and the RGD-containing sequence was used as a positive adhesion control. These PEG solutions (60 μL × 4 per sample solution) were sandwiched between two circular glass coverslips and placed under a long-wavelength UV light (365 nm, 3.9 mW/cm2) for 5 min to induce crosslinking. Following gelation, the top coverslip was removed, and each gel was placed into a well of a 24-well plate with 0.5 mL cell culture medium, changed three times over 24 h prior to cell seeding. NIH 3T3 fibroblasts were cultured as described earlier and seeded into wells at a concentration of 50,000 cells/cm2. At one day post-seeding, the culture medium in each well was removed and replaced with Live/Dead solution (2 μM calcein-AM, 4 μM ethidium-homodimer-1 in PBS). After 5 minutes of incubation in Live/Dead solution, photomicrographs of the cells were captured using a fluorescence microscope. The PEG gel samples were then transferred to a new 24-well plate, and adherent cells were removed from the PEG gel substrates via application of 250 μL M-PER lysis buffer (Mammalian Protein Extraction Reagent) to the wells for 30–60 min. The number of adherent cells in the cell lysates was quantified via a PicoGreen DNA assay according to manufacturer’s instructions, using a fluorescence microplate reader (Synergy HT, Biotek Instruments, Winooski, VT), and the standard curve generated from a dsDNA stock solution.
Statistics
Data are presented as mean ± standard deviation and were compared using ANOVA with Tukey’s HSD post-hoc test. P values less than or equal to 0.05 were considered statistically significant.
RESULTS
Polymer design and synthesis
Controlled ring-opening polymerization of β-lactams requires the use of a catalytic base and a co-initiator to ensure a coordinated start to polymer chain growth, which leads to low polydispersity. We employed LiN(SiMe3)2 as the base and imide I as the co-initiator. The co-initiator gives rise to the N-terminal β-amino acid residue in the polymer chain (Figure 2), and the use of I places a trityl-protected thiol group at the N-terminus. Deprotection provides a unique thiol in the polymer chain. In some of the applications described below this thiol is used for covalent attachment of nylon-3 molecules to the vinyl groups of diacrylated PEG via a photo-initiated thiol-ene reaction 20.
β-Lactams CH, CO, MM, DM and DH were used to prepare the nylon-3 derivatives discussed below. The cyclohexyl and cyclooctyl rings of CH and CO, respectively, give rise to hydrophobic subunits in polymers that are prepared from these β-lactams. MM (for “monomethyl”) and DM (for “dimethyl”) contain a substituent bearing a Boc-protected amino group. After polymerizations involving these β-lactams, the side chains can be deprotected; the resulting amino groups are protonated at neutral pH and below, which confers water-solubility on polymers that contain MM or DM subunits. Deprotection of a DH (for “dihydroxyl”) subunit after polymerization gives rise to two side chains each of which bears a hydroxyl group. Thus, the DH subunits are polar but uncharged and therefore distinct from the hydrophobic CH and CO subunits or the cationic MM and DM subunits.
All of the nylon-3 derivatives we examined contain either MM or DM subunits; thus, all are expected to bear positive charge in the presence of cell culture medium. This feature should promote hydration of immobilized polymer chains and facilitate interactions with cell surfaces. In addition to the cationic homopolymers, containing only MM or DM subunits, we examined a variety of random copolymers prepared from binary β-lactam mixtures: MM+CH, MM+CO, DM+CH and MM+DH (Figure 3). Several different stoichiometries were examined for each pairing. For the cationic/hydrophobic combinations, we prepared copolymers containing 10%, 20%, 30% or 40% of the hydrophobic subunit (CH or CO), and for the cationic/dihydroxyl combination we prepared copolymers containing 10%, 20%, 30%, 40%, 50% and 60% of the DH subunit. GPC analysis of the protected polymers indicated that these materials generally contained an average of 22–28 subunits, with Mw/Mn = PDI values in the range 1.2 to 1.4. Each of our five β-lactams is chiral, and each was used in racemic form; therefore, each of the 20 polymers in our nylon-3 library is heterochiral (mixture of diastereomers).
Figure 3.

Chemical structures of the polymers examined in this work.
Fabrication of substrates
Functionalized glass surfaces were used in our initial efforts to evaluate the ability of nylon-3 polymers to render a surface hospitable for NIH 3T3 cell attachment. A standard approach was used to modify the surface of commercially available amino-functionalized glass in a way that generates electrophilic sites, which are necessary for polymer immobilization (Figure 4). The amino-functionalized glass surface was treated with succinic anhydride to generate a surface that displays carboxyl groups, and these carboxyl groups were then converted to N-hydroxysuccinimide (NHS) esters.
Figure 4.

Preparation of functionalized glass surfaces.
Immobilization of our amine-containing polymers was achieved by allowing them to react with the NHS-functionalized glass. The cartoon in Figure 4 shows only one point of surface bonding per polymer chain, but it is quite possible that some or all polymer molecules are linked to the surface at more than one point. Surfaces for control studies were generated by allowing the NHS-functionalized glass to react with amino ethanol (“hydroxyl”; C1), ethylene diamine (“amine’; C2), amino-PEG-alcohol (“PEG-alcohol”; C4) or amino-PEG-amine (“PEG-amine”; C4). Another control surface was generated by treating the NHS-functionalized glass with 100 mM aqueous NaHCO3, which was expected to hydrolyze the NHS esters and generate a “carboxyl” surface (C3).
A two dimensional array of differentially functionalized surfaces bearing the 20 polymers in our nylon-3 library, along with the five control surfaces (each polymer and control in duplicate), was generated on a glass slide using a coverslip with 50 chambers. This array format enabled rapid comparative evaluation of the members of the polymer library for the ability to support cell attachment and proliferation.
Cell adhesion to polymer-functionalized substrates
NIH 3T3 fibroblasts, an immortalized cell type commonly used in adhesion and spreading experiments 21, were seeded onto glass substrates bearing the polymer array in both serum-containing and serum-free conditions. After 24 hours of culture on these substrates, the presence of viable and non-viable cells in each condition was evaluated quantitatively via measurement of fluorescence intensity of the live/dead viability dyes that had penetrated the cells (Figure 5). Photomicrographs were captured for qualitative assessment of cell spreading and morphology (Figure 6). The techniques used for surface immobilization of the nylon-3 copolymers enabled rapid, simultaneous screening of multiple polymer formulations (Figure 5A).
Figure 5.

(A) Example of polymer screening setup and fluorescence images obtained during high-throughput scanning of viable and non-viable cells adherent to glass functionalized with the library of polymer formulations (Pol-1 to Pol-20) or control surfaces (C1 to C5). (B) Adhesion of viable NIH 3T3 fibroblasts at one day post-seeding on polymer-functionalized substrates in serum-containing culture medium, as measured by fluorescent intensity of an internalized viability stain. *p<0.02 compared to C1 to C5, Pol-2 to Pol-4, Pol-8, and Pol-13 to Pol-20; ^p<0.04 compared to C1 to C5, Pol-2, and Pol-13 to Pol-19.
Figure 6.

Photomicrographs of viable fibroblasts adherent to control (C1 to C5) and polymer-functionalized substrates (Pol-1 to Pol-20) at one day post-seeding in serum-containing culture medium. Scale bar = 50 μm.
The number of viable cells adherent to the surfaces could be controlled by the composition of the immobilized nylon-3 derivative (Figure 5B). Specifically, polymers containing mixtures of MM+CO (Pol-5 to Pol-8) or DM+CH (Pol-9 to Pol-12) were the most supportive of fibroblast adhesion. Within these groups, cell adhesion increased with an increase in hydrophobic component (CO or CH) and a concomitant decrease in cationic component (MM or DM), with the highest levels of cell adhesion obtained on 60:40 MM:CO (Pol-5), 60:40 DM:CH (Pol-9), and 70:30 DM:CH (Pol-10). These surfaces supported not only greater cell adhesion, but also morphologically healthier cells, as seen in Figure 6, relative to surfaces bearing other nylon-3 copolymers. Fibroblasts cultured on Pol-5, Pol-6, or Pol-9 to Pol-12 were well-spread and homogeneously dispersed across the culture surfaces.
Polymers containing MM+CH (Pol-1 to Pol-4) or MM+DH (Pol-13 to Pol-19) were significantly less supportive of fibroblast adhesion with respect to both cell number (Figure 5B) and cell morphology (Figure 6), relative to the MM+CO and DM+CH polymers. With the exception of Pol-1, fibroblast adhesion to surfaces bearing MM+CH or MM+DH copolymers was not statistically different from adhesion to the control surfaces (C1-C5). Within the MM:CH or MM:DH series, cell adhesion trends did not significantly vary with changes in copolymer composition.
As illustrated by the photomicrographs in Figure 6, differences in cell morphology between surfaces bearing the MM+CH or MM+DH copolymers and the more cell-adhesive copolymers (Pol-5, Pol-6, and Pol-9 to Pol-12) were even more striking than the numerical differences in cell adhesion shown in Figure 5. Even relatively modest increases in quantitative cell adhesion measurements (Figure 5) corresponded to rather large differences in the density and quality of cell adhesion to the surfaces, as seen in Figure 6. Surfaces bearing MM+CO or DM+CH featured well-spread cells, but fibroblasts cultured on MM+CH or MM+DH substrates tended to display greatly altered morphology (Figure 6); in the latter cases the cells displayed spindled membrane protrusions and were heterogeneously grouped into clusters. Cell viability was not affected by any of the polymer formulations, as the number of non-viable cells comprised <10% of the population in each case and did not significantly vary across the different conditions. Overall, the results show that although our polymer library is small, a wide range of activities is evident within the library, including some cellular responses that are quite promising with respect to biomaterials applications.
The ability of these functionalized surfaces to support cell adhesion was examined in the absence of serum proteins, to complement the studies described above, which were conducted in the presence of serum. As expected, significantly fewer cells adhered to the surfaces in the absence of serum relative to the serum-containing conditions. However, somewhat unexpectedly, several of the copolymers supported both fibroblast adhesion and a well-spread morphology in the absence of serum. These polymer-specific trends in cell adhesion were similar to those observed in serum-containing conditions; specifically, nylon-3 copolymers containing MM+CO or DM+CH tended to be the most supportive of fibroblast adhesion in a serum-free environment, while the MM+DH copolymers were the least supportive (Figure 7). In a slight departure from the trend observed in the presence of serum, when serum was lacking, a favorable adhesive environment was provided by two of the MM+CH formulations (Pol-1 and Pol-2). In the absence of serum, morphological trends followed the quantitative adhesion findings, as previously observed in the presence of serum: well-spread cells were found on surfaces that supported high numbers of adherent cells (Pol-1, Pol-2, Pol-5 to Pol-8, Pol-15 and Pol-16), while cells on the less adhesive surfaces were generally rounded in appearance (Figure 8).
Figure 7.

Adhesion of viable fibroblasts at one day post-seeding on polymer-functionalized substrates in serum-free culture medium, as measured by fluorescent intensity of an internalized viability stain. *p<0.05 compared to C4 to C5, Pol-13 to Pol-19.
Figure 8.

Photomicrographs of viable fibroblasts adherent to control (C1 to C5) and polymer-functionalized substrates (Pol-1 to Pol-20) at one day post-seeding in serum-free culture medium. Scale bar = 50 μm.
Protein adsorption to polymer-functionalized substrates
Cell adhesion in the presence of serum is frequently mediated via adsorbed serum proteins 22. Thus, protein adsorption to the nylon-3-functionalized substrates was measured in order to investigate whether differential protein adsorption could be responsible for the trends in cell adhesion found under serum-containing conditions. Protein adsorption was reduced on the control (C1-C5) and dihydroxyl-modified substrates (MM+DH; Pol-13 to Pol-19) relative to the other substrate conditions (Figure 9), which correlated with the low cell adhesion and spreading observed on the control (C1-C5) and dihydroxyl-modified substrates. However, there was little variability in protein adsorption across the remaining surfaces (Pol-1 to Pol-12, Pol-19 and Pol-20), despite the fact that these surfaces support significantly different levels of cell adhesion.
Figure 9.

Amount of protein adsorbed to control (C1 to C5) and polymer-functionalized substrates (Pol-1 to Pol-20) after 30 and 120 minutes of exposure to serum-containing culture medium.
Comparison to standard cell-adhesive substrates
A subset of the nylon-3-functionalized surfaces was selected for further examination with respect to cell adhesion and spreading analyses in the presence or absence of serum. The most adhesive nylon-3 copolymers (Pol-5 and Pol-9), in addition to a representative low-adhesion copolymer (Pol-15) and two control surfaces (C1, C2), were investigated relative to four common cell culture surfaces: untreated polystyrene (PS), tissue culture-treated polystyrene (TCPS), collagen-coated glass (Coll), and amine-modified glass (NH2). In the presence of serum, both Pol-5 and Pol-9 supported a level of cell adhesion comparable to that achieved on the positive controls, TCPS and Coll (Figure 10A). Even at an early time point, 30 minutes post-seeding, spreading was evident in over 70% of the cells adherent to Pol-5 or Pol-9 (not shown). Cell adhesion to the non-adhesive polymer (Pol-15) and the immobilization controls (C1-C2) was similar to that achieved on amine-modified glass (NH2), while adhesion to unmodified PS was significantly lower than that found on any other condition (p<0.02). Substrates functionalized with Pol-5 or Pol-9 supported a cell morphology that was indistinguishable from the morphology observed on collagen, a native extracellular matrix component (Figure 10B).
Figure 10.

(A) Comparison of fibroblast adhesion on select polymer formulations (C1, C2, Pol-5, Pol-9, and Pol-15) against adhesion on standard cell culture surfaces: polystyrene (PS), tissue culture-treated polystyrene (TCPS), collagen-coated glass (Coll), and amine-functionalized glass (NH2). Cells were cultured for one day in serum-containing medium. *p<0.0001 compared to all conditions not marked with a symbol. (B) Representative photomicrographs of adherent fibroblasts corresponding to the conditions in (A). Scale bar = 50 μm.
When cells were seeded in the absence of serum, the overall number of adherent cells was diminished relative to seeding in the presence of serum. However, in the absence of serum, the two best nylon-3 copolymers (Pol-5 and Pol-9) demonstrated an ability to support substantially greater cell adhesion than any of the other conditions, including the positive controls (Figure 11A; p<0.04). Under serum-free conditions only the surfaces modified with collagen, Pol-5, or Pol-9 contained numerous well-spread cells, with the remaining surfaces populated by sparse, rounded fibroblasts (Figure 11B). Although the collagen-modified surface supported the expected high density of adherent cells in the presence of serum (Figure 10A), relatively few cells were found attached to the collagen-modified surface in the absence of serum (Figure 11A). This difference suggests that the integrin-binding events that enable cell adhesion and spreading on collagen may occur more slowly than the events that govern cell adherence to the polymer-modified surfaces, or that the interaction of serum proteins with the collagen coating facilitates greater cellular adhesion than does collagen alone.
Figure 11.

(A) Comparison of fibroblast adhesion in the absence of serum to select polymer formulations (C1, C2, Pol-5, Pol-9, and Pol-15) against adhesion on standard cell culture surfaces: polystyrene (PS), tissue culture-treated polystyrene (TCPS), collagen-coated glass (Coll), and amine-functionalized glass (NH2). Cells were cultured for one day in serum-free medium. *p<0.04 compared to all conditions not marked with a symbol. (B) Representative photomicrographs of adherent fibroblasts corresponding to the conditions in (A). Scale bar = 50 μm.
Translation to a biomaterial scaffold environment
PEG-based hydrogels provide a non-adhesive environment (often termed “blank slate”) that is attractive for studying the interaction between cells and discrete peptide or peptidomimetic moieties 23, 24. Moreover, PEG-based hydrogels have direct applicability as three-dimensional scaffold materials for numerous tissue engineering applications 25. We observed that covalent immobilization of selected nylon-3 copolymers to photo-crosslinked PEG hydrogels significantly impacted fibroblast adhesion in a manner that paralleled the findings on glass-based substrates. Specifically, the optimal copolymers (Pol-5 and Pol-9) supported extensive adhesion and spreading of fibroblasts on the PEG-based surfaces (Figure 12). PEG hydrogels bearing Pol-9 displayed a greater than two-fold increase in cell adhesion relative to PEG hydrogels bearing the adhesive Arginine-Glycine-Aspartic acid (RGD) peptide. This outcome is particularly notable, since RGD is widely used to promote cell adhesion to PEG-based materials 12. The cells on the hydrogel bearing Pol-9 appeared morphologically healthy, while fibroblasts on hydrogels bearing Pol-5 or Pol-15 tended to be more rounded and clustered together (not shown). The dramatic difference between the PEG hydrogel bearing Pol-9 and the PEG hydrogel bearing Pol-15, in terms of the extent of fibroblast adhesion and the morphology of adherent cells, shows that the nature of the copolymer immobilized on the hydrogel plays a crucial role in determining the quality of the material as a cell adhesion substrate.
Figure 12.

Adhesion of fibroblasts to photo-crosslinked PEG hydrogels functionalized with a positive control adhesive peptide sequence (Arg-Gly-Asp, or RGD) or functionalized with select nylon-3 copolymers (Pol-5, Pol-9, and Pol-15), as measured by DNA content of cells bound to the hydrogel surface. *p<0.05 compared to PEG; ^p<0.0001 compared to RGD.
DISCUSSION
This research was motivated by two hypotheses: (1) the protein-like backbone of nylon-3 polymers should render these materials protein-mimetic without being susceptible to proteolysis; (2) the range of side chain functionality available from recently developed β-lactam precursors should enable the discovery of nylon-3 copolymers that are attractive to cells. Validation of these hypotheses would introduce a new class of synthetically accessible and functionally diverse polymers as potential tools for generating cell-adhesive surfaces, which might ultimately be useful for tissue engineering applications. Synthetic molecules are valuable relative to natural molecules, such as collagen or other proteins, for construction of cell-adhesive surfaces due to challenges related to creating consistent, controlled, and tailored environments using natural materials. In addition, the increased antigenicity and risk of pathogenicity associated with natural materials are mitigated with synthetic materials 6. Discrete synthetic peptides are often used to generate cell-adhesive surfaces 12, 26, but these sequence-specific peptides must be prepared via a time-intensive step-wise process; in contrast, the nylon-3 copolymers we have explored are assembled in a single step.
We undertook a combinatorial approach to test the hypotheses outlined above. It was straightforward to prepare a library of nylon-3 copolymers with variable subunit identity and proportion. Immobilization of this library in a two-dimensional array on functionalized glass allowed rapid evaluation of these materials for promotion of fibroblast adhesion. A range of cell-adhesive propensities was detected in this screen, demonstrating that significant differences in cell adhesion can be achieved via relatively subtle changes in biomaterial chemistry. Subsequent analysis revealed that glass surfaces bearing certain nylon-3 copolymers were comparable or superior to a glass surface bearing collagen or to tissue culture-treated polystyrene in terms of both the adhesion and spreading of fibroblasts. These latter surfaces are widely regarded as excellent substrates for cell adhesion; thus, our screen enabled us to identify nylon-3 copolymers with very promising properties.
Two lines of reasoning suggest that attachment density of the polymers to the substrates is not the major determinant of the cell adhesion trends we have observed. First, polymers with the same proportion of reactive amino side chains can display very different cell adhesion propensities (e.g., Pol-5 vs. Pol-15). Thus, the extent to which the polymer-modified surfaces attract cells depends critically upon the non-reactive subunit. If attachment density were the sole controlling factor, then the proportion of subunits that bear amine-containing side chains, i.e., side chains that can react with the NHS esters on the surface, should be the major determinant of cell adhesion propensity, which is clearly not the case. Second, comparison of the two best polymer sets (MM:CO (Pol-5 to Pol-9) and DM:CH (Pol-10 to Pol-14)) with the controls shows that it is better to have polymer on the glass surface than not to have polymer, in terms of cell adhesion. However, within each series, cell adhesion is maximal for polymers that have the lowest proportion of subunits with amine-containing side chains. If greater polymer density were crucial, then the polymers with the highest concentration of amine-containing side chains would be best, which is clearly not the case. If lower polymer density were better, then no polymer at all should be best, which is clearly not the case. We cannot rule out the possibility that the density of immobilized polymer at the surface plays a contributing role in terms of cell adhesion, but our data indicate that polymer density cannot be the principal determining factor.
It is particularly noteworthy that some of the nylon-3 copolymers we evaluated can support the adhesion and spreading of fibroblasts in a serum-free environment. Under standard culture conditions, which include serum, cells adhere to synthetic substrates via an intermediate layer of adsorbed protein 22. Adhesion to selected (but not all) nylon-3-bearing surfaces in the absence of serum therefore raises the possibility that receptors in the cell membrane (e.g., integrins) somehow recognize molecular motifs within the synthetic materials, i.e., that motifs within the optimal polymers play a role analogous to that of well-established peptidic adhesion signals such as RGD. Another possibility is that the cells respond not to specific constellations of atoms but rather to biomimetic secondary or tertiary structures or higher-order assemblies formed by the nylon-3 copolymers. The documented response of cells to synthetic nanofibrous scaffolds, which is thought to represent mimicry of cellular response to collagen fibrils and to be based on feature size rather than molecular-level signals, provides a precedent for this hypothesis 3, 27. However, extrapolating this hypothesis to our nylon-3 copolymers is potentially problematic because these polymers have not been demonstrated to adopt specific higher-order structures. Further, the stereochemical heterogeneity of these polymers makes it unlikely that they form higher order structures of the necessary regularity for cell recognition. Yet another possible explanation for the results we observe under serum-free conditions is that specific membrane-bound extracellular matrix proteins on the cell interact with the polymer substrate, as has previously been described for membrane-bound fibronectin regulation of fibroblast adhesion 28. In the current work, the membrane-bound matrix proteins would presumably have to display adsorption preferences among the different nylon-3 copolymers we examined, leading to varied levels of cell adhesion and spreading on the various substrate compositions. This hypothesis would account for the attachment of cells to specific polymer formulations in serum-free conditions as well as the rapid spreading of a majority of these cells.
Further biological analyses will be required to decipher the mechanisms of attachment and spreading of NIH 3T3 cells under serum-free conditions to surfaces bearing nylon-3 copolymers. This task will be facilitated by use of the experimental strategies described here. The production of a library of systematically-altered, well-characterized copolymers, followed by rapid screening of these materials via surface immobilization and fluorescent cell staining, will enable the identification of specific chemical functionalities or motifs that regulate the polymer-cell interactions we have uncovered. Results from such studies, in turn, should facilitate the development of optimized polymers (which may have features not yet explored, such as block architectures or stereochemical homogeneity) for cell adhesion as well as elucidation of the mechanism of cell attachment to surfaces that display the synthetic polymers.
Our data show that cell adhesion to nylon-3-functionalized surfaces tends to increase with increasing proportion of the hydrophobic subunit within the MM+CO and DM+CH sets. Although such a trend might be interpreted to indicate that cell adhesion is regulated primarily or exclusively by protein adsorption, which in turn might be directly related to hydrophobic subunit proportion, several lines of evidence suggest that other factors are operative. Previous studies have shown that materials at the extremes of hydrophilicity or hydrophobicity tend to inhibit cell adhesion 22, 29, 30. For materials with a balance of hydrophobic and hydrophilic features, however, increases in substrate hydrophobicity can enable increased cell adhesion via increased protein adsorption 31. This trend appears to be reflected by some of our data, specifically the adhesion findings for the immobilization controls (C1-C5) and the MM+DH copolymers (Pol-13 to Pol-18). The MM+DH copolymer set contains the highest proportion of hydrophilic subunits among all the nylon-3 copolymers we examined, and our data indicate that these substrates permit only low protein adsorption and low cell adhesion (behavior comparable to that of the controls). However, an explanation based solely on hydrophilic/hydrophobic subunit proportion is not sufficient to explain differences in fibroblast adhesion among surfaces displaying other nylon-3 materials, the copolymers Pol-1 to Pol-12 and the two homopolymers, Pol-19 and Pol-20. Protein adsorption was constant among surfaces bearing these polymers, but cell adhesion and spreading varied considerably among these surfaces. Therefore, the variations among surfaces bearing these polymers must arise from a factor other than extent of serum protein adsorption. The relatively favorable adhesion and spreading of fibroblasts in the absence of serum on surfaces bearing selected nylon-3 copolymers provides further support for the hypothesis that the cells might somehow interact directly with the attached polymers, independent of adsorbed serum proteins. This possibility will be explored in future studies.
PEG hydrogels are commonly used not only as controlled, non-adhesive environments for exploring the functionality of discrete biological or biomimetic moieties 23, 24, but also as 3-D scaffold environments for tissue engineering applications 25. We attached the most favorable of the nylon-3 copolymers identified in our screen to PEG hydrogels in order to determine whether their cell-attractive properties would be manifested against a non-adhesive background, and to explore the feasibility of incorporating these new polymers into an engineered scaffold environment. Current approaches for rendering PEG hydrogels attractive to cells involve the incorporation of specific protein-derived adhesive sequences (such as RGD) 24, 32, or copolymerization of PEG with another material, either natural or synthetic 6, 33, 34, that manifests adhesive behavior. Copolymerization or blending of PEG with another synthetic polymer results in a material that is reliant upon non-specific protein adsorption to facilitate cell adhesion; such an approach is not compatible with the creation of a controlled, tailorable scaffold, and this strategy can potentially lead to scaffold immunogenicity 35. Our findings demonstrate that certain nylon-3 copolymers can functionally mimic specific cell-adhesive peptide motifs, such as RGD, when attached to PEG: the resulting hydrogels support cell adhesion and spreading more effectively than does a hydrogel bearing RGD, which is generally considered the “gold-standard” for promotion of cell attachment to PEG 12.
Overall, the results described here suggest that nylon-3 copolymers represent useful new materials for the development of cell-adhesive substrates. The ease with which these copolymers are synthesized, via ring-opening polymerization of β-lactams, makes them attractive relative to sequence-specific peptides, which require stepwise synthesis. Many features of nylon-3 structures can be readily varied (subunit composition, subunit stereochemistry, block vs. random copolymer architecture, nature of N-terminal group, length), and it seems likely that the experimental approaches outlined here will enable the discovery of additional useful examples.
Supplementary Material
Acknowledgments
This research was supported in part by the Nanoscale Science and Engineering Center at UW-Madison (DMR-0425880), the NSF Collaborative Research in Chemistry program (CHE-0404704), and a grant from the National Institutes of Health (1R21-EB005440). M.-r. L. was supported in part by a Korea Research Foundation Grant funded by the Korean Government (MOEHRD) (KRF-2006-214-C00053). We thank Prof. N. Abbott for helpful comments.
Footnotes
Supporting Information Available. Additional details describing the characterization of nylon-3 copolymers, fabrication of polymer-modified surfaces, and execution of cell adhesion and protein adsorption studies may be found in the Supporting Information document. This information is available free of charge via the Internet at http://pubs.acs.org/
References
- 1.Hersel U, Dahmen C, Kessler H. Biomaterials. 2003;24:4385–4415. doi: 10.1016/s0142-9612(03)00343-0. [DOI] [PubMed] [Google Scholar]
- 2.Ruoslahti E, Pierschbacher MD. Science. 1987;238:491–497. doi: 10.1126/science.2821619. [DOI] [PubMed] [Google Scholar]
- 3.Stevens MM, George JH. Science. 2005;310:1135–1138. doi: 10.1126/science.1106587. [DOI] [PubMed] [Google Scholar]
- 4.Ma PX. Adv Drug Deliv Rev. 2008;60:184–198. doi: 10.1016/j.addr.2007.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ratner BD, Bryant SJ. Annu Rev Biomed Eng. 2004;6:41–75. doi: 10.1146/annurev.bioeng.6.040803.140027. [DOI] [PubMed] [Google Scholar]
- 6.Rosso F, Giordano A, Barbarisi M, Barbarisi A. J Cell Physiol. 2004;199:174–180. doi: 10.1002/jcp.10471. [DOI] [PubMed] [Google Scholar]
- 7.Hartgerink JD, Beniash E, Stupp SI. Science. 2001;294:1684–1688. doi: 10.1126/science.1063187. [DOI] [PubMed] [Google Scholar]
- 8.Palmer LC, Newcomb CJ, Kaltz SR, Spoerke ED, Stupp SI. Chem Rev. 2008;108:4754–4783. doi: 10.1021/cr8004422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang S. Nat Biotechnol. 2003;21:1171–1178. doi: 10.1038/nbt874. [DOI] [PubMed] [Google Scholar]
- 10.Davis ME, Motion JP, Narmoneva DA, Takahashi T, Hakuno D, Kamm RD, Zhang S, Lee RT. Circulation. 2005;111:442–450. doi: 10.1161/01.CIR.0000153847.47301.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kretsinger JK, Haines LA, Ozbas B, Pochan DJ, Schneider JP. Biomaterials. 2005;26:5177–5186. doi: 10.1016/j.biomaterials.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 12.Lutolf MP, Hubbell JA. Nat Biotechnol. 2005;23:47–55. doi: 10.1038/nbt1055. [DOI] [PubMed] [Google Scholar]
- 13.Hashimoto K. Prog Polym Sci. 2000;25:1411–1462. [Google Scholar]
- 14.Zhang J, Kissounko DA, Lee SE, Gellman SH, Stahl SS. J Am Chem Soc. 2009;131:1589–1597. doi: 10.1021/ja8069192. [DOI] [PubMed] [Google Scholar]
- 15.Lee MR, Stahl SS, Gellman SH. Org Lett. 2008;10:5317–5319. doi: 10.1021/ol802274x. [DOI] [PubMed] [Google Scholar]
- 16.Graf R, Lohaus G, Börner K, Schmidt E, Bestian H. Angew Chem, Int Ed Engl. 1962;1:481–488. [Google Scholar]
- 17.Dener J, Fantauzzi P, Kshirsagar T, Kelly D, Wolfe A. Org Proc Res Dev. 2001;5:445–449. [Google Scholar]
- 18.Goodgame D, Hill S, Lincoln R, Quiros M, Williams D. Polyhedron. 1993;12:2753–2762. [Google Scholar]
- 19.Mowery BP, Lee SE, Kissounko DA, Epand RF, Epand RM, Weisblum B, Stahl SS, Gellman SH. J Am Chem Soc. 2007;129:15474–15476. doi: 10.1021/ja077288d. [DOI] [PubMed] [Google Scholar]
- 20.Salinas CN, Cole BB, Kasko AM, Anseth KS. Tissue Eng. 2007;13:1025–1034. doi: 10.1089/ten.2006.0126. [DOI] [PubMed] [Google Scholar]
- 21.Webb K, Hlady V, Tresco PA. J Biomed Mater Res. 1998;41:422–430. doi: 10.1002/(sici)1097-4636(19980905)41:3<422::aid-jbm12>3.0.co;2-k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilson CJ, Clegg RE, Leavesley DI, Pearcy MJ. Tissue Eng. 2005;11:1–18. doi: 10.1089/ten.2005.11.1. [DOI] [PubMed] [Google Scholar]
- 23.Hern DL, Hubbell JA. J Biomed Mater Res. 1998;39:266–276. doi: 10.1002/(sici)1097-4636(199802)39:2<266::aid-jbm14>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 24.Gonzalez AL, Gobin AS, West JL, McIntire LV, Smith CW. Tissue Eng. 2004;10:1775–1786. doi: 10.1089/ten.2004.10.1775. [DOI] [PubMed] [Google Scholar]
- 25.Nguyen KT, West JL. Biomaterials. 2002;23:4307–4314. doi: 10.1016/s0142-9612(02)00175-8. [DOI] [PubMed] [Google Scholar]
- 26.Drumheller PD, Hubbell JA. Anal Biochem. 1994;222:380–388. doi: 10.1006/abio.1994.1506. [DOI] [PubMed] [Google Scholar]
- 27.Ashammakhi N, Ndreu A, Nikkola L, Wimpenny I, Yang Y. Regen Med. 2008;3:547–574. doi: 10.2217/17460751.3.4.547. [DOI] [PubMed] [Google Scholar]
- 28.Grinnell F, Feld MK. Cell. 1979;17:117–129. doi: 10.1016/0092-8674(79)90300-3. [DOI] [PubMed] [Google Scholar]
- 29.Alves NM, Shi J, Oramas E, Santos JL, Tomas H, Mano JF. J Biomed Mater Res A. 2008 doi: 10.1002/jbm.a.32210. [DOI] [PubMed] [Google Scholar]
- 30.Kim SH, Ha HJ, Ko YK, Yoon SJ, Rhee JM, Kim MS, Lee HB, Khang G. J Biomater Sci Polym Ed. 2007;18:609–622. doi: 10.1163/156856207780852514. [DOI] [PubMed] [Google Scholar]
- 31.Campillo-Fernandez AJ, Unger RE, Peters K, Halstenberg S, Santos M, Sanchez MS, Duenas JM, Pradas MM, Ribelles JL, Kirkpatrick CJ. Tissue Eng Part A. 2008 doi: 10.1089/ten.tea.2008.0146. [DOI] [PubMed] [Google Scholar]
- 32.Weber LM, Hayda KN, Haskins K, Anseth KS. Biomaterials. 2007;28:3004–3011. doi: 10.1016/j.biomaterials.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 33.Park JS, Woo DG, Sun BK, Chung HM, Im SJ, Choi YM, Park K, Huh KM, Park KH. J Control Release. 2007;124:51–59. doi: 10.1016/j.jconrel.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 34.Gonen-Wadmany M, Oss-Ronen L, Seliktar D. Biomaterials. 2007;28:3876–3886. doi: 10.1016/j.biomaterials.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 35.Anderson JM, Rodriguez A, Chang DT. Semin Immunol. 2008;20:86–100. doi: 10.1016/j.smim.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.