

Abstract
Immunogenicity, manufacturing feasibility, and safety of a novel, autologous dendritic cell (DC)-based immunotherapy (AGS-004) was evaluated in ten human immunodeficiency virus type 1 (HIV-1)-infected adults successfully treated with antiretroviral therapy (ART). Personalized AGS-004 was produced from autologous monocyte-derived DCs electroporated with RNA encoding CD40L and HIV antigens (Gag, Nef, Rev, Vpr) derived from each subjects’s pre-ART plasma. Patients received monthly injections of AGS-004 in combination with ART. AGS-004 was produced within a mean of 6 weeks and yielded 4–12 doses/subject Full or partial HIV-specific proliferative immune responses occurred in 7 of 9 evaluable subjects. Responses were specific for the AGS-004 presented HIV antigens and preferentially targeted CD8+ cells. Mild adverse events included flu-like symptoms, fatigue, and injection site reactions. No evidence of autoimmunity, changes in viral load, or significant changes in absolute CD4+ and CD8+ T cell counts were observed. This pilot study supports the further clinical investigation of AGS-004.
Keywords: HIV, immunotherapy, vaccine, dendritic cells, acquired immunodeficiency syndrome
Introduction
Antiretroviral therapy (ART) improves morbidity and mortality associated with HIV-1 infection without improving the immune system’s ability to control HIV-1 viral replication, even after years of successful viral control. Immunotherapeutic strategies that enhance anti-HIV immune responses are needed to control viral replication in order to limit or delay exposure to ART [1]. Cytotoxic T lymphocyte (CTL) responses can only partially control HIV-1 replication [2] except in a very small percentage of patients with slow disease progression [3]. This ineffective HIV-specific CTL response is due to dysfunction of both CD4+ and CD8+ T cells [4–6]. Specifically, during disease progression, CD4+ T cells decrease and become poorly responsive to HIV antigens [7,8], while HIV-1-specific CD8+ T cells exhibit impaired effector function and selective depletion [9–11].
Immune therapy that promotes or restores potent CD8+ Tresponses may therefore be a rational treatment approach for HIV-infected patients, particularly if the treatment circumvents the need for direct priming, activation and expansion of the CD4+ T cell compartment, a potential source for viral replication. Monocyte-derived dendritic cells (DCs) stimulate and coordinate cell-mediated immunity through effects on CD8+ T cells [12].
Immunization with DCs presenting tumor-associated antigens have demonstrated immunogenicity, and in some patients antitumor effects were also observed [13,14]. Autologous DCs expressing HIV antigens have been evaluated for immunogenicity and anti-viral activity upon administration to HIV-infected patients either in with ART or without ART. Although well tolerated, only limited immunogenicity and/or viral control were observed [15–19].
To better address the inherent problem of HIV-1 extreme genetic diversity when using consensus or reference HIV protein sequences as immunogens, we developed a personalized immunotherapy using electroporation of DCs with autologous HIV antigen encoding RNAs to achieve antigen presentation, plus co-electroporation of cells with RNA encoding CD40L, the latter to achieve DC maturation. We have previously shown that ex vivo cytokine maturation of DCs followed by electroporation with RNA encoding CD40L protein, along with the RNA antigen payload, can improve the immunopotency of the final DC product [20,21]. Moreover, we have shown that DC matured by a CD40L dependent process and electroporated with RNAs encoding HIV antigens induce polyclonal immune responses in vitro [21,22]. In the present pilot clinical study, we evaluate an autologous DC-based immunotherapy (AGS-004) that uses autologous amplified HIV RNAs encoding Gag, Rev, Vpr and Nef as a source of HIV-1 antigens to stimulate CTL responses in HIV-1-infected patients treated with ART.
The choice of HIV antigens was based on data showing that Gag, Rev, Vpr and Nef are immunogenic and may contribute to viral load control [23–26]. To preserve full functionality of the DCs, a reduced quantity of Nef RNA was used compared to the other antigens [27] and the Vpr gene was truncated to remove its ability to suppress IL-12 and impact DC maturation [28, 29]. The DCs in the AGS-004 product are fully mature at the time of administration and are capable of exclusively inducing CD8+ CTL responses without a requirement for CD4+ T cell help [21,22]. Thus, AGS-004 was specifically designed to overcome viral variability and immune suppression mechanisms exerted by HIV-1 to inhibit DC maturation.
The present pilot study was designed to assess the immunogenicity, feasibility of production and delivery, and safety of AGS-004 in adult patients infected with HIV-1 receiving ART.
Materials and Methods
Study Subjects
Adults with documented HIV-1 infection with plasma HIV-1 RNA levels of ≤ 200 copies/mL and receiving their first ART regimen for at least 12 weeks prior to entry were enrolled at the McGill University Health Centre (Royal Victoria Hospital), Montreal, Quebec, between August 2006 and September 2007 (Clinical trial registry number NCT00381212).
All subjects had CD4+ T cell counts ≥ 350 cells/mm3 in the four weeks before study entry. Frozen plasma drawn approximately four weeks before starting ART (pre-ART plasma sample) was available for all eligible subjects. Subjects were free from co-infection with hepatitis B or C and had no prior lymph node irradiation or dissection. Subjects had not received previous HIV-1 immunotherapy or used systemic steroids or hydroxyurea. All subjects gave written informed consent, and the study was approved by the Royal Victoria Hospital Research Ethics Board.
AGS-004 Manufacturing
AGS-004 was produced according to current Good Manufacturing Practices (cGMP) at Argos. RNA was extracted and amplified from pre-ART plasma samples to generate HIV Gag, Rev, Vpr and Nef antigensas previously described [22]. CD40L RNA was transcribed in vitro from a CD40L-encoding plasmid using a co-transcriptional capping method (mMessage mMachine T7 Ultra, Ambion, Austin TX) [30]. The purified, sterile, endotoxin-free HIV antigen RNAs and polyadenylated CD40L RNA were formulated at a ratio of 0.25:1:1:1:4 μg for Nef, Gag, Rev, Vpr and CD40L in vitro transcribed RNAs, respectively[22].
Peripheral blood mononuclear cells (PBMCs) were obtained from each subject by standard leukapheresis (COBE Spectra Apheresis System, Caridian BCT, Lakewood, CO), and collected in a sterile, disposable, single-use cytapheresis bag that was transported to the Argos facility. PBMCs were isolated using Ficoll density gradient centrifugation. Monocytes were isolated from PBMCs through adherence to tissue culture plastic and cultured for six days with granulocyte macrophage-colony stimulating factor (GM-CSF) and interleukin-4 (IL-4) to generate immature DCs. DC maturation was initiated by overnight culture on day 6 with TNF-α, IFN-γ, and PGE2 as previously described [21].
CD40L RNA and autologous HIV-1 antigen RNA encoding Nef, Vpr, Gag, and Rev were electroporated into autologous DCs on day 7. The final AGS-004 product was suspended in 10% DMSO and 5% Dextrose for Injection in autologous plasma, quality control tested for sterility and immunophenotype (post thaw viability 78% ± 9%; CD40L expression 71% ± 17%) and cryopreserved as individual doses.
Study Design
Eligible subjects received intradermal injections of AGS-004 every 4 weeks for 4 treatments (Weeks 0, 4, 8 and 12). The target dose of AGS-004 was 1.0 × 107 DCs delivered in three injections of 0.2 mL each (0.6 mL total volume) to a single axillary lymph node area. At each treatment visit, blood was drawn prior to AGS-004 injection for routine hematology, clinical chemistry analysis, HIV-1 viral load and lymphocyte cell counts, autoimmunity, and immune monitoring. Subjects were observed for two hours following each injection and called on the day following injections to evaluate safety. A post-treatment leukapheresis occurred at week 14 to obtain cells for immune monitoring.
Study Endpoints and Assessments
The primary endpoint was the change from baseline in the proliferative capacity of CD8+ T cells in response to the four HIV RNA-encoded antigens expressed in AGS-004. Samples were assayed for T cell responses to AGS-004 HIV antigens using a previously reported assay [11] (National Immune Monitoring Laboratory Montreal, Canada). T cell reactivity to all four AGS-004 HIV antigens was measured using a CSFE proliferation assay. PBMC from subjects were labeled with CFSE and co-cultured with DCs electroporated with the RNA encoding the four HIV genes plus CD40L, analogous to the manufacturing process, as stimulators at a 40:1 ratio in RPMI 1640 medium plus 10% heat-inactivated AB serum. After 6 days of co-culture, cells were harvested and stained for immunophenotyping (CD3/CD4/CD8/CD28/CD45RA/CCR7). DCs electroporated with RNA encoding CD40L and enhanced green fluorescent protein (eGFP) served as a negative control stimulators. Data were acquired on an LSR2 flow cytometer (Becton Dickinson Biosciences, US), and analyzed using Becton Dickinson Diva software. Assays were performed in triplicate and data were reported as mean percentage of either CD3+/CD4+ or CD3+/CD8+ T cells that were CFSElow (indicating dilution of CFSE staining resulting from cell proliferation that occurred after stimulation).
Success of AGS-004 manufacturing was measured by the ability to produce AGS-004 and provide the first dose of AGS-004 to each subject within 12 weeks (84 days) of the initial leukapheresis.
Safety was evaluated by physical examinations, clinical laboratory hematology, absolute CD4+ and CD8+ T cell counts, and autoimmunity tests at treatment and post-treatment visits. Adverse event evaluations were made at each treatment visit and at the follow-up visit. Specifically, autoimmunity was assessed based on clinical and laboratory evaluations of antinuclear antibodies, rheumatoid factor, anti-double stranded DNA antibodies, total hemolytic complement (CH50), anti-thyroid antibodies, and indirect Coombs test at screening, at each dosing visit, and at the discontinuation visit. Injection site reactions were assessed for 2 hours following each injection and by a follow-up phone call 24 hours after each injection.
The study was designed to assess up to 10 evaluable subjects with the objective of having a total of 8 subjects for whom a complete set of data and biologic samples would be available for the assessment of the primary endpoint. Descriptive statistics were used. The primary endpoint was met if a change ≥2-fold from baseline in the proliferative capacity of CD8+ T cells was recorded at any time after dosing with AGS-004. A positive response was defined as the percentage proliferation of CD8+ T cells to HIV antigens minus the percentage proliferation to the GFP negative control plus 3 standard deviations. The study was considered positive if ≥ 4 subjects met the primary endpoint.
Results
Subjects
Twelve subjects were screened and RNA was successfully manufactured from 10 of the 12 pre-ART plasma samples. AGS-004 was successfully generated and administered to 10 subjects. Nine enrolled subjects were evaluable for the primary endpoint, received all four doses and had a mean duration of AGS-004 treatment of 84 ± 8 days. One subject was not evaluable to the inability to generate cells for immune monitoring at week 14. Demographics and baseline characteristics for each subject are shown in Table 1. All subjects were white males with a mean age of 39 years. Median CD4+ T cell nadir prior to initiating ART was 264 cells/mm3 and median pre-ART viral load was 8.2×105 copies/mL. HIV-1 viral load was undetectable (≤50 copies/mL) at baseline and throughout the duration of the study for all subjects.
Table 1.
Demographic and Baseline Data for All Subjects
| Subject | Age (yr) | ART* | CD4+ T CellCounts (cells/mm3) | CD8+ T Cell Counts (cells/mm3) | Pre-ART viral load** (copies/mL) | |
|---|---|---|---|---|---|---|
| Nadir | Baseline | Baseline | ||||
| 1 | 53 | PI, NRTI | 189 | 366 | 859 | 5.6×104 |
| 2 | 55 | PI, NRTI, | 200 | 359 | 346 | 2.2×104 |
| 3 | 44 | NRTI, NNRTI | 740 | 924 | 406 | 3.9×104 |
| 4 | 42 | PI, NRTI | 197 | 661 | 579 | 6.4×106 |
| 5 | 41 | PI, NRTI | 270 | 741 | 757 | 1.9×105 |
| 6 | 28 | PI, NRTI, NNRTI | 211 | 289 | 855 | 2.0×104 |
| 7 | 23 | PI, NRTI | 226 | 553 | 387 | 6.6×104 |
| 8 | 32 | NRTI, NNRTI | 146 | 360 | 684 | 1.9×105 |
| 9 | 39 | PI, NRTI | 212 | 464 | 594 | 1.1×104 |
| 10 | 30 | PI, NRTI | 250 | 348 | 455 | 1.2×106 |
| Mean (SD) | 39 (11) | NA | 264 (171) | 507 (209) | 592 (192) | 8.2 × 105 (2.0 × 106) |
PI protease inhibitor, NRTI nucleoside reverse transcriptase inhibitor, NNRTI non- nucleoside reverse transcriptase inhibitor.
Viral load at baseline and throughout the study was ≤ 50 copies/mL for all subjects.
Immunogenicity
CD8+ T cell proliferative responses are shown in Table 2 for the 9 evaluable subjects. Four subjects had increases in proliferation in response to DCs electroporated with RNA encoding the four autologous HIV antigens plus CD40L, representing the AGS-004 product, which met the criteria for a full response. Three additional subjects had increases representing partial responses to the AGS-004 product. The proliferative response to all four HIV antigens was specific to the CD8+ T cell pool as shown by the “heat” maps in Figure 1. The intensity of the proliferative response is represented by a linear distribution of colors and range from blue (no response) to red (greatest response) for CD8+ and CD4+ T cell pools at baseline, and weeks 4, 8 and 12. There was only one weak CD4+ proliferative response in a single subject at week 8. In contrast, CD8+ proliferative responses greater than the negative control plus 3 standard deviations were measured in 7/9 subjects.
Table 2.
Proliferative CD8+ T cell responses to all 4 antigens (GVRN) in AGS-004
| Subject | Study Phase | Control | GVRN | Difference | Response |
|---|---|---|---|---|---|
| 1 | Baseline | 3.63 | 3.42 | −0.21 | None |
| Week 4 | 3.99 | 2.96 | −1.03 | ||
| Week 8 | 3.34 | 2.49 | −0.85 | ||
| Week 12 | 3.53 | 2.93 | −0.6 | ||
| 2 | Baseline | 1.6 | 13.4 | 11.75 | Partial |
| Week 4 | 4.02 | 24.2 | 20.18 | ||
| Week 8 | 4.51 | 25.7 | 21.16 | ||
| Week 12 | 7.14 | 17.5 | 10.32 | ||
| 3 | Baseline | 2.38 | 8.8 | 6.42 | Partial |
| Week 4 | 2.88 | 11 | 8.14 | ||
| Week 8 | 3.54 | 12.7 | 9.11 | ||
| Week 12 | 4.55 | 10.7 | 6.15 | ||
| 4 | Baseline | 1.05 | 14 | 12.96 | Positive |
| Week 4 | 3.04 | 13.3 | 10.23 | ||
| Week 8 | 1.98 | 25.3 | 23.35 | ||
| Week 12 | 6.14 | 33 | 26.87 | ||
| 6 | Baseline | 1.49 | 1.34 | −0.15 | Positive |
| Week 4 | 0.56 | 0.67 | 0.11 | ||
| Week 8 | 1.04 | 0.8 | −0.24 | ||
| Week 12 | 0.76 | 0.55 | −0.21 | ||
| 7 | Baseline | 0.39 | 0.25 | −0.14 | Positive |
| Week 4 | 0.57 | 0.54 | −0.03 | ||
| Week 8 | 0.36 | 0.46 | 0.10 | ||
| Week 12 | 3.07 | 0.48 | −2.59 | ||
| 8 | Baseline | 1.1 | 0.98 | −0.12 | None |
| Week 4 | 1.23 | 1.2 | −0.03 | ||
| Week 8 | 5.84 | 5.47 | −0.37 | ||
| Week 12 | 3.6 | 2.5 | −1.10 | ||
| 9 | Baseline | 2.25 | 4.56 | 2.31 | Partial |
| Week 4 | 0.98 | 3.84 | 2.86 | ||
| Week 8 | 1.75 | 4.11 | 2.36 | ||
| Week 12 | . | . | N/A | ||
| 10 | Baseline | 3.3 | 4.32 | 1.02 | Positive |
| Week 4 | 3.69 | 5.25 | 1.56 | ||
| Week 8 | 7.92 | 9.98 | 2.06 | ||
| Week 12 | 4.09 | 5.42 | 1.33 |
Control plus 3 SD and GVRN values are the percent CFSElow CD8+ CTLs at baseline and at each treatment week. A positive primary endpoint is defined as a difference between the GVRN response on therapy versus baseline value at any time point that is ≥ 2x the baseline value. The time point that yields the positive primary endpoint is bolded. See text for additional information. N/A= not available
Figure 1.
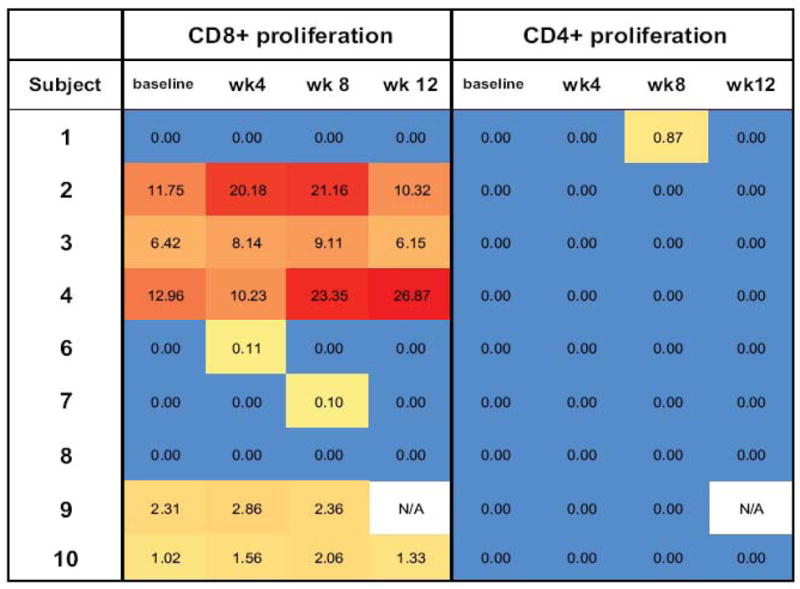
Heat map for proliferative response to AGS-004 HIV antigens for all evaluable subjects. Values represent the difference between eGFP control plus 3SD and HIV antigen induced CD8+ or CD4+ T cell proliferation at baseline, 4, 8 and 12 weeks derived from triplicate cultures. Colors represent a linear distribution of the data where blue corresponds to the lowest value (0.00) and red is the highest value (26.87), observed. N/A= not available
Feasibility
Ten subjects’ AGS-004 products were successfully manufactured within a mean of 6 weeks (range of 33–69 days). Manufacturing lots yielded 4 to 12doses of AGS-004 for each subject in one production cycle. Immunophenotyping demonstrated expression of the cell-surface activation markers CD80, CD83, and CD86 (data not shown) and confirm that DC electroporation with the four antigen-specific HIV RNAs did not impair DC maturation.
Safety
Seven of ten subjects had at least one treatment emergent adverse event as summarized in Table 3. Events considered possibly or probably related to AGS-004 treatment were reported in six subjects and were grade 1, including diarrhea, axillary pain, influenza-like illness, injection-site reactions, fatigue, headache and low rheumatoid factor increase. Injection site reactions evaluated 2 hours after treatment included reports in 4 patients of mild or Grade 1 levels of pain or soreness (n=5), mild erythema (n=7), mild induration (n=1) and itching (n=3) at the injection site 2 hours after injection. No subject discontinued due to any adverse event. In the 24 hr follow-up phone calls (not captured as adverse events) made by the site staff after each treatment, 5 subjects had reports of injection site reactions and 2 reported flu-like symptoms.
Table 3.
Treatment emergent adverse events reported in seven subjects.
| Number of subjects | Grade | Subject number* | |
|---|---|---|---|
| Fatigue | 5 | 1 | 2,3,5,6,10 |
| Injection site reactions** | 4 | 1 | 1,2,3,6 |
| Influenza-like illness | 2 | 1 | 5,6 |
| Headache | 1 | 1 | 10 |
| Diarrhea | 1 | 1 | 6 |
| Axillary pain | 1 | 1 | 10 |
| Rheumatoid factor increased | 1 | 1 | 5 |
| Nausea | 1 | 1 | 2 |
| Increased blood creatinine | 1 | 1 | 10 |
| Hematochezia | 1 | 1 | 5 |
| Eye infection | 1 | 1 | 2 |
| Insomnia | 1 | 1 | 3 |
| Squamous cell carcinoma | 1 | 2 | 1 |
| Gastroesophageal reflux disease | 1 | 2 | 10 |
| GI pain | 1 | 3 | 2 |
| Appendicitis | 1 | 3 | 9 |
| Cholecystitis | 1 | 3 | 2 |
| Anemia | 1 | 1 | 5 |
The autoimmunity panel and assessment of related clinical signs and symptoms (rash, cytopenia, and arthralgia) showed negative values throughout all study visits except for one subject who had a transient elevated rheumatoid factor at screening, weeks 12, and 18 without any clinical evidence of autoimmunity.
Two unrelated serious adverse events occurred in two subjects: (1) acute cholecystitis occurring seven months after the final AGS-004 injection resulting in cholecystectomy and (2) acute appendicitis two months after the completion of AGS-004 injection resulting in an appendectomy.
There were no changes in vital signs (blood pressure, heart rate, respiratory rate, and body temperature) during the 2 hour assessments following each AGS-004 injection that were considered clinically relevant. There were no clinically relevant changes in clinical laboratory tests during the course of the study and no subject treated with AGS-004 experienced any evidence of adenopathy at any time during the study.
No clinically relevant changes throughout the study in CD4+ and CD8+ T cell counts were observed. Figure 2 shows individual CD4+ T cell counts over time for each subject and the median cell counts. The CD4+/CD8+ ratio remained relatively constant from baseline through all treatment and follow-up assessments, with variations that were not considered clinically relevant. HIV-1 viral loads remained undetectable throughout the study.
Figure 2.

AbsoluteCD4 + T cell counts for all subjects throughout the study. Numbers adjacent to gray lines correspond to subject numbers in Tables 1 and 2. Black squares represent the median values at each time point.
Discussion
Immunotherapy with autologous DCs electroporated with RNA encoding CD40L and RNA encoding autologous HIV antigens appears to be clinically feasible, safe, and may be associated with CD8+ T cell proliferative responses in infected HIV-1 patients receiving ART. Although the study was limited by the small numbers of patients, we enrolled patients ranging in age from 23–55 years who had viral loads that were undetectable at entry and that remained undetectable for the entire study. There were no unexpected tolerability issues and no evidence of autoimmunity in the course of the study. The potential for autoimmunity to occur in response to this immunotherapeutic approach is possible but has not been substantiated by this study. Autoimmunity panels were negative for all but one subject with elevated rheumatoid factor that was not associated with any clinical evidence of autoimmunity. These results are consistent with DC-based immunotherapy approaches in general [31], those using RNA-encoding antigens [32] and those specifically targeting HIV-1 [15–17,19]. The most common adverse events occurring in this study were grade 1 fatigue, flu-like illness, and transient injection-site reactions. AGS-004 was administered intradermally rather than subcutaneously or by other routes in order to promote more efficient migration to lymph nodes as suggested by clinical and animal data [33,34]. There was no indication of adenopathy in any subject.
The induced immune responses preferentially targeted the CD8+ rather than the CD4+ T cell compartment as expected since the antigens are expressed from RNA translated in the cytosol, thereby leading to primarily MHC class I-restricted epitope presentation [35]. There were no clinically significant changes to the absolute CD4+ and CD8+ T cell counts over the course of the study which is consistent with a lack of non-specific T cell activation. This finding is significant since specific or non-specific T cell activation may be associated with greater viral rebound and reduced time to ART resumption following therapeutic immunization with the ALVAC-HIV product [36].
CD8+ T cell proliferative responses were elevated relative to baseline at 4 and/or 8 weeks for all subjects with full or partial responses. Responses in three subjects returned to baseline levels at 12 weeks. This may be due to multiple factors including migration of antigen-reactive cells from the peripheral blood or a change in the proliferative potential as a consequence of maturation of the immune response from a proliferative to a non-proliferative effector cell status. Significant inter-individual variability in the proliferative CD8+ T cell responses to AGS-004 was observed, and there was no clear relationship between responses and baseline patient characteristics (e.g., higher CD4+ T cell nadir, acute/early or chronic infection at the time of initiation of ART). It remains unknown whether or how the variability, elevation, and attenuation of proliferative responses correlate with improvement in immune function or control of viral replication [37].
This clinical study confirms previous proof-of-concept studies and in vitro findings that HIV-1 antigens encoded by RNA can be translated and presented by DC to induce MHC class I-restricted polyclonal immune responses [21,22]. The multiplex RT-PCR strategy used to amplify the target antigen RNA permits reliable strain-independent amplification of highly polymorphic target antigens from subjects without the requirement of knowing the individual’s viral sequence or needing to custom-design PCR primers for each individual. The functional characteristics of DCs electroporated with CD40L-encoding RNA and the four HIV-1 antigen-derived RNAs were maintained as determined by immunophenotyping for DC markers indicative of maturity. Thus, AGS-004 presents autologous HIV-1 viral sequences derived from a multitude of infectious quasispecies that arise under CTL pressure, and presents these antigens using fully-matured DCs. This novel approach may form the foundation for successful anti-HIV-1 immunotherapy. We also demonstrated the feasibility of generating a patient-derived, autologous HIV-1 immunotherapy and providing the first dose to each subject within a range of 33–69 days of the subject’s leukapheresis.
In summary, AGS-004, an autologous CD40L matured DC immunotherapy using autologous RNA encoding Gag, Rev, Vpr and Nef as the source of HIV-1 antigens, represents a novel and promising approach to the stimulation of the host immune system in HIV-1 infected subjects. Since controlled ART treatment interruption can be an effective tool in defining clinical responses to HIV-1 immunotherapy [38,39], a phase II multicenter trial to assess the ability of AGS-004 immunotherapy to improve immune control of HIV-1 replication following ART discontinuation is currently in progress in Canada and the US.
Acknowledgments
This project has been funded in part with Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract No. N01-AI-60019 and by CANVAC (HIV-001) and the Canadian trials Network (CTN) Study No. 229. Argos provided funding for statistical support, study monitoring, data management, and medical writing support. Statistical support was provided by Cemal Unal, PhD. Medical writing support was provided by Patrice Ferriola, PhD. We thank the study site personnel including Pierrette Bouchard; Mario Legault, and Marwan Samia at the clinical site, Christine Colven and Esther Villiard at Argos Therapeutics Inc., and Tom Baumgartner at the University of Montreal, Quebec, Canada for providing support in the conduct of the immune assays. J-P Routy, is a clinician-scientist supported by Fonds de la recherche en santé du Québec (FRSQ). R-P Sekaly is the Canada Research Chair in Human Immunology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kinloch-de Loes S. Role of therapeutic vaccines in the control of HIV-1. J Antimicrob Chemother. 2004;53:562–566. doi: 10.1093/jac/dkh132. [DOI] [PubMed] [Google Scholar]
- 2.Schmitz JE. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science. 1999;283:857–860. doi: 10.1126/science.283.5403.857. [DOI] [PubMed] [Google Scholar]
- 3.Addo MM, Draenert R, Rathod A, et al. Fully differentiated HIV-1 specific CD8+ T effector cells are more frequently detectable in controlled than in progressive HIV-1 infection. PLoS ONE. 2007;2:e321. doi: 10.1371/journal.pone.0000321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goulder PJ. Evolution and transmission of stable CTL escape mutations in HIV infection. Nature. 2001;412:334–338. doi: 10.1038/35085576. [DOI] [PubMed] [Google Scholar]
- 5.Barouch DH, Kunstman J, Kuroda MJ, et al. Eventual AIDS vaccine failure in a rhesus monkey by viral escape from cytotoxic T lymphocytes. Nature. 2002;415:335–339. doi: 10.1038/415335a. [DOI] [PubMed] [Google Scholar]
- 6.Vaccari M, Mattapallil J, Song K, et al. Reduced protection from simian immunodeficiency virus SIVmac251 infection afforded by memory CD8+ T cells induced by vaccination during CD4+ T-cell deficiency. J Virol. 2008;82:9629–9638. doi: 10.1128/JVI.00893-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McNeil AC, Shupert WL, Iyasere CA, et al. High-level HIV-1 viremia suppresses viral antigen-specific CD4(+) T cell proliferation. Proc Natl Acad Sci U S A. 2001;98:13878–13883. doi: 10.1073/pnas.251539598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Younes SA, Yassine-Diab B, Dumont AR, et al. HIV-1 viremia prevents the establishment of interleukin 2-producing HIV-specific memory CD4+ T cells endowed with proliferative capacity. J Exp Med. 2003;198:1909–1922. doi: 10.1084/jem.20031598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lichterfeld M, Yu X, Mui S, et al. Selective depletion of high-avidity human immunodeficiency virus type 1(HIV-1)-specific CD8+ T cells after early HIV-1 infection. J Virol. 2007;81:199–214. doi: 10.1128/JVI.01388-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zanussi S, Simonelli C, D’Andrea M, et al. CD8+ lymphocyte phenotype and cytokine production in long-term non-progressor and in progressor patients with HIV-1 infection. Clin Exp Immunol. 1996;105:220–224. doi: 10.1046/j.1365-2249.1996.d01-746.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trautmann L, Janbazian L, Chomont N, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med. 2006;12:1198–1202. doi: 10.1038/nm1482. [DOI] [PubMed] [Google Scholar]
- 12.Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. 2007;449:419–426. doi: 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- 13.Nicolette CA, Healey D, Tcherepanova I, et al. Dendritic cells for active immunotherapy: optimizing design and manufacture in order to develop commercially and clinically viable products. Vaccine. 2007;25(Suppl 2):B47–60. doi: 10.1016/j.vaccine.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 14.Osada T, Clay TM, Woo CY, Morse MA, Lyerly HK. Dendritic cell-based immunotherapy. Int Rev Immunol. 2006;25:377–413. doi: 10.1080/08830180600992456. [DOI] [PubMed] [Google Scholar]
- 15.Connolly NC, Whiteside TL, Wilson C, et al. Therapeutic immunization with human immunodeficiency virus type 1 (HIV-1) peptide-loaded dendritic cells is safe and induces immunogenicity in HIV-1-infected individuals. Clin Vaccine Immunol. 2008;15:284–292. doi: 10.1128/CVI.00221-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garcia F, Lejeune M, Climent N, et al. Therapeutic immunization with dendritic cells loaded with heat-inactivated autologous HIV-1 in patients with chronic HIV-1 infection. J Infect Dis. 2005;191:1680–1685. doi: 10.1086/429340. [DOI] [PubMed] [Google Scholar]
- 17.Lu W, Arraes LC, Ferreira WT, Andrieu JM. Therapeutic dendritic-cell vaccine for chronic HIV-1 infection. Nat Med. 2004;10:1359–1365. doi: 10.1038/nm1147. [DOI] [PubMed] [Google Scholar]
- 18.Whiteside TL, Piazza P, Reiter A, et al. Production of a dendritic cell-based vaccine containing inactivated autologous virus for therapy of patients with chronic human immunodeficiency virus type 1 infection. Clin Vaccine Immunol. 2009;16:233–240. doi: 10.1128/CVI.00066-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ide F, Nakamura T, Tomizawa M, et al. Peptide-loaded dendritic-cell vaccination followed by treatment interruption for chronic HIV-1 infection: a phase 1 trial. J Med Virol. 2006;78:711–718. doi: 10.1002/jmv.20612. [DOI] [PubMed] [Google Scholar]
- 20.DeBenedette MA, Calderhead DM, Ketteringham H, et al. Priming of a novel subset of CD28+ rapidly expanding high-avidity effector memory CTL by post maturation electroporation-CD40L dendritic cells is IL-12 dependent. J Immunol. 2008;181:5296–5305. doi: 10.4049/jimmunol.181.8.5296. [DOI] [PubMed] [Google Scholar]
- 21.Calderhead DM, DeBenedette MA, Ketteringham H, et al. Cytokine maturation followed by CD40L mRNA electroporation results in a clinically relevant dendritic cell product capable of inducing a potent proinflammatory CTL response. J Immunother. 2008;31:731–741. doi: 10.1097/CJI.0b013e318183db02. [DOI] [PubMed] [Google Scholar]
- 22.Tcherepanova I, Harris J, Starr A, et al. Multiplex RT-PCR amplification of HIV genes to create a completely autologous DC-based immunotherapy for the treatment of HIV infection. PLoS ONE. 2008;3:e1489. doi: 10.1371/journal.pone.0001489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Addo MM, Yu XG, Rosenberg ES, Walker BD, Altfeld M. Cytotoxic T-lymphocyte (CTL) responses directed against regulatory and accessory proteins in HIV-1 infection. DNA Cell Biol. 2002;21:671–678. doi: 10.1089/104454902760330219. [DOI] [PubMed] [Google Scholar]
- 24.Van Gulck ER, Ponsaerts P, Heyndrickx L, et al. Efficient stimulation of HIV-1-specific T cells using dendritic cells electroporated with mRNA encoding autologous HIV-1 Gag and Env proteins. Blood. 2006;107:1818–1827. doi: 10.1182/blood-2005-01-0339. [DOI] [PubMed] [Google Scholar]
- 25.Altfeld M, Addo MM, Eldridge RL, et al. Vpr is preferentially targeted by CTL during HIV-1 infection. J Immunol. 2001;167:2743–2752. doi: 10.4049/jimmunol.167.5.2743. [DOI] [PubMed] [Google Scholar]
- 26.Novitsky V, Cao H, Rybak N, et al. Magnitude and frequency of cytotoxic T-lymphocyte responses: identification of immunodominant regions of human immunodeficiency virus type 1 subtype C. J Virol. 2002;76:10155–10168. doi: 10.1128/JVI.76.20.10155-10168.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Quaranta MG, Mattioli B, Giordani L, Viora M. The immunoregulatory effects of HIV-1 Nef on dendritic cells and the pathogenesis of AIDS. FASEB J. 2006;20:2198–2208. doi: 10.1096/fj.06-6260rev. [DOI] [PubMed] [Google Scholar]
- 28.Majumder B, Janket ML, Schafer EA, et al. Human immunodeficiency virus type 1 Vpr impairs dendritic cell maturation and T-cell activation: implications for viral immune escape. J Virol. 2005;79:7990–8003. doi: 10.1128/JVI.79.13.7990-8003.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tcherepanova I, Starr A, Lackford B, et al. The Immunosuppressive Properties of the HIV Vpr protein are Linked to a Single Highly Conserved Residue, R90. PLoSOne. 2009;4:e5853. doi: 10.1371/journal.pone.0005853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tcherepanova IY, Adams MD, Feng X, et al. Ectopic expression of a truncated CD40L protein from synthetic post-transcriptionally capped RNA in dendritic cells induces high levels of IL-12 secretion. BMC Mol Biol. 2008;9:90. doi: 10.1186/1471-2199-9-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ridgway D. The first 1000 dendritic cell vaccinees. Cancer Invest. 2003;21:873–886. doi: 10.1081/cnv-120025091. [DOI] [PubMed] [Google Scholar]
- 32.Gilboa E, Vieweg J. Cancer immunotherapy with mRNA-transfected dendritic cells. Immunol Rev. 2004;199:251–263. doi: 10.1111/j.0105-2896.2004.00139.x. [DOI] [PubMed] [Google Scholar]
- 33.Hao S, Ye Z, Yang J. Intradermal vaccination of dendritic cell-derived exosomes is superior to a subcutaneous one in the induction of antitumor immunity. Cancer Biother Radiopharm. 2006;21:146–154. doi: 10.1089/cbr.2006.21.146. [DOI] [PubMed] [Google Scholar]
- 34.Morse M, Coleman R, Akabani G. Migration of human dendritic cells after injection in patients with metastatic malignancies. cancer Res. 1999;59:56–58. [PubMed] [Google Scholar]
- 35.Van Kaer L. Major histocompatibility complex class I-restricted antigen processing and presentation. Tissue Antigens. 2002;60:1–9. doi: 10.1034/j.1399-0039.2002.600101.x. [DOI] [PubMed] [Google Scholar]
- 36.Autran B, Murphy RL, Costagliola D, et al. Greater viral rebound and reduced time to resume antiretroviral therapy after therapeutic immunization with the ALVAC-HIV vaccine (vCP1452) AIDS. 2008;22:1313–1322. doi: 10.1097/QAD.0b013e3282fdce94. [DOI] [PubMed] [Google Scholar]
- 37.Appray V, Douek D, Price D. CD8+ T cell efficacy in vaccination and disease. Nat Med. 2008;14:623–628. doi: 10.1038/nm.f.1774. [DOI] [PubMed] [Google Scholar]
- 38.Jacobson JM, Turner BJ, Abrutyn E. Trials that matter: CD4+ T-lymphocyte count-guided interruption of antiretroviral therapy in HIV-infected patients. Ann Intern Med. 2007;146:682–683. doi: 10.7326/0003-4819-146-9-200705010-00014. [DOI] [PubMed] [Google Scholar]
- 39.Kutzler M, Jacobson JM. Treatment interruption as a tool to measure changes in immunologic response to HIV-1. Current Opinion in HIV & AIDS. 2008;3:131–135. doi: 10.1097/COH.0b013e3282f54cde. [DOI] [PubMed] [Google Scholar]