

Abstract
Numerous studies have demonstrated oxidative damage in the central nervous system in subjects with Alzheimer disease and in animal models of this dementing disorder. In the current study, we show that transgenic mice modeling Alzheimer disease—PDAPP mice with Swedish and Indiana mutations in human amyloid precursor protein (APP)—develop oxidative damage in brain, including elevated levels of protein oxidation (indexed by protein carbonyls and 3-nitrotyrosine) and lipid peroxidation (indexed by protein-bound 4-hydroxy-2-nonenal). This oxidative damage requires the presence of a single methionine residue at position 35 of the amyloid β-peptide (Aβ), since all indices of oxidative damage in brain were completely prevented in genetically and age-matched PDAPP mice with a M631L mutation in APP. No significant differences in levels of APP, Aβ(1-42), Aβ(1-40), or the ratio Aβ(1-42)/Aβ(1-40) were found, suggesting that the loss of oxidative stress in vivo in brain of PDAPP(M631L) mice results solely from the mutation of the Met35 residue to Leu in the Aβ peptide. However, a marked reduction in Aβ-immunoreactive plaques was observed in the M631L mice, which instead displayed small punctate areas of non-plaque immunoreactivity and a microglial response. In contrast to the requirement for Met at residue 35 of the Aβ sequence (M631 of APP) for oxidative damage, indices of spatial learning and memory were not significantly improved by the M631L substitution. Furthermore, a genetically matched line with a different mutation—PDAPP(D664A)—showed the reverse: no reduction in oxidative damage but marked improvement in memory. This is the first in vivo study to demonstrate the requirement for Aβ residue Met35 for oxidative stress in brain of a mammalian model of Alzheimer disease. However, in this specific transgenic mouse model of AD, oxidative stress is neither required nor sufficient for memory abnormalities.
Keywords: In vivo oxidative stress in brain, Alzheimer disease, amyloid β-peptide, methionine residue, transgenic mouse model of Alzheimer disease
Introduction
Oxidative stress is elevated in the brain from subjects with Alzheimer disease (AD) and its arguably earliest form, amnestic mild cognitive impairment (MCI) [1–11]. Proteomic analyses of brain proteins from subjects with AD and MCI have identified oxidatively modified proteins that were generally dysfunctional, and suggested to be consistent with the known biochemical, pathological, and behavioral alterations in both conditions [2, 3, 12–17].
Soluble amyloid β-peptide (1-42) [Aβ(1-42)] oligomers are thought to be the toxic species in brains of subjects with AD and MCI [18–21], leading to loss of learning and memory [19, 22]. Aβ(1-42) added to neurons in vitro results in elevated protein oxidation and lipid peroxidation as indexed by elevated levels of protein carbonyls, 3-nitrotyrosine (3-NT), and protein-bound 4-hydroxy-2-nonenal (HNE), each of which is blocked by antioxidants [23–26]. Likewise, human Aβ(1-42) in vivo, either in C. elegans [21] or in rodent brain [27–31], leads to oxidative stress.
The 42-mer Aβ(1-42), as well as the 40-mer [Aβ(1-40)], contains a single methionine residue at position 35 (corresponding to residue 631 of human APP). Our laboratory has suggested that oxidative stress associated with Aβ(1-42) is dependent on the formation of a sulfur-centered, transient free radical involving this Met residue that, in turn, leads to lipid peroxidation and protein oxidation in neurons [24, 32–35]. For example, substitution of norleucine (Nle) for Met (replacement of the S atom in Met by CH2) in human Aβ(1-42) [Aβ(1-42)M35Nle] creates a peptide of the same length and same hydrophobicity as native human Aβ(1-42) that abrogates the associated oxidative stress and neurotoxic properties of Aβ(1-42) in cultured neurons, but does not prevent fibril formation [32, 33, 35]. In C. elegans, the expression of the human Aβ(1-42) peptide is associated with oxidative damage [21, 32]; however, substitution of the codon for Met by that for Cys in DNA encoding human Aβ(1-42) prevents in vivo protein oxidation, while not affecting Aβ(1-42) deposition [32]. These results are consistent with the notion that mechanisms of oxidative stress involving Met35 of Aβ(1-42) apply in vitro and in vivo and do not involve deposition of the peptide.
In the current study, we tested the hypothesis that this same Met35 residue is critical for oxidative stress and the Alzheimer-related spatial memory deficits in a well characterized transgenic mouse model of Alzheimer disease. In this model, the PDAPP mouse model, mutations in human APP corresponding to the Swedish and Indiana familial forms of AD are expressed (APPSw,In) behind a platelet-derived growth factor (PDGF) β-chain promoter, resulting in Aβ accumulation, plaque formation, and memory deficits. In order to test this hypothesis, we generated transgenic mice matched for genetic background and APPSw,In expression level, carrying an additional mutation—M631L—that substituted leucine for methionine at residue 35 of the Aβ peptides derived from APP. Leucine was chosen because it has a naturally occurring codon (whereas norleucine does not) and exhibits length and hydrophobicity characteristics similar to those of methionine. We then compared the degree of oxidative damage and memory deficits in these PDAPP Alzheimer model mice with vs. without the M631L mutation. To complement these studies, we then assessed the oxidative damage in another PDAPP mutant mouse—D664A—that had been shown previously to prevent the memory deficits otherwise characteristic of Alzheimer model mice [36].
Materials and Methods
All chemicals and antibodies used in Oxyblot (slot-blot) studies were purchased from Sigma-Aldrich (St. Louis, MO) with exceptions noted. Dinitrophenylhydrazine (DNPH) and the primary antibody for the protein-bound 2,4-dinitrophenylhydrazone were purchased from Chemicon International (Temecula, CA). The primary antibody for protein-bound 4-hydroxy-2-trans-nonenal (HNE) was purchased from Alpha Diagnostics International, Inc. (San Antonio, TX). Nitrocellulose membranes were purchased from Bio-RAD (Hercules, CA).
Animals
Only male animals were used in the studies reported here. All animal studies were approved by the IACUC of the Buck Institute for Age Research, and carried out in the Institute’s AAALAC-accredited vivarium. Derivation of transgenic mice has been described previously [36]. The PDAPP mice, J20 line, were kindly provided by Prof. Lennart Mucke, and kept in their original C57Bl/J6 background. The PDAPP(D664A) mice have been described and characterized previously [36]. The PDAPP(M631L) mice were derived directly in the same genetic background as the other lines, i.e., C57Bl/J6, using the same approach described previously for the PDAPP(D664A) mice [36]. A mutation was introduced in the PDGF β-chain promoter-driven human APP minigene carrying the Swedish and Indiana mutations that mutated Met631 (APP695 numbering) to Leu to generate PDAPP(M631L) transgenic mice by direct injection into C57BL/6J embryos. Mice from the PDAPP(D664A) B21 transgenic line were described previously [36]. PDAPP(M631L) transgenic mice express the PDAPP(Sw, Ind, M631L) transgene to levels 12.5% higher than those of the PDAPP(Sw, Ind) transgene in the PDAPP(J20) line. Groups of male non-Tg littermates from each transgenic line were used as controls. All transgenic lines were maintained by heterozygous crosses with C57BL/6J breeders (Jackson Laboratories, Bar Harbor, ME). All transgenic animals were heterozygous with respect to the transgene. Animals showing no motivation to swim (percent of time floating >60 percent, i.e., “floaters”) were not found in these studies reported here. Thus, no animals were excluded (see Methods, Behavioral Testing, and Supporting Text). Experimental groups of animals were: Tg PDAPP, n=11; Tg PDAPP(M631L), n=10; non-Tg PDAPP, n=4; non-Tg PDAPP(M631L), n=4. All animals used in the behavioral studies were males. Non-transgenic littermates were used as controls in all studies.
OxyBlots
Sample Preparation
One-half of the brain was homogenized using a Wheaton glass homogenizer (~100 passes) in 1 ml of Media I buffer (0.32 M sucrose, 0.10 mM Tris HCl (pH 8.0), 0.10 mM MgCl2, 0.08 mM EDTA, 10 μg/ml leupeptin, 0.5 μg/ml pepstatin, and 11.5 μg/ml aprotinin; pH 8.0). Homogenates were vortexed, aliquotted into Eppendorf tubes, and sonicated with a Fisher 550 Sonic Dismembrator (Pittsburgh, PA) for 10 s at 20 percent power. Protein concentrations were determined according to the Pierce BCA method (Pierce, Rockford, IL).
Protein Carbonyls
Protein carbonyls are a marker of protein oxidation and were determined as previously described [37]. Samples (5 μL) were derivatized at room temperature for 20 min in 10 mM 2,4-dinitrophenylhydrazine (DNPH) and 5 μL of 12 percent sodium dodecyl sulfate (SDS). Samples were neutralized with 7.5 μL of neutralization solution (2 M Tris in 30 percent glycerol). Derivatized samples (250 ng) were then blotted onto a nitrocellulose membrane under vacuum pressure using a slot-blot apparatus (Bio-RAD, Hercules, CA). Membranes were blocked with 3% bovine serum albumin (BSA) in Wash Blot (a PBS solution containing 0.04% (v/v) Tween-20, and 0.10 M NaCl) for 1.5 h and incubated with a 1:100 dilution of rabbit polyclonal anti-DNP primary antibody in Wash Blot for 2 h at room temperature on a rocker. Blots were rinsed three times for 5 min each in Wash Blot, and subsequently incubated with a 1:8000 dilution of anti-rabbit IgG alkaline phosphatase secondary antibody in Wash Blot for 1 h at room temperature on a rocker. The membrane was washed three times for 5 min each in Wash Blot and developed using a solution of nitrotetrazolium blue chloride (NBT; 0.2 mM) and 5-bromo-4-chloro-3-indolyl phosphate dipotassium (BCIP; 0.4 mM) in ALP buffer (0.1 M Tris, 0.1 M NaCl, 5 mM MgCl2; pH 9.5). Dried blots were quantified using Scion Image software (PC version of Macintosh compatible NIH software). Controls, using samples with no primary antibody, or samples pretreated with NaBH4 to reduce protein carbonyls, resulted in no staining (see Results).
4-Hydroxy-2-trans-nonenal (HNE)
Levels of protein-bound HNE are used as a marker of lipid peroxidation and were determined as previously described [38]. Samples (5 μL) were incubated at room temperature for 20 min in 5 μL of 12% SDS and 10 μl of Laemmli buffer (0.125 M Tryzma base, pH 6.8, 4% (v/v) SDS, and 20% (v/v) Glycerol), and diluted with 7.5 μL PBS. Samples (250 ng) were then blotted onto a nitrocellulose membrane under vacuum pressure using a slot-blot apparatus. A 1:200 dilution of rabbit polyclonal anti-HNE primary antibody was used. Blots were developed and quantified as described above for protein carbonyls. Controls in which the primary antibody was preabsorbed with HNE gave no staining (data not shown).
3-Nitrotyrosine (3-NT)
3-NT levels are used as another marker of oxidative damage [5, 37]. Samples (5 μL) were incubated at room temperature for 20 min in 5 μL of 12% SDS and 10 μl of Laemmli buffer, and diluted with 7.5 μL PBS. Samples (250 ng) were blotted onto a nitrocellulose membrane (as described above) and a 1:2000 dilution of rabbit polyclonal anti-3-NT primary antibody was added. Blots were developed and quantified as described above for protein carbonyls. Controls in which samples were treated with dithionite to reduce 3-NT residues to amines resulted in no staining (data not shown).
ELISA Measurements of Aβ(1-40) and Aβ(1-42)
The other halves of the brains of the mice used for markers of oxidative stress were used for measurements of Aβ(1-40) and Aβ(1-42) levels by ELISA. Aβ40 and 42 were measured in 5M guanidine brain homogenates following the manufacturer’s protocol (Invitrogen). Briefly, mouse hemi-brains were weighed frozen and dissolved in 50mM Tris buffer, pH 8 containing 5M guanidine to achieve 100mg brain/ml buffer. Brains were kept on ice and sonicated several times until homogeneous (buffer was added in stages). Homogenates were rotated at room temperature for 3–5 hours and stored at −20°. Aβ(1-40) and Aβ(1-42) levels were then measured by ELISA (Invitrogen). Samples were diluted 1:50 in the provided standard diluent buffer and that same amount of guanidine was also included in the standard curve.
Immunohistochemistry
Amyloid plaques/Aβ and microglia were labeled, and plaques counted, in six PDAPP and six TgPDAPPM631L mice at eight months of age. Mice were anesthetized, perfused with saline, and a sagittal hemi-brain from each mouse submersion fixed in 2% paraformaldehyde. The fixed brains were processed into paraffin and 10μm coronal sections cut from the area where the hippocampus is readily apparent (Bregma -2.18) and mounted on charged glass slides (Platinum line, Mercedes Medical). Two slides were used for each antibody label for each individual mouse, two sections per slide. Both the 6E10 and the anti-Iba 1 require that antigen retrieval be performed in 10mM sodium citrate, pH 6 for 5min. at 98°C. For the mouse monoclonal 6E10, endogenous mouse IgG was blocked with chicken anti-mouse (Vector) at 1:200 for 1 h in TBST (20mM Tris base, 137mM NaCl, pH7.8, 0.05% Tween 20). Blocking of non-specific antibody binding and all antibody incubations were done in 5% normal donkey serum in TBST; and all washes were done in TBST. Both primary antibodies were used at 1:500 overnight at 4°C. The secondary antibodies were anti-mouse and anti-rabbit AlexaFluor 488 raised in donkey (Invitrogen). The labeled sections were viewed and imaged at 20 and 40X on Nikon Eclipse E800 fluorescent microscope using the FITC filter and Nikon Act 1 software. Plaques were counted only from the molecular layer of the hippocampus within the granular cell layer of the CA regions and above the dentate gyrus.
Behavioral Testing
The Morris water maze [39] (MWM) was used to test spatial memory. All animals had normal motor and visual skills as determined by sensorimotor tasks performed prior to testing. All groups were assessed for swimming ability with a straight water alley (15 by 200 cm) containing a submerged (1 cm) 12 × 12 cm platform 2 days before testing. The procedure described by Morris et al. [39] was followed. Details are included in Supporting Text.
Statistical analyses for behavioral testing
Please see Supporting Text.
Results
Many lines of PDAPPM631L were produced (>10) in order to derive two with high-level APP expression, which is needed both for the phenotype and for accurate comparison to the PDAPP (J20) line (which has similar APP expression levels). The two highest-expressing lines, A1058 and A1059, are shown for measures of Aβ(1-40 and Aβ(1-42) below. However, the higher of these two lines, A1058, is quite similar in APP expression to the PDAPP (J20) line, and therefore was used for the studies reported in this manuscript.
Oxidative Stress
Compared to non-transgenic (Non Tg PDAPP) littermate mice, brains isolated from mice with the Swedish and Indiana mutations in human APP [Tg PDAPP(J20)] displayed elevated indices of oxidative stress (Figs. 1a–c). Levels of protein oxidation [indexed by protein carbonyls (p<0.003, Fig. 1a) and 3-NT (p<0.002, Fig. 1b)] and lipid peroxidation [indexed by protein-bound HNE (p<0.02, Fig. 1c)] were elevated in brains from Tg PDAPP(J20) mice. To determine the specificity of the marker for protein carbonyls, J20 samples were pre-treated with NaBH4, which reduces carbonyls to alcohols, thereby preventing covalent attachment of DNPH. Fig. 2 shows that NaBH4 treatment prevents staining by anti-DNP-protein antibody, in marked contrast to that of the J20 samples, demonstrating specificity of the antibody for protein carbonyls. This result confirms our previous reports of this same finding [16,40].
Figure 1.

In vivo oxidative modification of brain in transgenic and non-transgenic mice: role of Met-35 of Aβ. Oxidative stress is indexed by: (a) Protein Carbonylation. Results show a significant increase in the levels of protein carbonyls in Tg PDAPP(J20) mice (n=10) compared to age-matched Non Tg PDAPP controls (n=5; *P<0.003). Tg PDAPP(M631L) mice (n=10) did not show a significant increase in protein carbonyls compared to their respective Non Tg PDAPP(M631L) controls (n=5; P<0.35), but did, however, have significantly reduced protein carbonyl levels compared to age-matched Tg PDAPP(J20) mice (**P<0.0004). Non-transgenic control values were set to 100 percent, to which transgenic values were compared. These data are presented as mean ± S.E.M. Statistical analyses employed Student’s two-tailed t-test. (b) Lipid peroxidation as indexed by protein-bound HNE. A significant increase in the levels of protein-bound HNE in Tg PDAPP(J20) mice (n=10) compared to age-matched Non Tg PDAPP(J20) controls (n=5; *P<0.02) was found; however, Tg PDAPP(M631L) mice (n=10) did not show a significant change in protein-bound HNE levels compared to Non Tg PDAPP(M631L) controls (n=5; P<0.95). Moreover, there were significantly reduced protein-bound HNE levels in Tg PDAPP(M631L) mice compared to age-matched Tg PDAPP(J20) mice (**P<0.04). Non-transgenic control values were set to 100 percent, to which transgenic values were compared. These data are presented as mean ± S.E.M. Statistical analyses employed Student’s two-tailed t-test. (c) Protein-resident 3-NT in brain. In agreement with both protein carbonyl and protein-bound HNE parameters, a significant increase in Tg PDAPP(J20) mouse (n=10) 3-NT levels was found compared to age-matched Non Tg PDAPP(J20) controls (n=5; *P<0.002). In contrast, Tg PDAPP(M631L) mice (n=10) did not show a significant change in 3-NT levels compared to Non Tg PDAPP(M631L) controls (n=5; P<0.76), while a significant reduction in Tg PDAPP(M631L) 3-NT levels compared to age-matched Tg PDAPP(J20) mice was observed (**P<0.0001). Non-transgenic control values were set to 100 percent, to which transgenic values were compared. These data are presented as mean ± S.E.M. Statistical analyses employed Student’s two-tailed t-test.
Figure 2. Specificity of the assay for protein carbonyls.

Duplicate brain samples from PDAPP (J20) mice (n=2) were subjected to analysis of protein carbonyls without (Left) and with (Right) prior treatment with the strong reducing agent, NaBH4, as described previously [16,40], which reduces carbonyls to alcohols. As in our earlier studies, prior treatment of brain samples with NaBH4 abrogates staining with anti-DNP-protein antibodies, demonstrating the specificity of the assay for protein carbonyls.
In contrast to the oxidative stress present in brains of PDAPP (J20) mice, replacement of methionine 35 of Aβ by Leu (corresponding to residue 631 of APP [Tg PDAPP(M631L)]), which does not alter hydrophobicity or side-chain length, completely prevented the elevation in these oxidative stress parameters in brain [protein carbonyls: p<0.35; 3-NT: p<0.76; protein-bound HNE: p<0.95, (Figs. 1a–c)]. To ensure that these results were not due to a difference in expression of APP or accumulation of Aβ(1-42) or Aβ(1-40), levels of APP expression were compared by Western blotting, and levels of both peptides were determined by ELISA assays. The ratio of Aβ(1-42)/Aβ(1-40) was also determined. No significant differences in expression of Aβ(1-40) and Aβ(1-42) were found in brains of Tg PDAPP(M631L) mice compared to those of Tg PDAPP(J20) mice (Fig. 3). If anything, the ratio of Aβ(1-42) to Aβ(1-40) was elevated in brains of the Tg PDAPP(M631L) mice. Plaque load was determined by 6E10 immunohistochemistry. There was a marked reduction in Aβ-immunoreactive plaques in the M631L mice (Fig. 4), and the M631L plaques were smaller than plaques in the J20. Accompanying this plaque reduction was a clear increase in punctate 6E10 labeling, along with a microglial response (Fig. 4), the latter demonstrated by Iba-1 staining and the observation of microglial processes characteristic of activation (Fig. 4).
Figure 3. PDAPP(M631L) mice express APP, and produce Aβ(1-40) and Aβ(1-42), at levels similar to the PDAPP mice.
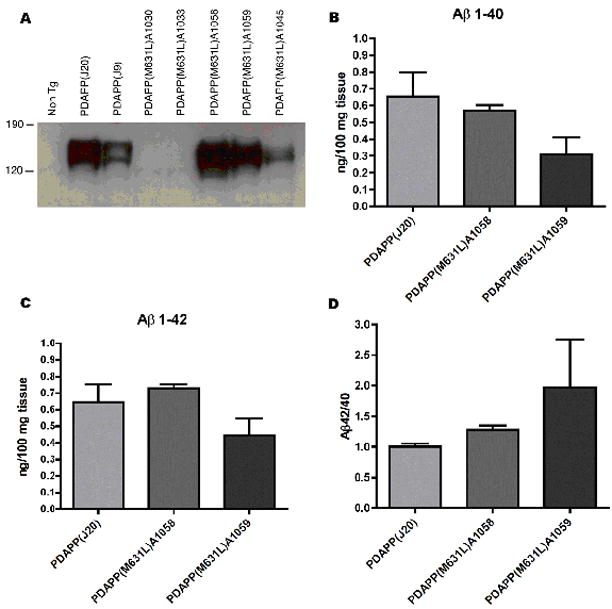
APP expression and Aβ quantitation were performed as previously described [36]. No significant differences were found in APP expression, Aβ(1-40) production, or Aβ(1-42) production between the PDAPP J20 line and the PDAPP(M631L) A1058 line, which were the lines used in this study.
Figure 4. Immunohistochemical staining of amyloid plaques and microglia.
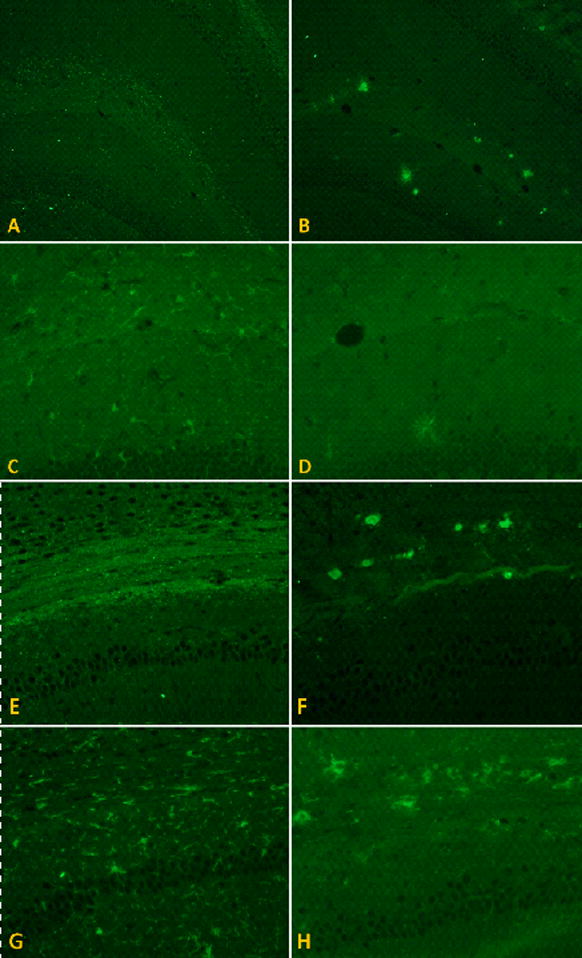
Coronal paraformaldehyde-fixed paraffin embedded sections were labeled with antibodies against Aβ(6E10) and ionized calcium binding protein 1 (Iba 1), a marker for microglia. In the molecular layer of the hippocampus just superior to the dentate gyrus in the M631L mice (A) there were few plaques but distinctive punctate staining; in the J20 (B), however, greater plaque formation was seen in the same area (6E10 images at 10x), but no punctate staining. The punctate pattern of Aβ distribution was associated with microglial activation in the M631L (C), which was not observed in the J20, the latter of which instead showed amoeboid phagocytic microglia surrounding plaques (D) (Iba 1 images at 20X). The punctate labeling in the M631L mice was even more marked in the corpus callosum (E) and the white matter tracts lateral to the hippocampus and surrounding the thalamus, associated with a proportionately greater microglial response (G); again, the J20 did not demonstrate punctate staining, but instead formed plaques in the corpus callosum (F), surrounded by phagocytic microglia (H) (all 20X).
To complement these studies, we assessed the oxidative damage in another PDAPP mutant mouse—D664A—that had been shown previously to prevent the memory deficits otherwise characteristic of PDAPP mice [36]. Unlike the Tg PDAPP(M631L) mice, the D664A mutation did not result in any reduction in protein carbonyls, 3-nitrotyrosine, or protein-bound 4-hydroxy-2-nonenal (Supp. Fig. 1).
Behavioral Measures
Prior to behavioral analyses, mice are evaluated to ensure that their visual and somatic motor systems are intact, and that they are capable of swimming. None of the mice evaluated in this study displayed any problems completing these preliminary tests.
In analyses of behavior employing the Morris water maze, two indices were examined. The latency index, which reflects the time necessary to find a hidden escape platform, is a measure of visuospatial learning and is characteristically abnormal in Alzheimer model transgenic mice, as well as other mice with hippocampal dysfunction. The M631L mutation led to no improvement over the PDAPP mice in latency index: performance of Tg PDAPP and Tg PDAPP(M631L) mice was indistinguishable, and in both cases significantly impaired with respect to that of the Non Tg PDAPP group (p<0.001 and p<0.001, respectively, Bonferroni’s post test applied to a significant effect of genotype F(3,78)=7.05, p=0.0013, repeated measures two-way ANOVA) (Fig. 5a). Transgenic PDAPP(M631L) mice also showed significantly increased floating with respect to all other groups, which may reflect helplessness associated with memory dysfunction (p<0.001, p<0.01, and p<0.001 with respect to Non Tg PDAPP, Non Tg PDAPP(M631L), and Tg PDAPP, respectively, Bonferroni’s post test applied to a significant effect of genotype, F(3,78)=9.22, p=0.0003, repeated measures two-way ANOVA), which were not significantly different from each other (Fig. 5b).
Figure 5. Effect of the M631L mutation on behavior as assessed with the Morris water maze in PDAPP mice.
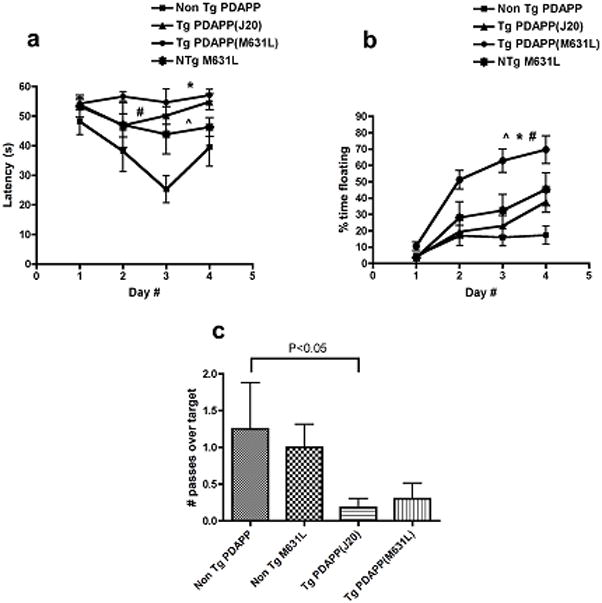
(a) Spatial training. Mean latencies to reach a hidden platform were significantly different for the Tg PDAPP, the Tg PDAPP(M631L) and the Non Tg PDAPP(M631L) groups with respect to Non Tg PDAPP littermates (#P<0.01, *P<0.01 and ^P<0.05, respectively, as a result of Bonferroni’s post test applied to a significant effect of genotype F(3,78)=7.05, P=0.0013, repeated measures two-way ANOVA). (b) Floating. Percent time spent floating increased significantly as a function of day number during training (F(3,78)=40.14, P<0.0001, two-way ANOVA). Tg PDAPP(M631L) mice spent a significantly larger percent of trial time floating than all other groups (*P<0.001, #P<0.01 and ^P<0.001 with respect to Non Tg PDAPP, Non Tg PDAPP(M631L) and Tg PDAPP respectively, Bonferroni’s post test applied to a significant effect of genotype F(3,78)=9.22, P=0.0003, repeated measures two-way ANOVA). “Floaters” were excluded from all other analyses. (c) Probe trial. Retention of the former platform site was impaired in Tg PDAPP mice with respect to the non-Tg group (*P<0.05 as a result of Dunnet’s multiple comparison test applied to a significant effect of genotype (P=0.03) in one-way ANOVA). No significant difference was observed for the comparison between Tg PDAPP and Tg PDAPP(M631L) (p=0.63). Data are means ± SEM.
The second behavioral index assessed was the number of passes mice made over the original position of the platform after acquisition training was completed and the platform was subsequently removed; this index measures memory retention. As described previously [36, 41, 42], transgenic PDAPP mice showed a significant reduction in crosses over the former platform position (target) as compared to Non Tg littermates (p<0.05 as a result of Dunnet’s multiple comparison test applied to a significant effect of genotype (p=0.03) in one-way ANOVA). The PDAPP(M631L) mice showed, just as for the latency index, no significant difference between the PDAPP and the PDAPP(M631L) mice (p=0.63, two-tailed unpaired t-test), suggesting that the M631L mutation did not improve retention (Fig. 5c).
Discussion
To our knowledge, this is the first study to demonstrate the in vivo requirement for the single Met residue at position 35 of human Aβ in the production of Alzheimer-associated oxidative stress in mammalian brain. Substitution of Met by Leu at residue 631 of Swedish and Indiana human mutant APP (and therefore position 35 of the derivative Aβ peptides) completely abolished the oxidative stress observed when Met was present (Figs. 1a–c). Differential expression of APP, Aβ(1-42), or Aβ(1-40) was shown not to be the cause of this lack of oxidative stress in brains of Tg PDAPP(M631L) mice (Fig. 3). However, the M35L mutation in Aβ peptides did influence the plaque formation, since the M631L mice displayed a reduction in immunoreactive plaques; instead, they showed non-plaque punctate staining accompanied by a microglial response. These findings suggest that Met35 may be involved in Aβ plaque formation. Moreover, the results are consistent with the notion that any non-specific or compensatory changes secondary to the presence of a human transgene are unlikely to be responsible for the lack of oxidative stress in brain isolated from the Tg PDAPP(M631L) mouse.
Methionine has important cellular functions, such as shielding the active sites of enzymes against oxidation [43], promotion of helical secondary structure of proteins [44], aiding activity of certain repair or chaperone proteins [45], and participation in maintenance of the redox state of cells [46]. In addition, within the Aβ peptide, the current studies indicate that Met35 plays a critical role in the oxidative damage associated with Alzheimer disease.
We previously demonstrated that oxidative stress associated with Aβ(1-42) in neurons is via catalytic processes [35]. This previous study demonstrated two important aspects of Aβ-mediated oxidative stress in neurons: (a) A small amount of Met-centered free radical on Aβ(1-42) can be greatly amplified. The oxidative damage to neurons induced by Aβ(1-42) results in part from the chain reaction produced in the lipid phase of the membrane initiated by abstraction of allylic H atoms from unsaturated acyl chains on lipids by the radical cation on the S atom of Met, i.e., the sulfuranyl radical [35]. This chain reaction continues as long as there are such allylic H atom sites available, and products of lipid peroxidation, such as HNE, are produced. These, in turn, react with Cys, His, and Lys residues of proteins to cause massive changes in structure [47] and function [38,48,49]. (b) The sulfuranyl free radical formed on the single Met residue of Aβ(1-42), when it abstracts the allylic H atom from an unsaturated acyl chain of phospholipids, forms an acid, whose pKa is -5. Hence, any base, including water, can remove the H+ from this acid, forming again Met itself, which can undergo a one-electron oxidation again to the sulfuranyl free radical. That is, the process is catalytic. Of course, HNE can only be formed by a radical located within the bilayer, since the reactivity of the radicals is too high for diffusion from outside the cell into the bilayer to find an allylic H atom on unsaturated chains of lipids. Yet, Abeta does indeed lead to HNE formation [38,50].
The notion proposed by the Butterfield laboratory that a one-electron oxidation of Met35 of Aβ(1-42) to form a sulfuranyl free radical that in turn leads to lipid peroxidation and protein oxidation in neurons in brain of subjects with AD and MCI [33–35] has been supported by other laboratories. For example, pulse radiolysis studies revealed a sulfur-centered free radical on Aβ(1-40), but not on the peptide with the non-toxic reverse sequence, Aβ(40-1) [51]. Our studies showing lack of neurotoxicity, protein oxidation, and lipid peroxidation in brain cells exposed to Aβ(1-42) when the single Met residue of this peptide was substituted by norleucine [32, 35] or Cys [32] have been confirmed by others [52–54]. The idea of a one-electron oxidation of Met35 within Aβ(1-42) [32–34] has also been supported by the experimental and theoretical research of others [55,56]. The latter study suggested that Cu2+ was bound weakly to Met in Aβ(1-42). This is consistent with our earlier hypothesis [33] and consistent with the results of studies with the divalent cation chelator, clioquinol [57,58]. Previously, researchers had shown that this agent could dissolve plaques from transgenic mice containing human mutant APP [57], even though the KD of Cu2+ for clioquinol reportedly was nine orders of magnitude greater than that for Aβ(1-42) [58]. Hence, we suggested [33] that a weakly bound Cu2+ on Met could account for two observations: (a) an electron transfer reaction involving a one-electron oxidation of Met to form the sulfuranyl radical, while Cu2+ is simultaneously reduced to Cu+, the latter capable of undergoing Fenton-type chemistry to produce highly reactive hydroxyl free radicals; and (b) the clioquinol data, i.e., a weakly bound Cu2+ could easily be removed from Met by clioquinol, while it is unlikely that clioquinol could remove Cu2+ bound to the three His residues of Aβ(1-42) (positions 6, 13, and 14) with a KD in the attomolar range.
Oxidative stress has been demonstrated in brain of another transgenic mouse model of AD [59], as well as a knock-in mouse model of AD [27]. Although some have questioned the relevance of oxidative stress associated with Aβ(1-42) in vivo [60], it is clear from the present study that oxidative stress does not occur in brain in vivo if the single Met residue of Aβ(1-42) is not present. The importance of the current study is that the results establish unequivocally the critical nature of the Met35 residue of Aβ for oxidative stress in brain in vivo in a mammalian species. We posit that similar importance of Met35 in Aβ(1-42) occurs in the brains of subjects with MCI and AD. Clearly, genetic manipulation of Met in Aβ(1-42) in humans is currently not possible, so our results point to clinical translational possibilities for therapy to potentially treat, slow the progression of, or prevent MCI and AD. Specifically, blocking the oxidative damage dependent on the Met residue of Aβ(1-42) in brains of subjects with MCI and AD offers a highly focused therapeutic approach. Studies to test this notion are in progress.
In contrast to the clear role of Met at residue 35 of Aβ(1-40 and/or 1-42) in oxidative damage, spatial learning and memory in mice with the Tg PDAPP Swe/Ind double mutant APP did not demonstrate a dependence on Met35 of Aβ. In Morris water maze studies, neither the latency period nor the number of times the Tg PDAPP(M631L) mice swam over the platform site differed significantly from that of the Tg PDAPP mice. Hence, in this mouse model, Met35 and its associated oxidative damage are not required for the spatial memory loss that is characteristic of the model (and which also characterizes human Alzheimer disease).
Furthermore, a different mutation in the same model—PDAPP(D664A)—prevented spatial memory abnormalities, yet failed to show any effect on oxidative damage indices, demonstrating that such oxidative damage is not sufficient for the behavioral phenotype, either. However, it is important to note that it is still possible that we have, to date, missed a behavioral effect of the M631L mutation: mice were analyzed for markers of oxidative stress at nine months of age, but for behavior necessarily at an earlier age (6 months), so earlier or later differences in behavior, if present, would have been missed by the studies reported here. The possibility that later deficits might be mollified by the M631L mutation is compatible with recently published studies showing that the PDAPP(D664A) mice do show a trend toward Morris water maze abnormalities at 13 months of age that they do not show at earlier ages [41]; and while it is not clear that this trend is the result of oxidative damage, the PDAPP(D664A) mice do indeed show such damage. Therefore, subsequent studies will assess transgenic mice with both the D664A and M631L mutations, to determine whether they resist the trend toward late deterioration in Morris water maze performance.
Finally, immunoreactive Aβ deposits in the brains of PDAPPM631L mice clearly revealed marked differences from the frank plaque deposits of PDAPP(J20) mice. Moreover, there is evidence of microglial activation in the M631L mice. Hence, it is conceivable that the latter influenced behavior in the PDAPPM631L mice, potentially contributing to the lack of behavioral improvement relative to that of PDAPP(J20) mice.
In contrast to the lack of effect of the M631L mutation on memory in Alzheimer model mice, in the C. elegans AD model, in which human Aβ(1-42) was expressed in muscle (as opposed to full-length APP in neurons), substitution of the Met of Aβ(1-42) by Cys abrogated the paralytic phenotype of the worm without changing the deposition of the modified peptide [32]. Also in potential contrast to the present results, aged beagle dogs (whose Aβ sequence is identical to that of humans) fed a high antioxidant diet and given a program of behavioral enrichment showed decreased oxidative stress in brain, improved learning and memory, and decreased levels of Aβ(1-42) [61]. While the fundamental differences of these previous studies make direct comparisons to the current studies difficult—for example, comparison of mutant APP expression in mouse brain to Aβ expression in nematode muscle may be a non-productive exercise—such comparisons may nonetheless offer some clues to pathogenesis and strategies for future studies: first, the mouse model employed full-length APP with familial AD-associated mutations, whereas the nematode model expressed only the Aβ peptide. Thus, the lack of improvement in the mouse model may have been due to the other, non-Aβ peptides derived from APP—sAPPβ, Jcasp, and C31—all of which have been implicated in aspects of the AD phenotype [36, 62–64]. Second, the mouse, dog, and nematode data are all compatible with the notion that multiple mechanisms are involved in AD pathogenesis, such that the threshold mechanism is likely to differ from model to model. If this proves to be the case, then the results from these different systems argue that it will be critical to test potential new therapeutics in more than one system, and ultimately it will be crucial to determine which model or combination of models provides the greatest predictive value for human Alzheimer disease.
Supplementary Material
Acknowledgments
This research was supported in part by NIH grants to D.A.B. [AG-05119] and to D.E.B. [NS-45093, NS-33376, and AG-12282], as well as support from the Joseph Drown Foundation (to D.E.B), the Stephen D. Bechtel Foundation (to V.G.), and the Alzheimer’s Association (to D.E.B. and V.G.). We thank Prof. Lennart Mucke for the J20 line of PDAPP mice and for the PDGF β-chain promoter-driven APPSw,In minigene.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Butterfield DA. β-Amyloid-associated free radical oxidative stress and neurotoxicity: implications for Alzheimer’s disease. Chem Res Toxicol. 1997;10:495–506. doi: 10.1021/tx960130e. [DOI] [PubMed] [Google Scholar]
- 2.Butterfield DA, Poon HF, St Clair D, Keller JN, Pierce WM, Klein JB, Markesbery WR. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the development of Alzheimer’s disease. Neurobiol Dis. 2006;22:223–232. doi: 10.1016/j.nbd.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 3.Butterfield DA, Reed T, Newman SF, Sultana R. Roles of amyloid beta-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer’s disease and mild cognitive impairment. Free Radic Biol Med. 2007;43:658–677. doi: 10.1016/j.freeradbiomed.2007.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Butterfield DA, Reed T, Perluigi M, De Marco C, Coccia R, Cini C, Sultana R. Elevated protein-bound levels of the lipid peroxidation product, 4-hydroxy-2-nonenal, in brain from persons with mild cognitive impairment. Neurosci Lett. 2006;397:170–173. doi: 10.1016/j.neulet.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 5.Butterfield DA, Reed TT, Perluigi M, De Marco C, Coccia R, Keller JN, Markesbery WR, Sultana R. Elevated levels of 3-nitrotyrosine in brain from subjects with amnestic mild cognitive impairment: implications for the role of nitration in the progression of Alzheimer’s disease. Brain Res. 2007;1148:243–248. doi: 10.1016/j.brainres.2007.02.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hensley K, Hall N, Subramaniam R, Cole P, Harris M, Aksenov M, Aksenova M, Gabbita SP, Wu JF, Carney JM, et al. Brain regional correspondence between Alzheimer’s disease histopathology and biomarkers of protein oxidation. J Neurochem. 1995;65:2146–2156. doi: 10.1046/j.1471-4159.1995.65052146.x. [DOI] [PubMed] [Google Scholar]
- 7.Keller JN, Schmitt FA, Scheff SW, Ding Q, Chen Q, Butterfield DA, Markesbery WR. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology. 2005;64:1152–1156. doi: 10.1212/01.WNL.0000156156.13641.BA. [DOI] [PubMed] [Google Scholar]
- 8.Lyras L, Cairns NJ, Jenner A, Jenner P, Halliwell B. An assessment of oxidative damage to proteins, lipids, and DNA in brain from patients with Alzheimer’s disease. J Neurochem. 1997;68:2061–2069. doi: 10.1046/j.1471-4159.1997.68052061.x. [DOI] [PubMed] [Google Scholar]
- 9.Markesbery WR. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic Biol Med. 1997;23:134–147. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- 10.Smith MA, Richey Harris PL, Sayre LM, Beckman JS, Perry G. Widespread peroxynitrite-mediated damage in Alzheimer’s disease. J Neurosci. 1997;17:2653–2657. doi: 10.1523/JNEUROSCI.17-08-02653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Varadarajan S, Yatin S, Aksenova M, Butterfield DA. Review: Alzheimer’s amyloid β-peptide-associated free radical oxidative stress and neurotoxicity. J Struct Biol. 2000;130:184–208. doi: 10.1006/jsbi.2000.4274. [DOI] [PubMed] [Google Scholar]
- 12.Reed T, Perluigi M, Sultana R, Pierce WM, Klein JB, Turner DM, Coccia R, Markesbery WR, Butterfield DA. Redox proteomic identification of 4-hydroxy-2-nonenal-modified brain proteins in amnestic mild cognitive impairment: insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer’s disease. Neurobiol Dis. 2008;30:107–120. doi: 10.1016/j.nbd.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 13.Reed TT, Pierce WM, Jr, Turner DM, Markesbery WR, Butterfield DA. Proteomic identification of nitrated brain proteins in early Alzheimer’s disease inferior parietal lobule. J Cell Mol Med. 2008 doi: 10.1111/j.1582-4934.2008.00478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sultana R, Boyd-Kimball D, Cai J, Pierce WM, Klein JB, Merchant M, Butterfield DA. Proteomics analysis of the Alzheimer’s disease hippocampal proteome. J Alzheimers Dis. 2007;11:153–164. doi: 10.3233/jad-2007-11203. [DOI] [PubMed] [Google Scholar]
- 15.Sultana R, Boyd-Kimball D, Poon HF, Cai J, Pierce WM, Klein JB, Markesbery WR, Zhou XZ, Lu KP, Butterfield DA. Oxidative modification and down regulation of Pin1 in Alzheimer’s disease hippocampus: A redox proteomics analysis. Neurobiol Aging. 2006;27:918–925. doi: 10.1016/j.neurobiolaging.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 16.Sultana R, Boyd-Kimball D, Poon HF, Cai J, Pierce WM, Klein JB, Merchant M, Markesbery WR, Butterfield DA. Redox proteomics identification of oxidized proteins in Alzheimer’s disease hippocampus and cerebellum: an approach to understand pathological and biochemical alterations in AD. Neurobiol Aging. 2006;27:1564–1576. doi: 10.1016/j.neurobiolaging.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 17.Sultana R, Reed T, Perluigi M, Coccia R, Pierce WM, Butterfield DA. Proteomic identification of nitrated brain proteins in amnestic mild cognitive impairment: a regional study. J Cell Mol Med. 2007;11:839–851. doi: 10.1111/j.1582-4934.2007.00065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem. 2005;280:17294–17300. doi: 10.1074/jbc.M500997200. [DOI] [PubMed] [Google Scholar]
- 19.Selkoe DJ. Soluble oligomers of the amyloid β-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192:106–113. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Viola KL, Velasco PT, Klein WL. Why Alzheimer’s is a disease of memory: the attack on synapses by Aβ oligomers (ADDLs) J Nutr Health Aging. 2008;12:51S–57S. doi: 10.1007/BF02982587. [DOI] [PubMed] [Google Scholar]
- 21.Drake J, Link CD, Butterfield DA. Oxidative stress precedes fibrillar deposition of Alzheimer’s disease amyloid β-peptide (1–42) in a transgenic Caenorhabditis elegans model. Neurobiol Aging. 2003;24:415–420. doi: 10.1016/s0197-4580(02)00225-7. [DOI] [PubMed] [Google Scholar]
- 22.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 23.Ansari MA, Joshi G, Huang Q, Opii WO, Abdul HM, Sultana R, Butterfield DA. In vivo administration of D609 leads to protection of subsequently isolated gerbil brain mitochondria subjected to in vitro oxidative stress induced by amyloid β-peptide and other oxidative stressors: relevance to Alzheimer’s disease and other oxidative stress-related neurodegenerative disorders. Free Radic Biol Med. 2006;41:1694–1703. doi: 10.1016/j.freeradbiomed.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boyd-Kimball D, Mohmmad Abdul H, Reed T, Sultana R, Butterfield DA. Role of phenylalanine 20 in Alzheimer’s amyloid β-peptide (1-42)-induced oxidative stress and neurotoxicity. Chem Res Toxicol. 2004;17:1743–1749. doi: 10.1021/tx049796w. [DOI] [PubMed] [Google Scholar]
- 25.Pappolla MA, Chyan YJ, Poeggeler B, Bozner P, Ghiso J, LeDoux SP, Wilson GL. Alzheimer β protein mediated oxidative damage of mitochondrial DNA: prevention by melatonin. J Pineal Res. 1999;27:226–229. doi: 10.1111/j.1600-079x.1999.tb00619.x. [DOI] [PubMed] [Google Scholar]
- 26.Sultana R, Ravagna A, Mohmmad-Abdul H, Calabrese V, Butterfield DA. Ferulic acid ethyl ester protects neurons against amyloid β-peptide(1-42)-induced oxidative stress and neurotoxicity: relationship to antioxidant activity. J Neurochem. 2005;92:749–758. doi: 10.1111/j.1471-4159.2004.02899.x. [DOI] [PubMed] [Google Scholar]
- 27.Abdul HM, Sultana R, St Clair DK, Markesbery WR, Butterfield DA. Oxidative damage in brain from human mutant APP/PS-1 double knock-in mice as a function of age. Free Radic Biol Med. 2008;45:1420–1425. doi: 10.1016/j.freeradbiomed.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boyd-Kimball D, Sultana R, Poon HF, Lynn BC, Casamenti F, Pepeu G, Klein JB, Butterfield DA. Proteomic identification of proteins specifically oxidized by intracerebral injection of amyloid β-peptide (1-42) into rat brain: implications for Alzheimer’s disease. Neuroscience. 2005;132:313–324. doi: 10.1016/j.neuroscience.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 29.Mohmmad Abdul H, Sultana R, Keller JN, St Clair DK, Markesbery WR, Butterfield DA. Mutations in amyloid precursor protein and presenilin-1 genes increase the basal oxidative stress in murine neuronal cells and lead to increased sensitivity to oxidative stress mediated by amyloid β-peptide (1-42), H2O2 and kainic acid: implications for Alzheimer’s disease. J Neurochem. 2006;96:1322–1335. doi: 10.1111/j.1471-4159.2005.03647.x. [DOI] [PubMed] [Google Scholar]
- 30.Resende R, Moreira PI, Proenca T, Deshpande A, Busciglio J, Pereira C, Oliveira CR. Brain oxidative stress in a triple-transgenic mouse model of Alzheimer disease. Free Radic Biol Med. 2008;44:2051–2057. doi: 10.1016/j.freeradbiomed.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 31.Zhu M, Gu F, Shi J, Hu J, Hu Y, Zhao Z. Increased oxidative stress and astrogliosis responses in conditional double-knockout mice of Alzheimer-like presenilin-1 and presenilin-2. Free Radic Biol Med. 2008;45:1493–1499. doi: 10.1016/j.freeradbiomed.2008.08.027. [DOI] [PubMed] [Google Scholar]
- 32.Yatin SM, Varadarajan S, Link CD, Butterfield DA. In vitro and in vivo oxidative stress associated with Alzheimer’s amyloid β-peptide (1-42) Neurobiol Aging. 1999;20:325–330. doi: 10.1016/s0197-4580(99)00056-1. discussion 339–342. [DOI] [PubMed] [Google Scholar]
- 33.Butterfield DA, Boyd-Kimball D. The critical role of methionine 35 in Alzheimer’s amyloid β-peptide (1-42)-induced oxidative stress and neurotoxicity. Biochim Biophys Acta. 2005;1703:149–156. doi: 10.1016/j.bbapap.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 34.Kanski J, Aksenova M, Schoneich C, Butterfield DA. Substitution of isoleucine-31 by helical-breaking proline abolishes oxidative stress and neurotoxic properties of Alzheimer’s amyloid β-peptide. Free Radic Biol Med. 2002;32:1205–1211. doi: 10.1016/s0891-5849(02)00821-3. [DOI] [PubMed] [Google Scholar]
- 35.Varadarajan S, Kanski J, Aksenova M, Lauderback C, Butterfield DA. Different mechanisms of oxidative stress and neurotoxicity for Alzheimer’s Aβ (1--42) and Aβ (25--35) J Am Chem Soc. 2001;123:5625–5631. doi: 10.1021/ja010452r. [DOI] [PubMed] [Google Scholar]
- 36.Galvan V, Gorostiza OF, Banwait S, Ataie M, Logvinova AV, Sitaraman S, Carlson E, Sagi SA, Chevallier N, Jin K, Greenberg DA, Bredesen DE. Reversal of Alzheimer’s-like pathology and behavior in human APP transgenic mice by mutation of Asp664. Proc Natl Acad Sci U S A. 2006;103:7130–7135. doi: 10.1073/pnas.0509695103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Butterfield DA, Stadtman ER. Protein Oxidation Processes in Aging Brain. Adv Cell Aging Gerontol. 1997;2:161–191. [Google Scholar]
- 38.Lauderback CM, Hackett JM, Huang FF, Keller JN, Szweda LI, Markesbery WR, Butterfield DA. The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer’s disease brain: the role of Aβ1-42. J Neurochem. 2001;78:413–416. doi: 10.1046/j.1471-4159.2001.00451.x. [DOI] [PubMed] [Google Scholar]
- 39.Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 40.Aksenov MY, Aksenova MV, Butterfield DA, Geddes JW, Markesbery WR. Protein oxidation in the brain in Alzheimer’s disease. Neuroscience. 2001;103:373–383. doi: 10.1016/s0306-4522(00)00580-7. [DOI] [PubMed] [Google Scholar]
- 41.Galvan V, Zhang J, Gorostiza OF, Banwait S, Huang W, Ataie M, Tang H, Bredesen DE. Long-term prevention of Alzheimer’s disease-like behavioral deficits in PDAPP mice carrying a mutation in Asp664. Behav Brain Res. 2008;191:246–255. doi: 10.1016/j.bbr.2008.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saganich MJ, Schroeder BE, Galvan V, Bredesen DE, Koo EH, Heinemann SF. Deficits in synaptic transmission and learning in amyloid precursor protein (APP) transgenic mice require C-terminal cleavage of APP. J Neurosci. 2006;26:13428–13436. doi: 10.1523/JNEUROSCI.4180-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stadtman ER. Cyclic oxidation and reduction of methionine residues of proteins in antioxidant defense and cellular regulation. Arch Biochem Biophys. 2004;423:2–5. doi: 10.1016/j.abb.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 44.Vogt W. Oxidation of methionyl residues in proteins: tools, targets, and reversal. Free Radic Biol Med. 1995;18:93–105. doi: 10.1016/0891-5849(94)00158-g. [DOI] [PubMed] [Google Scholar]
- 45.Bose-Basu B, DeRose EF, Kirby TW, Mueller GA, Beard WA, Wilson SH, London RE. Dynamic characterization of a DNA repair enzyme: NMR studies of [methyl-13C]methionine-labeled DNA polymerase β. Biochemistry. 2004;43:8911–8922. doi: 10.1021/bi049641n. [DOI] [PubMed] [Google Scholar]
- 46.Schoneich C. Redox processes of methionine relevant to β-amyloid oxidation and Alzheimer’s disease. Arch Biochem Biophys. 2002;397:370–376. doi: 10.1006/abbi.2001.2621. [DOI] [PubMed] [Google Scholar]
- 47.Subramaniam R, Roediger F, Jordan B, Mattson MP, Keller JN, Waeg G, Butterfield DA. The lipid peroxidation product, 4-hydroxy-2-trans-nonenal, alters the conformation of cortical synaptosomal membrane proteins. J Neurochem. 1997;69:1161–1169. doi: 10.1046/j.1471-4159.1997.69031161.x. [DOI] [PubMed] [Google Scholar]
- 48.Sayre LM, Zelasko DA, Harris PL, Perry G, Salomon RG, Smith MA. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer’s disease. J Neurochem. 1997;68:2092–2097. doi: 10.1046/j.1471-4159.1997.68052092.x. [DOI] [PubMed] [Google Scholar]
- 49.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 50.Mark RJ, Lovell MA, Markesbery WR, Uchida K, Mattson MP. A role for 4-hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid beta-peptide. J Neurochem. 68:255–264. doi: 10.1046/j.1471-4159.1997.68010255.x. [DOI] [PubMed] [Google Scholar]
- 51.Kadlcik V, Sicard-Roselli C, Mattioli TA, Kodicek M, Houee-Levin C. One-electron oxidation of β-amyloid peptide: sequence modulation of reactivity. Free Radic Biol Med. 2004;37:881–891. doi: 10.1016/j.freeradbiomed.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 52.Clementi ME, Pezzotti M, Orsini F, Sampaolese B, Mezzogori D, Grassi C, Giardina B, Misiti F. Alzheimer’s amyloid β-peptide (1-42) induces cell death in human neuroblastoma via bax/bcl-2 ratio increase: an intriguing role for methionine 35. Biochem Biophys Res Commun. 2006;342:206–213. doi: 10.1016/j.bbrc.2006.01.137. [DOI] [PubMed] [Google Scholar]
- 53.Dai XL, Sun YX, Jiang ZF. Attenuated cytotoxicity but enhanced β–fibril of a mutant amyloid β-peptide with a methionine to cysteine substitution. FEBS Lett. 2007;581:1269–1274. doi: 10.1016/j.febslet.2007.02.038. [DOI] [PubMed] [Google Scholar]
- 54.Murray IV, Sindoni ME, Axelsen PH. Promotion of oxidative lipid membrane damage by amyloid β proteins. Biochemistry. 2005;44:12606–12613. doi: 10.1021/bi050926p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Crouch PJ, Barnham KJ, Duce JA, Blake RE, Masters CL, Trounce IA. Copper-dependent inhibition of cytochrome c oxidase by Aβ (1-42) requires reduced methionine at residue 35 of the Aβ peptide. J Neurochem. 2006;99:226–236. doi: 10.1111/j.1471-4159.2006.04050.x. [DOI] [PubMed] [Google Scholar]
- 56.Gomez-Balderas R, Raffa DF, Rickard GA, Brunelle P, Rauk A. Computational studies of Cu(II)/Met and Cu(I)/Met binding motifs relevant for the chemistry of Alzheimer’s disease. J Phys Chem A. 2005;109:5498–5508. doi: 10.1021/jp050843i. [DOI] [PubMed] [Google Scholar]
- 57.Cherny RA, Atwood CS, Xilinas ME, Gray DN, Jones WD, McLean CA, Barnham KJ, Volitakis I, Fraser FW, Kim Y, Huang X, Goldstein LE, Moir RD, Lim JT, Beyreuther K, Zheng H, Tanzi RE, Masters CL, Bush AI. Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron. 2001;30:665–676. doi: 10.1016/s0896-6273(01)00317-8. [DOI] [PubMed] [Google Scholar]
- 58.Huang X, Cuajungco MP, Atwood CS, Hartshorn MA, Tyndall JD, Hanson GR, Stokes KC, Leopold M, Multhaup G, Goldstein LE, Scarpa RC, Saunders AJ, Lim J, Moir RD, Glabe C, Bowden EF, Masters CL, Fairlie DP, Tanzi RE, Bush AI. Cu(II) potentiation of Alzheimer Aβ neurotoxicity. Correlation with cell-free hydrogen peroxide production and metal reduction. J Biol Chem. 1999;274:37111–37116. doi: 10.1074/jbc.274.52.37111. [DOI] [PubMed] [Google Scholar]
- 59.Smith MA, Hirai K, Hsiao K, Pappolla MA, Harris PL, Siedlak SL, Tabaton M, Perry G. Amyloid-beta deposition in Alzheimer-transgenic mice is associated with oxidative stress. J Neurochem. 1998;70:2212–2215. doi: 10.1046/j.1471-4159.1998.70052212.x. [DOI] [PubMed] [Google Scholar]
- 60.Nunomura A, Moreira PI, Lee HG, Zhu X, Castellani RJ, Smith MA, Perry G. Neuronal death and survival under oxidative stress in Alzheimer and Parkinson diseases. CNS Neurol Disord Drug Targets. 2007;6:411–423. doi: 10.2174/187152707783399201. [DOI] [PubMed] [Google Scholar]
- 61.Opii WO, Joshi G, Head E, Milgram NW, Muggenburg BA, Klein JB, Pierce WM, Cotman CW, Butterfield DA. Proteomic identification of brain proteins in the canine model of human aging following a long-term treatment with antioxidants and a program of behavioral enrichment: relevance to Alzheimer’s disease. Neurobiol Aging. 2008;29:51–70. doi: 10.1016/j.neurobiolaging.2006.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lu DC, Rabizadeh S, Chandra S, Shayya RF, Ellerby LM, Ye X, Salvesen GS, Koo EH, Bredesen DE. A second cytotoxic proteolytic peptide derived from amyloid β-protein precursor. Nat Med. 2000;6:397–404. doi: 10.1038/74656. [DOI] [PubMed] [Google Scholar]
- 63.Madeira A, Pommet JM, Prochiantz A, Allinquant B. SET protein (TAF1β, I2PP2A) is involved in neuronal apoptosis induced by an amyloid precursor protein cytoplasmic subdomain. FASEB J. 2005;19:1905–1907. doi: 10.1096/fj.05-3839fje. [DOI] [PubMed] [Google Scholar]
- 64.Nikolaev A, McLaughlin T, O’Leary DD, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–989. doi: 10.1038/nature07767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.