

Abstract
Recent evidence suggests that abnormal activation of cyclin-dependent kinase 5 (cdk5) is a critical prodeath signal in stroke. However, the mechanism(s) by which cdk5 promotes death is unclear. Complicating the role of cdk5 are the observations that cdk5 can exist in multiple cellular regions and possess both prosurvival and prodeath characteristics. In particular, the critical role of cytoplasmic or nuclear cdk5 in neuronal jury, in vivo, is unclear. Therefore, we determined where cdk5 was activated in models of ischemia and how manipulation of cdk5 in differing compartments may affect neuronal death. Here, we show a critical function for cytoplasmic cdk5 in both focal and global models of stroke, in vivo. Cdk5 is activated in the cytoplasm and expression of DNcdk5 localized to the cytoplasm is protective. Importantly, we also demonstrate the antioxidant enzyme Prx2 (peroxiredoxin 2) as a critical cytoplasmic target of cdk5. In contrast, the role of cdk5 in the nucleus is context-dependent. Following focal ischemia, nuclear cdk5 is activated and functionally relevant while there is no evidence for such activation following global ischemia. Importantly, myocyte enhancer factor 2D (MEF2D), a previously described nuclear target of cdk5 in vitro, is also phosphorylated by cdk5 following focal ischemia. In addition, MEF2D expression in this paradigm ameliorates death. Together, our results address the critical issue of cdk5 activity compartmentalization, as well as define critical substrates for both cytoplasmic and nuclear cdk5 activity in adult models of stroke.
Introduction
Stroke results from a transient or permanent reduction in blood flow to the brain. The mechanisms involved in ischemic neuronal death are not fully defined. In this regard, accumulating evidence has implicated cyclin-dependent kinases (CDKs) with stroke damage (Rashidian et al., 2007). CDKs are best recognized for their role in regulating cell cycle progression (Pines, 1993). However, they are now noted for their roles in other biological processes (Gold and Rice, 1998). Specifically, some specialized members are predominately active in the nervous system. Cdk5, a proline-directed serine/threonine kinase, is one such member. Cdk5 does not use cyclins to promote activation. Instead, it uses p35 and p39 which are predominantly expressed in CNS (Dhavan and Tsai, 2001). Moreover, in contrast to other CDKs, there is little functional evidence for involvement of cdk5 in classical cell cycle regulation. Instead, cdk5 is considered a key element in neuronal development and function (Dhavan and Tsai, 2001; Li et al., 2001; Johansson et al., 2005). Cdk5 is also believed to be critical for neuronal survival following exposure to apoptotic stimuli (Li et al., 2002, 2003; Wang et al., 2006).
Interestingly, in contrast to its normal function, there is increasing evidence that suggests a pathogenic role of cdk5 in neurodegenerative disorders, such as Parkinson's disease (PD) (Smith et al., 2003), Alzheimer's disease (AD) (Cruz and Tsai, 2004), amyotrophic lateral sclerosis (ALS) (Nguyen and Julien, 2003), Niemann-Pick type C (NPC) (Bu et al., 2002), and stroke (Wang et al., 2003; Rashidian et al., 2005). This pathogenic face is proposed to be unmasked by calpain-mediated cleavage of the p35 to a more stable active p25 form. There is evidence of increasing levels of p25 in the nucleus, which could alter cdk5 substrate specificity under some neurotoxic conditions (Patrick et al., 1999; Kusakawa et al., 2000; O'Hare et al., 2005). p35 contains a myristoylation motif and associates with membranous compartments of the cell, while p25 lacks this motif and localizes cdk5 to soluble fractions (Kusakawa et al., 2000). Moreover, several studies have demonstrated that cdk5 can be both nuclear and cytoplasmic (Ino and Chiba, 1996; O'Hare et al., 2005; Fu et al., 2006). Recent evidence suggests that cell localization might be important for its prosurvival/prodeath role. We have recently shown that inhibition of cytoplasmic cdk5 sensitizes neurons to DNA damage, suggesting a prosurvival role of cytoplasmic cdk5 under normal conditions (O'Hare et al., 2005). In contrast, nuclear cdk5, in an in vitro model of excitotoxicity is death promoting, through inhibition of the survival properties of myocyte enhancer factor 2D (MEF2D) (O'Hare et al., 2005; Tang et al., 2005). This would suggest that cytoplasmic activity is normal while nuclear activity is pathogenic. However, this simplistic view is likely incorrect. For example, cdk5 can phosphorylate tau in the cytoplasm, a presumptive prodeath stress (Cruz and Tsai, 2004). These observations suggest that cdk5 activity is much more complex. Moreover, the role of cdk5 localization in adult models of injury, particularly in stroke is unclear. Understanding of where cdk5 is activated and contributes to pathogenesis is critical, particularly in light of its prosurvival function(s).
Presently, we determine the localization of cdk5 activity in multiple models of stroke in vivo and provide critical downstream targets for such activity. We provide evidence that cytoplasmic cdk5 activity is essential for death in multiple paradigms of ischemia (focal and global) in vivo, while nuclear cdk5 activity is only activated following focal ischemia. Critically, we also identify peroxiredoxin 2 (Prx2) as a common critical cytoplasmic target and the transcription factor MEF2D as a relevant nuclear target in stroke. These observations highlight the complexity of cdk5 activation and define at least two critical cdk5 signaling axes in stroke.
Materials and Methods
Animals.
All animal experiments were approved by the University of Ottawa Animal Care Committee and conformed to the guidelines set forth by the Animal Care Council of Canada and Canadian Institutes of Health Research.
Viral construction.
Recombinant adeno-associated virus (AAV) vectors were constructed by subcloning cDNA sequences of DNcdk5-Flag (Gong et al., 2003; Smith et al., 2003), Wtcdk5-GFP-NLS(NES), DNcdk5-GFP-NLS(NES) (O'Hare et al., 2005), and Prx2-Flag and its mutants (Prx2T89A, Prx2T89E) in the AM/CBA-pl-WPRE-bGH plasmid. Then, viruses were generated as previously described (Zolotukhin et al., 2002). The adenoviruses (AV) expressing Prx2, Prx2T89A, Prx2T89E, and MEF2D-S444 were engineered by subcloning these sequences into pAdTrack vectors under control of a CMV promoter. These vectors also expressed green fluorescent protein (GFP) by a separate CMV promoter. Finally, these constructs were used to generate AV, as described previously (He et al., 1998).
P35 knock-out mice.
P35−/− mice were generated by breeding heterozygote p35 mutants and genotyped as described previously (Hallows et al., 2003).
Cell culture.
Primary cerebellar granule neuron (CGN) cultures were prepared from 7–9 d postnatal mice as previously described (Rashidian et al., 2005).
Glutamate model of neuronal death.
Five day plated CGN cultures in 96-well plates were infected with AV with multiplicity of infection of 20 (MOI = 20) for survival assay. On day 7, cultures were exposed to 50 μm glutamate for 60–70 min, in the presence or absence of MK801, and then washed with conditional medium and incubated for 90 min. Cultures were then fixed with 4% paraformaldehyde (PFA) and stained with Hoescht 33342 (0.25 μg/ml; Sigma-Aldrich) for nuclei staining. Nuclei from live cells appeared intact and noncondensed or nonfragmented while nuclei from dead cells appeared condensed or fragmented. For the survival assay, the numbers of live GFP-positive cells were assessed in each entire well. Three wells for each treatment were counted and averaged. To calculate percentage of live CGNs after glutamate treatment, we compared the number of live GFP-positive neurons obtained in the absence of MK801 with the number of live GFP-positive neurons obtained in the presence of MK801 for each virus. MK801 is an NMDA receptor blocker, which inhibits >90% of death induced by glutamate treatment in this paradigm. We used the MK801/glutamate-treated cultures as control for any minor nonspecific death induced by media change. Survival is expressed as percentage of live CGNs as described by (number of live GFP-positive neurons in the absence of MK801/number of live GFP-positive neurons in the presence of MK801) × 100.
For detecting phosphorylation of WtPrx2 and Prx2T89A, 5 d plated CGN cultures in 4-well plates were infected with AV (MOI = 20). On day 7, cultures were exposed to 20 μm glutamate for 20 min and then washed with conditional medium and incubated. Cultures were collected for Western blot analyses 0, 30, 60 and 90 min following incubation.
Viral injection.
Male Wistar rats weighing 80–100 g were unilaterally injected with AAV vectors carrying DNcdk5-Flag, Wtcdk5-GFP-NLS(NES), DNcdk5-GFP-NLS(NES), WtPrx2-Flag, Prx2T89A-Flag, Prx2T89E-Flag, or GFP as control 2 weeks before induction of ischemia. AAV was diluted by mixing 2 μl of virus stock (1010 genomes/μl) with 1 μl of 20% mannitol and administered into the hippocampus (from bregma: −3.6 mm anteroposterior, +2.1 mm lateral, −2.75 mm deep; in 4VO model) or striatum (from bregma: +0.9 mm anteroposterior, +2.8 mm lateral, −5.8 mm deep; in endothelin model) using an infusion pump (Harvard Infusion Pump) over a 30 min period (0.1 μl per minute) as described previously (Rashidian et al., 2005). AV carrying MEF2D-S444, a cdk5 phosphorylation mutant of MEF2D or GFP was delivered into striatum, using above coordinates. Unilateral injection of virus was given to each animal (3 μl; 3 × 107 particles/μl) one week before injection of endothelin-1 as described previously (Smith et al., 2003).
Global ischemia induced by 4-vessel occlusion.
Five minute 4-vessel occlusion (4VO) was induced in male Wistar rats weighing 180–220 g. One day before global ischemia insult, the common carotid arteries were exposed by a ventral midline neck incision and looped with suture. Both vertebral arteries were permanently occluded by electrocauterization. The animals were allowed to recover for 24 h. The next day, the carotid arteries were exposed and the rats were allowed to recover from anesthesia till they reacted to a tail-pinching stimulus. At this time, both common carotid arteries were quickly occluded for 5 min using clips. During the occlusion, body temperature was monitored with a rectal probe and maintained at 37°C. After removing clips, rats were kept in a 37°C incubator for 1 h and then moved to individual cages. Only rats that were unresponsive during the procedure and following reperfusion were included in the study. Brains were collected and stained for hematoxylin and eosin (H&E) 4 d after surgery. Cell counts from the left and right hippocampi were averaged and expressed as counts/mm for CA1 region.
Focal ischemia induced by injection of endothelin-1.
Endothelin-1 was injected into the striatum of the male Wistar rats (from bregma: +0.9 mm anteroposterior, +2.9 mm lateral, −6 mm deep) weighting 180–220 g or 8–12 weeks old male mice (from bregma: +0.5 mm anteroposterior, +2.1 mm lateral, −2.9 mm deep). Endothelin-1 was dissolved in sterile distilled water to a final concentration of 400 pm/μl. One microliter (in rats) or 2 × 1 μl (in mice; double injection, 0.4 mm apart) of the endothelin-1 solution was injected over a period of 3 min into the striatum using an infusion pump. Brains were collected 4 d after injection and 14 μm-coronal sections of striatum were stained with cresyl violet (CV). The infarct volume was measured using the method described previously (Swanson et al., 1990). Briefly, for every fifth section, the area of damage was marginated and quantified using image analyses (Northern Eclipse). Infarct volume was calculated by multiplying the area of damage by number of sections and thickness of sections (14 μm).
Immunohistochemistry.
Fourteen micrometer coronal sections at the level of striatum were collected as freefloating in 0.01 m PBS + 0.02% sodium azide. Then sections were permeabilized and blocked in buffer A (50 mm Tris, pH 7.5 + 100 mm NaCl + 0.3% Triton X-100) plus 3% BSA overnight. Next, sections were washed in buffer A and incubated in anti-pT89Prdx2 antibody (Qu et al., 2007) coincubated with a blocking peptide for specificity at 4°C overnight. Then slides were visualized using Alexa Fluor 488 anti-rabbit IgG (1:2000; Invitrogen).
Western blot analyses.
To detect expression of DNcdk5, Wtcdk5-GFP-NLS(NES) and DNcdk5-GFP-NLS(NES), Prx2 and its variants, MEF2D and pS444MEF2D in brain, a 2 mm punch was obtained and analyzed by Western blot using anti-Flag (1:4000; Sigma-Aldrich) or anti-cdk5 (1:1000; Santa Cruz Biotechnology), anti-GFP (1:5000; Abcam), anti-Prdx2 (1:500; Abcam), anti-pT89Prx2 (1:500; Qu et al., 2007), anti-MEF2D (1:2000; BD Transduction Laboratories), and anti-pS444MEF2D (1:500) (Gong et al., 2003) antibodies, respectively. Histone H1 (1:1000; Santa Cruz Biotechnology) and Raf-1 (1:1000; Santa Cruz Biotechnology) were used as nuclear and cytoplasmic markers, respectively. To detect viral expressed Prx2 variants in CGNs, 2-d-infected cultures were collected and subjected to Western blot using anti-Flag antibody. β-Actin (1:3000; Sigma-Aldrich) was used as loading control for all Western blots.
Cdk5 kinase assay.
Cdk5 kinase assay was performed on hippocampal CA1 or striatal tissue samples. One-hundred micrograms of protein was extracted from samples and incubated overnight with anti-cdk5 antibody (2 μg/sample; Santa Cruz Biotechnology). Thiry microliters of 50% slurry of Protein A-Sepharose (Sigma-Aldrich) was then added to immunoprecipitates, and samples were incubated for 2 h at 4°C. The immune complexes were next pelleted and washed in PBS and incubated with 2 μg of histone H1 (Boehringer) and 1 μCi [32P] ATP at 30°C for 30 min. Kinase activity was determined by SDS-PAGE and autoradiography.
Extraction of nuclear and cytoplasmic proteins.
Hippocampal CA1 or striatal proteins were obtained using a 2 mm punch and nuclear and cytoplamic proteins were extracted as described before (Wang et al., 2002). Western blot analyses for histone H1, as a nuclear marker, and Raf-1, as a cytoplasmic marker, were performed to evaluate purity of fractions.
Results
Differential roles of cytoplasmic and nuclear cdk5 in multiple models of stroke
We have previously provided evidence that cdk5 plays an essential role in a focal model of stroke induced by exposure to the vasoconstrictor endothelin-1. It is also functionally relevant in a 5 min global model of ischemia (Rashidian et al., 2005) (see also supplemental Material 1, available at www.jneurosci.org), but not following longer duration (10 min) of 4VO. In the latter case, cdk4 appears to play a more central role (Rashidian et al., 2005). Importantly, the mechanism(s) by which cdk5 might promote ischemic death is unknown. As the first step in deciphering the cdk5 localization promoting death in vivo, we examined whether nuclear or cytoplasmic cdk5 was essential for death in two models of global and focal ischemia. To determine this, we constructed and used recombinant AAV expressing a wild-type (Wt) or kinase-dead dominant-negative (DN) mutant of cdk5. DNcdk5 carries a point mutation within its catalytic site and is inactive. Presumably, it can competitively inhibit endogenous cdk5 activity by binding to its activators or its substrates. Wtcdk5 and DNcdk5 were fused to GFP (for visualization) as well as either a nuclear exclusion signal (NES) for expression exclusively in the cytoplasm [implicated as Wt(DN)cdk5-NES] or a nuclear localization signal (NLS) for targeting to the nucleus [implicated as Wt(DN)cdk5-NLS]. We have shown previously that these fusion constructs target to either cytoplasm or nucleus (O'Hare et al., 2005). We also presently show by GFP fluorescence imaging that this pattern of exclusive expression in the nucleus or cytoplasm also occurs in the brain in vivo (Fig. 1a). For the global model of ischemia, recombinant AAV vectors carrying these constructs or GFP, as control, were injected unilaterally into hippocampus 2 weeks before induction of 5 min 4VO. Expression of the constructs was confirmed by Western blot analyses (Fig. 1b). Survival of CA1 neurons was assessed 4 d after 4VO by staining of coronal sections of the brain with H&E. Live CA1 neurons are characterized by round soma and intact nuclei, while dead neurons appear shrunken with pyknotic nuclei (supplemental Material 2, available at www.jneurosci.org). Neuronal counts for the CA1 region showed significant increase in survival in the DNcdk5-NES-injected side compared with the noninjected side (44% survival vs 16%) (Fig. 1b). In comparison, overexpression of all other constructs, including DNcdk5-NLS, did not result in any significant survival (Fig. 1b). These results suggest that activation of cytoplasmic, but not nuclear, cdk5 leads to neuronal death following 4VO. To confirm that cytoplasmic cdk5 activation may be more critical in transmitting the death signal, we assessed cdk5 kinase activity in both the cytoplasm and nucleus after 4VO. Cdk5 was immunoprecipitated from nuclear and cytoplasmic extracts of CA1 at different time points following 4VO. Fractionation was confirmed using histone H1 as nuclear marker and Raf-1 as cytoplasmic marker (Fig. 1c). The samples were then subjected to in vitro kinase assay using histone H1 as substrate. As Figure 1c indicates, cytoplasmic cdk5 activity dramatically increased 3 h following reperfusion, with a maximum level at 12 h (2.5 folds). In contrast, cdk5 activity did not increase in the nucleus. This suggests that abnormal activation of cytoplasmic cdk5 has an essential role in neuronal death following 4VO.
Figure 1.
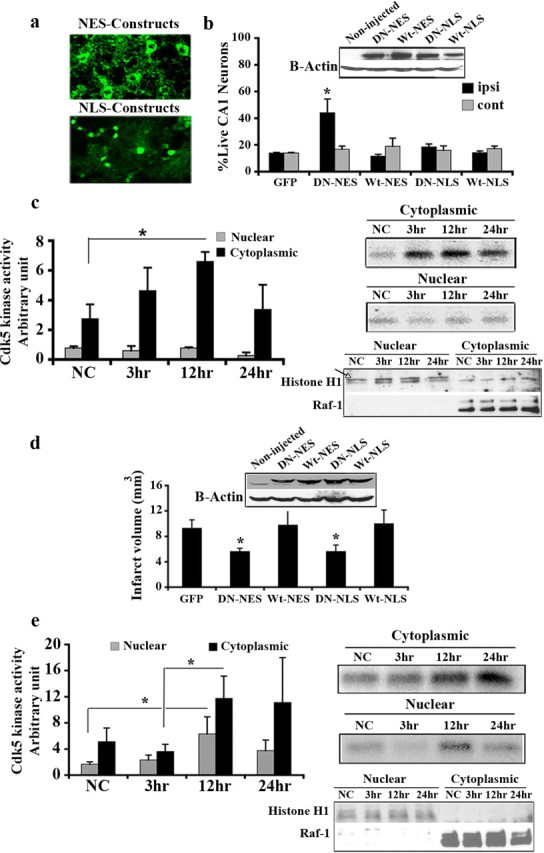
Cytoplasmic cdk5 is mediator of neuronal death following global ischemia, while in focal ischemia both nuclear and cytoplasmic cdk5 signal for death. a, AAV-mediated expression of Wt(DN)cdk5-NLS in the nuclei and Wt(DN)cdk5-NES in the cytoplasm was detected using GFP fluorescence in the striatum. b, Quantification of surviving CA1 neurons expressing GFP (n = 5), DNcdk5-NES (n = 5), Wtcdk5-NES (n = 5), DNcdk5-NLS (n = 5), and Wtcdk5-NLS (n = 5) 4 d after 4VO. Expression of constructs in CA1 was shown by Western blot analysis. c, Cdk5 kinase assay on nuclear and cytoplasmic proteins from hippocampus using histone H1 as substrate at the different time points after 4VO. The graph presents densitometry values from n = 4 experiments. d, Infarct volume of focal ischemic brains expressing GFP (n = 7), DNcdk5-NES (n = 7), Wtcdk5-NES (n = 8), DNcdk5-NLS (n = 7), and Wtcdk5-NLS (n = 7) 4 d after injection of endothelin-1. Expression of constructs in striatum was shown by Western blot analysis. e, Cdk5 kinase assay on nuclear and cytoplasmic proteins from striatum using histone H1 as substrate at the different time points after injection of endothelin-1. The graph presents densitometry values from n = 5 experiments. Fractionation was assessed using histone H1 as nuclear marker and Raf-1 as cytoplasmic marker in both c and e. “NC” represents nonstroked control animals; ipsi is injected and cont is noninjected side. Data are presented as mean ± SEM (*Student's t-test, p < 0.05).
The above results were quite surprising to us given previous results from our laboratory (O'Hare et al., 2005) as well as others (Gong et al., 2003), that, at least in in vitro cultured neurons, nuclear cdk5 is critical for excitotoxic death. This suggests that the role of cdk5 in vivo in the adult animals might differ from that of immature neurons grown in the dish. To examine this possibility, we also performed similar protection experiments as described above in another adult model, a focal ischemia model induced by endothelin-1. Recombinant AAV vectors carrying the DNcdk5 or Wtcdk5 constructs described above or GFP, as control, were injected into the striatum 2 weeks before injection of endothelin-1. Expression of constructs was confirmed by Western blot analyses (Fig. 1d). Regions of striatum with infarct were distinguished by CV staining 4 d after injection, as described previously (Rashidian et al., 2005). In contrast to the 4VO model, there was a very significant decrease (40%) in damage in both DNcdk5-NLS and DNcdk5-NES expressing brains, compared with GFP-expressing brains (Fig. 1d). Overexpression of Wtcdk5 in cytoplasm or nucleus, however, was not protective (Fig. 1d). This indicates that the participation of nuclear and cytoplasmic cdk5 differs in various models of stroke. In a focal model, both nuclear and cytoplasmic cdk5 signal for death in excitotoxic type of death. We also assessed cdk5 kinase activity in the cytoplasm and nucleus after injection of endothelin-1 similar to that performed after global insult. As Figure 1e indicates, cdk5 activity increased 12 h after injection, compared with negative controls in both cytoplasm and nucleus. This activation is slightly slower than that observed following 4VO. However, this delay can be explained by the fact that in the focal ischemia model induced by endothelin, occlusion and reperfusion are likely much more gradual than with global ischemia.
Together, these results suggest the central nature of cytoplasmic cdk5 as a key mediator of ischemic death in multiple models of ischemia, in vivo. However, the activity of nuclear cdk5 does not always participate in the death process.
Prx2 is a substrate for cytoplasmic cdk5 in ischemic death
We have recently reported that Prx2, a cytoplasmic peroxidase, is a cdk5 target (Qu et al., 2007). This antioxidant enzyme is phosphorylated at Thr89 and inactivated by cdk5. Therefore, we examined whether Prx2 may act as a downstream target for cytoplasmic cdk5 in our models of ischemia.
In vitro model of ischemia
We first examined the importance of Prx2 in in vitro model of death induced by glutamate in CGNs. CGNs provide a relatively homogenous population of neurons and are efficiently killed by stroke-related insults, such as glutamate treatment. We have previously reported that cdk5 is essential for excitotoxic neuronal death induced by glutamate in CGNs (Rashidian et al., 2005). We first evaluated the effect of glutamate on Thr89 phosphorylation using the phospho-specific antibody pT89 in CGN cultures from Wt and p35−/− mice. The increase in pT89Prx2 was observed in cultures by immunofluorescence and was quantified over a number of neurons by image analyses (Fig. 2a). Quantification indicated a maximum of 50% increase in pT89Prx2 signal in glutamate-treated Wt CGNs compared with nontreated controls. In contrast, p35−/− cultures showed only 10% statistically nonsignificant increase upon glutamate treatment, compared with controls (Fig. 2b). These results indicate that Prx2 is phosphorylated by cdk5 upon glutamate exposure. To determine whether this phosphorylation is relevant in this paradigm, we infected CGNs with AV expressing WtPrx2, a mutant of Prx2, which cannot be phosphorylated at the Thr89 (Prx2T89A), or a phospho-mimicking form, which we have shown to be less active (Prx2T89E), before glutamate treatment. Expression of these constructs was confirmed by Western blot analyses (Fig. 2c). The wells treated with MK801/glutamate served as controls. This was to control for nonspecific death caused by washing (which typically accounts for <10% of cells; supplemental Material 3, available at www.jneurosci.org). As shown in Figure 2d, neurons exposed to glutamate expressing WtPrx2 and Prx2T89A showed ∼90% survival versus 40% survival in GFP-expressing controls. Expression of Prx2T89E showed dramatically less protection than the other Prx2 constructs. Interestingly, Western blot analysis revealed that at least some of the virally expressed WtPrx2 is phosphorylated on Thr89 following glutamate treatment, consistent with observations from endogenous Prx2 (supplemental Material 4, available at www.jneurosci.org). The resistant form Prx2T89A, on the other hand, did not show increase in phosphorylation status (data not shown). Together, this data suggests that phosphorylation of Prx2 on Thr89 by cdk5 in the glutamate model triggers death pathways in CGN cultures.
Figure 2.

Prx2 is phosphorylated at Thr89 by cdk5 in glutamate model and causes neuronal death. a, Phosphorylation of Prx2 is attenuated in p35-deficient CGNs compared with cultures from Wt littermates following glutamate (Glu) excitotoxicity. Cultures were fixed with 4% PFA after glutamate exposure and immunostained for pT89Prx2. b, Quantification of surviving CGNs after glutamate exposure. CGNs from each mouse (Wt n = 7; p35−/− n = 4) were plated on 3 wells and intensity of fluorescent signal in soma of neurons was measured by image analyses from 60 neurons in 4 random fields/well. c, AV-mediated expression of WtPrx2, Prx2T89A, Prx2T89E in CGNs was shown by Western blot analysis using anti-Flag antibody. d, Overexpression of Prx2 protects CGNs against glutamate. Five day plated CGNs were infected with AV-expressing Wt, phosphorylation resistant (T89A), and phospho-mimicking (T89E) mutants of Prx2 or only GFP as control and exposed to glutamate 2 d after infection. Fixed cultures were then counted for live GFP expressing neurons. Graph is representative of n = 3 experiments. Data are presented as mean ± SEM (*Student's t-test, p < 0.05).
In vivo models of ischemia
To confirm the above results in vivo, we next examined whether Prx2 may be important in the global model of ischemia. We generated AAV expressing WtPrx2, Prx2T89A, and Prx2T89E, and targeted these constructs to the hippocampus before 4VO. Expression of these constructs was confirmed by Western blot analysis (Fig. 3a). Evaluation of CA1 neurons showed that expression of WtPrx2 and Prx2T89A significantly protected this region from ischemia (38% and 28% live, respectively) compared with expression of Prx2T89E (15% live) or GFP (14% live) (Fig. 3a). The above evidence indicates that Prx2 may be important for ischemic damage. To show a connection between Prx2 and cdk5, we determined whether Prx2 is phosphorylated on Thr89 following ischemia in a cdk5-dependent manner. We first analyzed for total Prx2 and pT89Prx2 by Western blot at different time points following insult. As Figure 3b shows, both total Prx2 and pT89Prx2 levels were increased at 3 h, with maximum levels at 12 h after reperfusion. Moreover, the specific activity of phosphorylation (pT89Prx2 to total Prx2) also showed 2.8-fold increase compared with control animals. To ensure that this phosphorylation is cdk5-dependent, DNcdk5 was expressed unilaterally in hippocampus before 4VO. As demonstrated in Figure 3c, expression of DNcdk5 attenuated Prx2 phosphorylation. Together, the above data suggests that Prx2 is a functionally relevant cytoplasmic cdk5 target following global ischemia.
Figure 3.

Prx2 is phosphorylated by cdk5 in global ischemia and leads to neuronal death. a, Quantification of surviving CA1 neurons expressing GFP (n = 5), Wt (n = 11), as well as T89A (n = 10) or T89E (n = 6) forms of Prx2 after 4VO. AAV-mediated expression of WtPrx2, Prx2T89A, Prx2T89E in hippocampus was detected by Western blot analysis using anti-Flag antibody. b, Analysis of pPrx2 level in hippocampus at the time points after 4VO by Western blot. The membrane is representative of n = 6 experiments and graph presents densitometry values of pPrx2 relative to Prx2. c, Inhibition of cdk5 attenuates pPrx2. Rats were unilaterally injected with AAV expressing DNcdk5 into hippocampus and subjected to 4VO. Total proteins from both sides of the hippocampus were analyzed for pPrx2 by Western blot at 24 h after 4VO. Expression of DNcdk5 has been shown using anti-Flag antibody. The membrane is representative of n = 3 experiments and graph presents densitometry values of pPrx2 relative to Prx2. ipsi, Virus-injected side; cont, noninjected side. “NC” represents nonstroked control animals. Data are presented as mean ± SEM (*Student's t-test, p < 0.05).
Similar results with Prx2 were also observed in a focal model of ischemia. We virally overexpressed WtPrx2, mutants Prx2T89A and Prx2T89E, or GFP in striatum before injection of endothelin-1. Expression of these constructs was confirmed by Western blot analysis (Fig. 4a). As Figure 4a shows, expression of Prx2T89A, the phosphorylation resistant mutant, provided significant protection from ischemia (58% smaller infarct compared with expression of GFP alone). In contrast, expression of Prx2T89E, the phospho-mimicking mutant, did not offer any protection. Interestingly, unlike in the global model, we did not see significant protection with WtPrx2. As it has been shown in supplemental Material 5, available at www.jneurosci.org, viral expressed WtPrx2 is phosphorylated at Thr89 24 h after injection of endothelin-1. The reason why WtPrx2 protects following global but not focal insult is unknown but may have to do with timing/degree of phosphorylation of the exogenously expressed Prx2. Alternatively, other death signals might play a role in the focal, but not global model, including signaling in the nucleus (see below).
Figure 4.
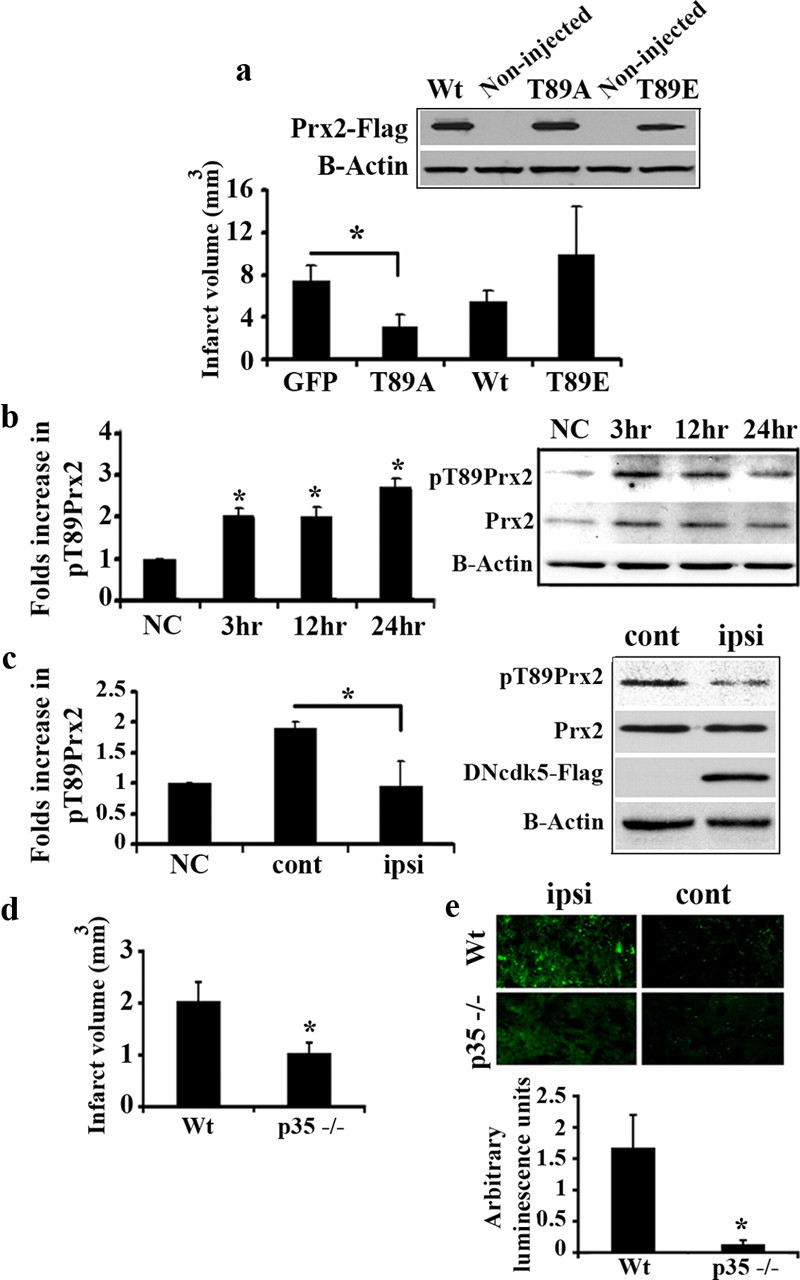
Prx2 is phosphorylated by cdk5 in focal ischemia and causes neuronal death. a, Quantification of infarct volume in striatum overexpressing GFP (n = 5), Wt (n = 7), as well as T89A (n = 5) or T89E (n = 4) forms of Prx2 following injection of endothelin-1. AAV-mediated expression of constructs in striatum was detected by Western blot analysis using anti-Flag antibody. b, Analysis of pPrx2 level in striatum at the time points following endothelin-1 injection by Western blot. The membrane is representative of n = 3 experiments and graph presents densitometry values of pPrx2 relative to Prx2. c, Inhibition of cdk5 attenuates pPrx2. Rats were unilaterally injected with AAV expressing DNcdk5 and then endothelin-1 into both sides of striatum. Total proteins from both sides of the striatum were analyzed for pPrx2 by Western blot at 24 h after ischemia. Expression of DNcdk5 has been shown using anti-Flag antibody. The membrane is representative of n = 3 experiments and graph presents densitometry values of pPrx2 to Prx2. ipsi, Virus injected side; cont, noninjected side. “NC” represents nonstroked control animals. d, p35-deficient mice are resistant to focal ischemia. p35−/− (n = 5) or Wt (n = 7) mice were injected with endothelin-1 in striatum and the infarct volume was measured. e, Increased pPrx2 signal is attenuated in p35 deficient mice in focal ischemia. Sections from mice brains (Wt n = 5; p35−/− n = 3) at striatum level were probed with anti-pT89Prx2 antibody and rabbit Alexa Fluor 488 antibody (green) as secondary antibody. Relative amount of luminescence was assessed by normalizing the average intensity of signal emitted (20 counts/brain) in the endothelin-injected side to that of the noninjected side. Data are presented as mean ± SEM (*Student's t-test, p < 0.05).
To determine whether endogenous Prx2 is phosphorylated in this focal model, striatal protein samples were analyzed for total Prx2 and pT89Prx2 by Western blot. As Figure 4b shows, both total Prx2 and pT89Prx2 levels were increased. Similar to the global model, the levels of pT89Prx2 relative to total Prx2 were elevated 2.7 fold 24 h after stroke. To test whether phosphorylation of Prx2 was cdk5-mediated, we examined the effects of cdk5 inhibition in two ways. First, we expressed DNcdk5 and measured its effect on Prx2 phosphorylation. As shown in Figure 4c, such expression attenuated phosphorylation induced by endothelin-1. Second, we also examined the effects of p35 deficiency. We first measured the extent of damage induced by endothelin-1. As Figure 4d demonstrates, p35−/− mice displayed significantly smaller infarct (50%) compared with wild-type mice. Finally, we performed immunofluorescence analysis for phospho-Prx2 on the coronal sections obtained at the level of striatum. In p35−/− sections, the increase in pT89Prx2 signal observed upon ischemia was significantly attenuated compared with the ones obtained from wild type (Fig. 4e). Thus, p35 deficiency effectively prevents Prx2 phosphorylation and reduces damage following stroke insult. Together, the above data suggests that Prx2 is a critical cytoplasmic cdk5 target, which is phosphorylated and inactivated following focal ischemic injury.
MEF2 is a substrate for nuclear cdk5 in ischemic death
Our above results identify the cytoplasmic cdk5-Prx2 pathway as a critical signaling axis of ischemic neuronal death. In addition, our data demonstrates that under selective conditions, nuclear cdk5 also appears to play a role. To further examine the mechanisms by which this nuclear cdk5 activity promotes neuronal death, we investigated the survival promoting transcription factor MEF2D. Previous evidence indicates that MEF2D is a direct target for cdk5-mediated neurotoxicity, at least in cultured neurons. Cdk5 has been reported to phosphorylate MEF2D on Ser444, which leads to its inactivation (Gong et al., 2003). Accordingly, we next determined whether this nuclear target plays an important role in excitotoxicity induced by endothelin-1. We first performed Western blot analysis on protein samples extracted from endothelin-1-injected striatum using a specific anti-pS444MEF2D antibody. As shown in Figure 5a, MEF2D is phosphorylated at Ser444 following ischemia with the maximum levels at 24 h. In addition, the level of total MEF2D is decreased in a time-dependent manner (Fig. 5a). This is in agreement with previous studies, which showed that phosphorylation of MEF2D by cdk5 promotes its degradation (Tang et al., 2005). In contrast to focal ischemia, neither the levels of pS444MEF2D, nor total MEF2D change following global ischemia (Fig. 5a). This is consistent with the notion that nuclear cdk5 activity is not induced with the latter insult. To confirm that the phosphorylation observed following focal insult depends on cdk5 activity, we examined the effect of cdk5 inhibition by expression of DNcdk5. As Figure 5b shows, expression of DNcdk5 attenuated pS444MEF2D. To further decipher the role of MEF2D phosphorylation in our model, we virally expressed a mutant form of MEF2D, which cannot be phosphorylated at Ser444 (MEF2D-444), or GFP into the striatum of rats, which were subsequently subjected to focal ischemia. As shown in Figure 5d, the phosphorylation-resistant MEF2D significantly protected against damage and resulted in a smaller infarct (66% smaller) compared with GFP-injected control brains.
Figure 5.

MEF2D is phosphorylated at Ser444 by cdk5 in focal ischemia. a, Analysis of pMEF2D level at the time points following endothelin-1 injection or 4VO. Total proteins extracted from striatum (focal ischemia) or hippocampus (4VO) were subjected to Western blot analysis using anti-pS444MEF2D and then MEF2D antibodies. The pMEF2D band has been marked in blots. The membrane is representative of n = 3 experiments and graph presents densitometry values of pMEF2D relative to MEF2D. b, Inhibition of cdk5 attenuates pMEF2D in focal ischemia. Rats were unilaterally injected with AAV expressing DNcdk5 into striatum and 14 d later endothelin-1 was injected into both sides. Proteins from both sides of the striatum were analyzed for pMEF2D by Western blot at 24 h after ischemia. Expression of DNcdk5 has been shown using anti-Flag antibody. The membrane is representative of n = 3 experiments and graph presents densitometry values of pMEF2D relative to MEF2D. c, Expression of AV-mediated MEF2D-444 in striatum was detected by Western blot analysis. d, Quantification of infarct volume in brains overexpressing GFP (n = 4) and MEF2D-444 (n = 5) following focal ischemia. Data are presented as mean ± SEM (*Student's t-test, p < 0.05).
Overall, these results suggest that in an in vivo model of excitotoxic type of neuronal death cytoplasmic Prx2 and nuclear MEF2D are targeted and modulated by cdk5.
Discussion
Although cdk5 has received considerable attention as a potential mediator of neuronal damage, evidence for its functional importance in adult models of injury, including stroke, has only recently begun to emerge (Smith et al., 2003; Wang et al., 2003; Rashidian et al., 2005). However, the essential role of cdk5, with respect to its localization and targets, in regulating ischemic damage is not completely clear. Our present work is significant because we have shown the following: (1) we have defined the cytoplasmic and nuclear compartments in which cdk5 acts to promote ischemic death; (2) we show that the involvement of these cellular compartments is different depending upon the particular model. This highlights the important notion that one mechanistic paradigm does not describe the mode of action of cdk5; (3) we define Prx2 and MEF2D as critical substrates for cdk5.
Differential roles of cdk5: importance of cytoplasmic cdk5 in ischemic damage
Cdk5 displays two opposing and seemingly contradictory functions. It is a key regulator of neuronal morphology and its activity is essential during development (Dhavan and Tsai, 2001). In contrast, several lines of evidence have suggested that it could also be essential in neuronal death (Bu et al., 2002; Nguyen and Julien, 2003; Smith et al., 2003; Cruz and Tsai, 2004; Rashidian et al., 2005). The manner by which a key regulator of proper neuronal function can also possess a pathogenic role has been of considerable interest. While several possibilities might account for this contradiction, cell localization may be critical. In this regard, cdk5 activity was initially thought to be predominately localized membranous compartments. Presumably, these membrane-bound cdk5 complexes fulfilled the “normal” function of cdk5. In support of this, we have shown that inhibition of cytoplasmic cdk5 actually promotes death following DNA damage in vitro (O'Hare et al., 2005). In contrast, a pathogenic form of cdk5 was proposed to occur through calpain-mediated cleavage of the activator to a shorter more stable form. The hyperactivity of this complex was presumed to be deleterious. This cleavage also deletes the myristoylation sequence allowing cdk5 to traffic abnormally to the nucleus (Patrick et al., 1999; Kusakawa et al., 2000). In support of this, a recent report indicated that nuclear cdk5 activity increases in response to glutamate and that a nuclear target of cdk5, MEF2D, is functionally relevant for excitotoxic death, at least in vitro (Tang et al., 2005). These observations have led to a simplistic model by which normal cdk5 activity is membrane bound while pathogenic cdk5 is nuclear. However, the situation is likely much more complex. For example, recent reports have suggested that cdk5/p35 complex can normally be soluble and displays higher activity than membranous complex (Sato et al., 2007). Moreover, p35 can be specifically imported into nucleus (Fu et al., 2006). In addition, whether the model described above holds true in all cases, particularly following adult injury, is unclear. Indeed, one important possibility is that cdk5 complexes may also play a role in death/survival in the cytoplasm. Surprisingly, our results indicate that cytoplasmic cdk5 appears to play a crucial role in promoting death following ischemia. This contrasts with our previous findings that cytoplasmic cdk5 functions to promote survival following DNA damage (O'Hare et al., 2005) or that nuclear cdk5 activity is critical in promoting excitotoxic death (O'Hare et al., 2005; Tang et al., 2005). These latter studies were performed in vitro and suggest that the function of cdk5 is critically context-dependent and may differ in the adult. These data are also critical in that they show that cdk5 in the nucleus does not necessarily promote death. Indeed, under select conditions (i.e., global ischemia) we could not detect robust nuclear cdk5 activation. In models of focal ischemia, in contrast, activation and requirement of cdk5 activity is found in both nuclear and cytoplasmic fractions. The reasons for these differences are unclear, but likely are mediated through the specifics of the signaling environment for each ischemic model. It is clear that the activation of cdk5 is not solely mediated through formation of p25 in either global or focal model (data not shown). Indeed, we have noticed that p25 is formed in both fractions, but that there is no correlation between the timing of the formation of p25 and the induction of cdk5 activity (data not shown). This indicates that other signals (type of cells, other activators) are involved to determine the extent of cdk5 activation.
Cytoplasmic target for cdk5 in stroke
How does cdk5 induce its death signal within cytoplasm? Presently, we describe a new cytoplasmic target for cdk5 in stroke, the antioxidant enzyme Prx2. Oxidative stress is a critical mediator of death in stroke, and management of ROS has been shown to improve stroke in a number of stroke contexts (Margaill et al., 2005). Prx2, a cytoplasmic antioxidant enzyme with peroxidase activity, is abundantly expressed in neurons (Jin et al., 2005) and likely a major candidate in the defense of the neurons against oxidative stress. We have recently provided evidence that Prx2 is a substrate for cdk5 (Qu et al., 2007). We showed that Prx2 can be phosphorylated by cdk5 on Thr89 and that this modification leads to its inactivation (Qu et al., 2007). Presently, we demonstrate upregulation of Prx2 phosphorylation on Thr89 following focal and global ischemia in a manner dependent upon cdk5. We also show that modulation of Prx2 levels regulates death providing evidence for a functional importance of this activity. Interestingly, Prx2 has been implicated in other neurodegenerative situations. For example, Prx2 was found coaggregated with GPx1 and SOD-1 in neuronal Lewy body-like hyaline inclusions of spinal cord of familial ALS (Kato et al., 2004). We also demonstrated that Prx2 is phosphorylated in PD (Qu et al., 2007). Therefore, modulation of Prx2 levels may be a common and critical factor in the management of free radical stress that is critical for death regulation.
In addition to Prx2, there are likely other cytoplasmic targets of cdk5 in stroke. Potential targets include tau (Patrick et al., 1999; Cruz and Tsai, 2004). For example, tau has been proposed to be phosphorylated at Ser199/202 by cdk5 following transient forebrain ischemia (Morioka et al., 2006). However, this observation was supported by using a relatively nonspecific CDK inhibitor, olomoucine. Therefore, it is unclear whether other CDKs may account for this phosphorylation activity. In addition, the actually role of tau phosphorylation in this case, is unclear. A second target also includes the NMDA receptor NR2A subunit. Cdk5 is reported to phosphorylate this subunit leading to increased calcium influx and neuronal death molecular cascades (Li et al., 2001). Understanding of whether/how these multiple targets interact will be of interest in future studies.
Nuclear target for cdk5 in stroke
In addition to its role in the cytoplasm, cdk5 may also have additional roles in the nucleus. In fact, several targets, which are predominately nuclear, have been described. These include the tumor suppressor p53 and the transcription factor MEF2D. For example, under select apoptotic conditions, cdk5 has been shown to increase p53 levels (Lee et al., 2007). Cdk5 has also been shown in inactivate survival factors such as MEF2D. In this case, cdk5 phosphorylates MEF2D on Ser444 suppressing its transcriptional activity. This Ser444 phosphorylation has been shown to occur in in vitro models of death following peroxide and excitotoxicity exposure in a manner dependent on cdk5 (Gong et al., 2003; Tang et al., 2005). However, its role in adult models of stroke in vivo was unknown. Presently, we show that MEF2D is also phosphorylated on Ser444 in a focal ischemic model where nuclear cdk5 activity is observed, but not following global insult where nuclear cdk5 activity is not detected. Consistent with this observation, we also show that restoration of active form of MEF2D is protective following focal ischemia. While our results suggest the possibility that modulation of MEF2D by itself is sufficient to account for the death-promoting effects of MEF2D in the nucleus, there may be additional nuclear targets of cdk5 in stroke. In this regard, it will be interesting to examine other potential targets of cdk5, such as p53, which has also been implicated in stroke (Crumrine et al., 1994).
In summary, we have described the critical nature of cytoplasmic cdk5-Prx2 activity in modulating ischemic neuronal death and context-dependent involvement of a nuclear cdk5-MEF2D pathway. These results also indicate the complex nature of cdk5 signaling in neuronal injury, suggesting that the nuclear cdk5 activity is not necessarily required for death.
Footnotes
This work was supported by grants from Canadian Institutes of Health Research, Heart and Stroke Foundation of Ontario, the Canadian Stroke Network, The Centre for Stroke Recovery (D.S.P.), and National Institutes of Health (Grants AG023695, NS048254, and ES015317; to Z.M.). We thank Dr. Inez Vincent for providing p35-deficient mice.
References
- Bu B, Li J, Davies P, Vincent I. Deregulation of cdk5, hyperphosphorylation, and cytoskeletal pathology in the Niemann-Pick type C murine model. J Neurosci. 2002;22:6515–6525. doi: 10.1523/JNEUROSCI.22-15-06515.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crumrine RC, Thomas AL, Morgan PF. Attenuation of p53 expression protects against focal ischemic damage in transgenic mice. J Cereb Blood Flow Metab. 1994;14:887–891. doi: 10.1038/jcbfm.1994.119. [DOI] [PubMed] [Google Scholar]
- Cruz JC, Tsai LH. Cdk5 deregulation in the pathogenesis of Alzheimer's disease. Trends Mol Med. 2004;10:452–458. doi: 10.1016/j.molmed.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Dhavan R, Tsai LH. A decade of CDK5. Nat Rev Mol Cell Biol. 2001;2:749–759. doi: 10.1038/35096019. [DOI] [PubMed] [Google Scholar]
- Fu X, Choi YK, Qu D, Yu Y, Cheung NS, Qi RZ. Identification of nuclear import mechanisms for the neuronal Cdk5 activator. J Biol Chem. 2006;281:39014–39021. doi: 10.1074/jbc.M512663200. [DOI] [PubMed] [Google Scholar]
- Gold MO, Rice AP. Targeting of CDK8 to a promoter-proximal RNA element demonstrates catalysis-dependent activation of gene expression. Nucleic Acids Res. 1998;26:3784–3788. doi: 10.1093/nar/26.16.3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong X, Tang X, Wiedmann M, Wang X, Peng J, Zheng D, Blair LA, Marshall J, Mao Z. Cdk5-mediated inhibition of the protective effects of transcription factor MEF2 in neurotoxicity-induced apoptosis. Neuron. 2003;38:33–46. doi: 10.1016/s0896-6273(03)00191-0. [DOI] [PubMed] [Google Scholar]
- Hallows JL, Chen K, DePinho RA, Vincent I. Decreased cyclin-dependent kinase 5 (cdk5) activity is accompanied by redistribution of cdk5 and cytoskeletal proteins and increased cytoskeletal protein phosphorylation in p35 null mice. J Neurosci. 2003;23:10633–10644. doi: 10.1523/JNEUROSCI.23-33-10633.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ino H, Chiba T. Intracellular localization of cyclin-dependent kinase 5 (CDK5) in mouse neuron: CDK5 is located in both nucleus and cytoplasm. Brain Res. 1996;732:179–185. doi: 10.1016/0006-8993(96)00523-9. [DOI] [PubMed] [Google Scholar]
- Jin MH, Lee YH, Kim JM, Sun HN, Moon EY, Shong MH, Kim SU, Lee SH, Lee TH, Yu DY, Lee DS. Characterization of neural cell types expressing peroxiredoxins in mouse brain. Neurosci Lett. 2005;381:252–257. doi: 10.1016/j.neulet.2005.02.048. [DOI] [PubMed] [Google Scholar]
- Johansson JU, Lilja L, Chen XL, Higashida H, Meister B, Noda M, Zhong ZG, Yokoyama S, Berggren PO, Bark C. Cyclin-dependent kinase 5 activators p35 and p39 facilitate formation of functional synapses. Brain Res Mol Brain Res. 2005;138:215–227. doi: 10.1016/j.molbrainres.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Kato S, Saeki Y, Aoki M, Nagai M, Ishigaki A, Itoyama Y, Kato M, Asayama K, Awaya A, Hirano A, Ohama E. Histological evidence of redox system breakdown caused by superoxide dismutase 1 (SOD1) aggregation is common to SOD1-mutated motor neurons in humans and animal models. Acta Neuropathol. 2004;107:149–158. doi: 10.1007/s00401-003-0791-1. [DOI] [PubMed] [Google Scholar]
- Kusakawa G, Saito T, Onuki R, Ishiguro K, Kishimoto T, Hisanaga S. Calpain-dependent proteolytic cleavage of the p35 cyclin-dependent kinase 5 activator to p25. J Biol Chem. 2000;275:17166–17172. doi: 10.1074/jbc.M907757199. [DOI] [PubMed] [Google Scholar]
- Lee JH, Kim HS, Lee SJ, Kim KT. Stabilization and activation of p53 induced by Cdk5 contributes to neuronal cell death. J Cell Sci. 2007;120:2259–2271. doi: 10.1242/jcs.03468. [DOI] [PubMed] [Google Scholar]
- Li BS, Sun MK, Zhang L, Takahashi S, Ma W, Vinade L, Kulkarni AB, Brady RO, Pant HC. Regulation of NMDA receptors by cyclin-dependent kinase-5. Proc Natl Acad Sci U S A. 2001;98:12742–12747. doi: 10.1073/pnas.211428098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li BS, Zhang L, Takahashi S, Ma W, Jaffe H, Kulkarni AB, Pant HC. Cyclin-dependent kinase 5 prevents neuronal apoptosis by negative regulation of c-Jun N-terminal kinase 3. EMBO J. 2002;21:324–333. doi: 10.1093/emboj/21.3.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li BS, Ma W, Jaffe H, Zheng Y, Takahashi S, Zhang L, Kulkarni AB, Pant HC. Cyclin-dependent kinase-5 is involved in neuregulin-dependent activation of phosphatidylinositol 3-kinase and Akt activity mediating neuronal survival. J Biol Chem. 2003;278:35702–35709. doi: 10.1074/jbc.M302004200. [DOI] [PubMed] [Google Scholar]
- Margaill I, Plotkine M, Lerouet D. Antioxidant strategies in the treatment of stroke. Free Radic Biol Med. 2005;39:429–443. doi: 10.1016/j.freeradbiomed.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Morioka M, Kawano T, Yano S, Kai Y, Tsuiki H, Yoshinaga Y, Matsumoto J, Maeda T, Hamada J, Yamamoto H, Fukunaga K, Kuratsu J. Hyperphosphorylation at serine 199/202 of tau factor in the gerbil hippocampus after transient forebrain ischemia. Biochem Biophys Res Commun. 2006;347:273–278. doi: 10.1016/j.bbrc.2006.06.096. [DOI] [PubMed] [Google Scholar]
- Nguyen MD, Julien JP. Cyclin-dependent kinase 5 in amyotrophic lateral sclerosis. Neurosignals. 2003;12:215–220. doi: 10.1159/000074623. [DOI] [PubMed] [Google Scholar]
- O'Hare MJ, Kushwaha N, Zhang Y, Aleyasin H, Callaghan SM, Slack RS, Albert PR, Vincent I, Park DS. Differential roles of nuclear and cytoplasmic cyclin-dependent kinase 5 in apoptotic and excitotoxic neuronal death. J Neurosci. 2005;25:8954–8966. doi: 10.1523/JNEUROSCI.2899-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402:615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- Pines J. Cyclins and cyclin-dependent kinases: take your partners. Trends Biochem Sci. 1993;18:195–197. doi: 10.1016/0968-0004(93)90185-p. [DOI] [PubMed] [Google Scholar]
- Qu D, Rashidian J, Mount MP, Aleyasin H, Parsanejad M, Lira A, Haque E, Zhang Y, Callaghan S, Daigle M, Rousseaux MW, Slack RS, Albert PR, Vincent I, Woulfe JM, Park DS. Role of Cdk5-mediated phosphorylation of Prx2 in MPTP toxicity and Parkinson's disease. Neuron. 2007;55:37–52. doi: 10.1016/j.neuron.2007.05.033. [DOI] [PubMed] [Google Scholar]
- Rashidian J, Iyirhiaro G, Aleyasin H, Rios M, Vincent I, Callaghan S, Bland RJ, Slack RS, During MJ, Park DS. Multiple cyclin-dependent kinases signals are critical mediators of ischemia/hypoxic neuronal death in vitro and in vivo. Proc Natl Acad Sci U S A. 2005;102:14080–14085. doi: 10.1073/pnas.0500099102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashidian J, Iyirhiaro GO, Park DS. Cell cycle machinery and stroke. Biochim Biophys Acta. 2007;1772:484–493. doi: 10.1016/j.bbadis.2006.11.009. [DOI] [PubMed] [Google Scholar]
- Sato K, Zhu YS, Saito T, Yotsumoto K, Asada A, Hasegawa M, Hisanaga S. Regulation of membrane association and kinase activity of Cdk5–p35 by phosphorylation of p35. J Neurosci Res. 2007;85:3071–3078. doi: 10.1002/jnr.21438. [DOI] [PubMed] [Google Scholar]
- Smith PD, Crocker SJ, Jackson-Lewis V, Jordan-Sciutto KL, Hayley S, Mount MP, O'Hare MJ, Callaghan S, Slack RS, Przedborski S, Anisman H, Park DS. Cyclin-dependent kinase 5 is a mediator of dopaminergic neuron loss in a mouse model of Parkinson's disease. Proc Natl Acad Sci U S A. 2003;100:13650–13655. doi: 10.1073/pnas.2232515100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10:290–293. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- Tang X, Wang X, Gong X, Tong M, Park D, Xia Z, Mao Z. Cyclin-dependent kinase 5 mediates neurotoxin-induced degradation of the transcription factor myocyte enhancer factor 2. J Neurosci. 2005;25:4823–4834. doi: 10.1523/JNEUROSCI.1331-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CX, Song JH, Song DK, Yong VW, Shuaib A, Hao C. Cyclin-dependent kinase-5 prevents neuronal apoptosis through ERK-mediated upregulation of Bcl-2. Cell Death Differ. 2006;13:1203–1212. doi: 10.1038/sj.cdd.4401804. [DOI] [PubMed] [Google Scholar]
- Wang F, Corbett D, Osuga H, Osuga S, Ikeda JE, Slack RS, Hogan MJ, Hakim AM, Park DS. Inhibition of cyclin-dependent kinases improves CA1 neuronal survival and behavioral performance after global ischemia in the rat. J Cereb Blood Flow Metab. 2002;22:171–182. doi: 10.1097/00004647-200202000-00005. [DOI] [PubMed] [Google Scholar]
- Wang J, Liu S, Fu Y, Wang JH, Lu Y. Cdk5 activation induces hippocampal CA1 cell death by directly phosphorylating NMDA receptors. Nat Neurosci. 2003;6:1039–1047. doi: 10.1038/nn1119. [DOI] [PubMed] [Google Scholar]
- Zolotukhin S, Potter M, Zolotukhin I, Sakai Y, Loiler S, Fraites TJ, Jr, Chiodo VA, Phillipsberg T, Muzyczka N, Hauswirth WW, Flotte TR, Byrne BJ, Snyder RO. Production and purification of serotype 1, 2, and 5 recombinant adeno-associated viral vectors. Methods. 2002;28:158–167. doi: 10.1016/s1046-2023(02)00220-7. [DOI] [PubMed] [Google Scholar]