

Abstract
Bacterial AlkB and three human AlkB homologues (ABH1, ABH2, and ABH3) are Fe2+/2-oxoglutarate-dependent oxygenases that directly repair alkylation-damaged DNA. Here, we show that ABH1 unexpectedly has a second activity, cleaving DNA at abasic (AP) sites such as those arising spontaneously from alkylation-dependent depurination reactions. The DNA cleavage activity of ABH1 does not require added Fe2+ or 2-oxoglutarate, is not inhibited by EDTA, and is unaffected by mutation of the putative metal-binding residues, indicating that this activity arises from an active site distinct from that used for demethylation. AP-specific DNA cleavage was shown to occur by a lyase mechanism, rather than by hydrolysis, with the enzyme remaining associated with the DNA product. ABH1 can cleave at closely spaced AP sites on opposite DNA strands yielding double-strand breaks in vitro and this reaction may relate to the physiological role of this unexpected AP lyase activity.
Keywords: Demethylase, Lyase, Dioxygenase, Abasic site
1. Introduction
All living organisms possess a variety of DNA repair enzymes to retain the integrity of their genetic information [1,2]. In 2002, a novel mechanism for direct repair of alkylated DNA was discovered in Escherichia coli [3–5]; i.e., AlkB couples the oxidative decarboxylation of -oxoglutarate to the hydroxylation of 1-methyl adenine (1-meA) or 3-methyl cytosine (3-meC), with the unstable intermediate decomposing to restore the normal base and release formaldehyde (Supplemental Fig. S1). Humans possess eight AlkB homologues, referred to as ABH1-ABH8 [6], and the related FTO dioxygenase associated with fat mass and obesity [7]. ABH1 exhibits low levels of 3-meC demethylase activity when using single stranded (ss)-DNA and ss-RNA, but unlike AlkB it does not act on 1-meA or on methylated bases in double stranded (ds)-DNA [8]. ABH2 and ABH3 catalyze the same oxidative demethylation reaction as AlkB, albeit with ABH3 preferring ss-DNA and also acting on RNA whereas ABH2 acts mainly on ds-DNA [3,9]. The enzymatic properties of ABH4-ABH8 have not been reported, but human ABH8 was recently linked to the progression of bladder cancer [10]. The FTO gene product demethylates 3-methyl thymine and 3-methyl uracil and its inactivation protects mice from obesity [7,11–13]. The crystal structures of AlkB, ABH2, and ABH3 have been solved [14–16] and reveal a common double-stranded β-sheet core, but with fundamental changes in the oligonucleotide-binding regions [17].
ABH1 is the human homologue with closest similarity to AlkB [6]; however, it is longer by 173 residues (71 at the amino terminus, 42 at the carboxyl terminus, and the remainder within the sequence). The full-length sequence of ABH1 is strongly conserved in humans, mouse, and chicken (70–83% identity), suggesting that the extensions have functional significance. ABH1(−/−) knockout mice have been created, but the only phenotype thus far reported relates to deficiencies in placental trophoblast lineage differentiation [18]. The tissue distribution of ABH1 is a matter of some dispute. One study used Northern blot analysis to show highest mRNA expression in heart and muscle tissue [8]. Other authors used real time PCR and microarray analysis to show that ABH1 is most highly expressed in spleen [19,20]. Three lines of evidence (i.e., transient expression of an ABH1-EYFP fusion protein followed by fluorescence microscopy, immunocytochemical studies using anti-ABH1 antibodies, and subcellular fractionation) suggest a mitochondrial location of the protein [8]. Only very low levels of 3-meC demethylase activity were detected using recombinant ABH1, where this activity was shown to depend on Fe2+ plus 2-oxoglutarate, and it was abolished in the putative Fe2+-ligand alanine mutants [8]. The weak demethylase activity of ABH1 coupled with the sequence conservation of the ABH1 extensions raise the plausible hypothesis that this protein might have additional roles in vivo.
In this study, we further investigate the properties of ABH1. Using human ABH1 heterologously expressed in E. coli and Sf9 insect cells, we demonstrate the ability of this protein to cleave DNA at apurinic/apyrimidinic (AP) or abasic sites. We show that this enzymatic reaction occurs independently of Fe2+ or 2-oxoglutarate, and that it uses a lyase mechanism to produce a DNA nick on the 3′ side of the abasic site leaving a 3′-phospho-α,β-unsaturated aldehyde and a 5′ phosphate. ABH1 acts on both ss-DNA and ds-DNA and is able to produce double-strand breaks (DSB) when the abasic sites are in close proximity on opposed DNA strands.
2 Materials and methods
2.1 Constructions of plasmids and mutants
Plasmid pBAR67, encoding a His6-tagged version of human ABH1 [3], was kindly provided by B. Sedgwick. The H231A, D233A, and H287A mutants were constructed by site-directed mutagenesis using the Quickchange site-directed mutagenesis kit (Stratagene) with the primers listed in Supplemental Table S1 and using pBAR67 as a template. Each mutation was confirmed by sequencing (Davis Sequencing). To produce ABH1 in Sf9 cells, the gene was cloned into the baculovirus vector pAcHLT-B (BD Biosciences). First, ABH1 was amplified with primers ABH1_pAcHLTb-for (5′-ggcagcaccatggggaagatggc-3′) and ABH1_pAcHLTb-rev (5′-gttagcagccggtacctcgaggtctc-3) to introduce an NcoI site at the start codon and a KpnI site after the stop codon (underlined). The PCR fragment was cloned into pGEM-t easy, the sequence was confirmed by sequencing and the fragment was subcloned into pAcHLT-B with the above mentioned restriction sites.
2.2 Overexpression of ABH1 and purification of ABH1
The His6-tagged fusion of ABH1 was overproduced in E. coli BL21(DE3)-CodonPlus RIPL (Stratagene) containing pBAR67 or the derived mutant plasmids. Cells were grown at 25 °C until reaching OD600 of 0.7 and protein production was induced by addition of 0.1 mM isopropyl-β-D-1-thiogalactoside. His6-tagged ABH1 was purified in a single step by using Ni-bound nitrilotriacetic acid (Ni-NTA) Sepharose (GE Healthcare) chromatography to achieve about ~95% purity (Supplemental Fig. S2). The His6-ABH1-containing fractions were immediately subjected to buffer exchange into 20 mM Tris, pH 8, by using a disposable desalting column (GE Healthcare). When necessary, His6-ABH1 containing samples were concentrated by using an Amicon Ultra-4 centrifugal filter unit. For additional purification, Ni-NTA Sepharose-purified ABH1 was loaded onto a Q-Sepharose column equilibrated with 20 mM Tris, pH 8, buffer and eluted with a linear gradient to 20 mM Tris, pH 8, buffer containing 1 M NaCl in five column volumes. Alternatively, the Ni-NTA Sepharose-purified His6-ABH1 was subjected to gel filtration chromatography (Superdex-75, GE Healthcare) in 20 mM Tris (pH 8) buffer containing 20 mM NaCl. For ABH1 purification from Sf9 cells, recombinant baculovirus particles containing the ABH1 gene were generated according to the manufacturer’s instructions (BD Biosciences). The recombinant baculoviruses were plaque purified, amplified, and used in over-expression studies. Sf9 cells were transfected with recombinant ABH1-baculovirus or wild type virus, respectively, and grown for 70 h at 27 °C. Cells were harvested and lysed by sonication in buffer A (25 mM Tris [pH 8], 150 mM KCl, 10% glycerol). His6-tagged ABH1 was bound to Ni-NTA agarose beads by incubation for 1 h at 4 °C. Nonspecifically bound protein was removed by washing the beads three times with buffer A containing 30 mM imidazole. The samples were washed three times with 20 mM Tris (pH 8) to remove salt and imidazole and the bead-bound ABH1 was directly used in activity assays.
2.3 DNA substrates used in assays
Plasmid pUC18 DNA was treated with N-methyl-N′-nitro-N-nitrosoguanidine (MNNG, 5 mg/ml in 10% dimethylsulfoxide) and dithiothreitol (10 mM) for 10 min at room temperature. The reaction was stopped by addition of sodium thiosulfate (10 mM). The methylated DNA was isolated from reagents by using PCR purification columns (Qiagen) and the concentration was determined spectrophotometrically. The CpG methyltransferase M.SssI (New England Biolabs) was used to introduce 5-methylcytosine (5-meC) sites into pUC18 according to the instructions of the manufacturer. Oligonucleotide substrates were purchased from Integrated DNA Technologies and are listed in Table 1. Equimolar amounts of the complementary oligonucleotides were annealed by incubating at 95 °C and slowly cooling to room temperature. To create abasic sites, deoxyuridine (dU)-containing ds-DNA was treated with uracil-DNA N-glycosylase (UDG, 2 U per 5 μM ds-DNA) for 30 min to 1 h at 37 °C. The AP-containing oligonucleotides were purified with PCR purification columns (Qiagen) and the final DNA concentration was determined spectrophotometrically. In case of the 30-mers, the last step was omitted and the oligonucleotides were used in the activity assays without further purification. To produce AP-sites, oligo2 (Table 1) annealed to oligo2c was treated with UDG for 15 min at 37 °C. Tetrahydrofuran (THF)-containing oligonucleotides (Table 1) were purchased from Midland.
Table 1.
Oligonucleotides used as DNA substrates
| Name | sequence |
|---|---|
| Oligo1 | 5′-agt aga cag cta cca tgc ctg cac gaa Uta agc aat tcg taa tca tgg tca tag cta gta-3′ |
| Oligo1c | 5′-tac tag cta tga cca tga tta cga att gct tag ttc gtg cag gca tgg tag ctg tct act-3′ |
| Oligo2 | 5′-agt aga cag cta cca tgc ctg cac gaa gUt agc aat tcg taa tca tgg tca tag cta gta-3′ |
| Oligo2c | 5′-tac tag cta tga cca tga tta cga att gct agU ttc gtg cag gca tgg tag ctg tct act-3′ |
| Oligo2-THF | 5′-agt aga cag cta cca tgc ctg cac gaa g(THF)t agc aat tcg taa tca tgg tca tag cta gta-3′ |
| Oligo2c-THF | 5′-tac tag cta tga cca tga tta cga att gct ag(THF) ttc gtg cag gca tgg tag ctg tct act-3′ |
| Oligo3 | 5′-agt aga caa gUt acc atg cct gca cga agt t-3′ |
| Oligo3c | 5′-aac ttc gtg cag gca tgg tag Utt gtc tac t-3′ |
| Oligo3c2 | 5′-aac ttc gtg cag gca tgg tag ctt gtc tac t-3′ |
2.4. Electrophoretic mobility shift assays
ABH1 was incubated with polynucleotides (150 – 250 ng) in a total volume of 10 μl in 20 mM Tris, pH 8, buffer for 15 min at 37 °C. Samples with plasmid substrates were examined by using 1% agarose gels and those with oligonucleotides were analyzed by using native 6% polyacrylamide gels. The enzyme:DNA complexes were separated from the unbound DNA by electrophoresis in Tris-borate EDTA buffer and the DNA was visualized with EtBr (5 μg/ml).
2.5 Non-radioactive DNA cleavage assays
ABH1 (10 μM) was incubated with plasmid substrates in 20 mM Tris (pH 8) for 3 h at 37 °C in a total reaction volume of 50 μl. Proteinase K (4 U per reaction, Invitrogen) was added to the samples to stop the reaction and the mixtures were incubated for 15 min at 65 °C to allow for protein digestion. The DNA samples were further purified by using DNA spin columns (Qiagen). Samples were loaded onto a 1% agarose gel, separated by electrophoresis, and visualized by EtBr staining. For DSB cleavage assays, ABH1 (5 μM) was incubated with AP-oligo2 annealed to AP-oligo2c (0.5 – 1 μM) for varied times (15–120 min) in 20 mM Tris, pH 8. Activity was stopped by addition of proteinase K (1 U) plus sodium dodecyl sulfate (SDS, 0.5%) and incubation at 65 °C for 15 min. The samples were directly loaded onto a 13.5% polyacrylamide gel, electrophoretically separated, and visualized as described before. Activity assays of Ape1, EndoIII, and EndoVIII (all from NEB) were carried out similarly.
2.6 32P-labeled substrate cleavage assays
For the 5′ labeling reaction, oligonucleotides (1 or 10 μM) were incubated with T4 polynucleotide kinase and [γ-32P]-ATP for 1 h at 37 °C and annealed to equimolar amounts of the complementary strand by heating the sample to 95 °C and slowly cooling to room temperature. The dU-containing substrates were treated with UDG either prior to or during incubation with ABH1 as indicated for the respective experiments. Activity assays were carried out in a total volume of 10 μL in 20 mM Tris, pH 8, and UDG-buffer (in some cases containing 1 mM ascorbate, 200 μM (NH4)2FeSO4, and 1 mM 2-oxoglutarate) for the times indicated in the experiments. The assays with 5 and 10 μM oligonucleotides contained 1 μM labeled oligonucleotide and were supplemented with the respective non-labeled ds- or ss-oligonucleotide to the final concentration. All reactions were terminated by addition of proteinase K and incubation at 65 °C for up to 1 h. When necessary, the samples were further purified by phenol-chloroform isolation and ethanol precipitation. The samples were loaded onto different % polyacrylamide denaturing gels (7 M urea) and the substrate and product bands were separated by electrophoresis. The gels were either dried and exposed to a Typhoon 9410 phosphorimager (GE Healthcare) or exposed to a Hyblot CL film (Denville Scientific Inc.). To calculate specific activity or the amount of product formed, autoradiographs were scanned and analyzed using the program ImageJ [21].
2.7 Trapping of intermediates
For the NaBH4 trapping of intermediates, 5 μM ABH1 and 0.1 μM oligonucleotide substrate labeled at the 5′ end were incubated in a total volume of 20 μl in the presence of 20 mM NaBH4 for 30 min at 37 °C. The reactions with EndoIII (20 U) were carried out identically. The reactions were stopped by addition of SDS-polyacrylamide gel electrophoresis (PAGE) loading dye and heated for 3′ at 95 °C. The trapped intermediates were visualized on a 10% SDS-PAGE gel exposed to a Hyblot CL film.
3. Results
3.1 DNA binding and AP-specific cleavage activity
Highly purified human ABH1 from recombinant E. coli cells (Supplemental Fig. S2) was examined for its ability to bind to nucleic acids by electrophoretic mobility shift assays (Fig. 1A). Highly enriched ABH1 binds to DNA (untreated, methylated by MNNG-treatment, or containing 5-meC) and to RNA. To our surprise, we observed that extended incubation (3 h at 37 °C) of plasmid DNA with ABH1 results in complete degradation of MNNG-methylated, but not 5-meC-containing or non-methylated DNA (Fig. 1B). The results indicated that ABH1 has DNA cleavage activity that targets aberrantly methylated DNA. Because certain methylated bases (for instance 7-meG, 7-meA, 3-meG, and 3-meA) are labile and undergo spontaneous depurination reactions [22], we considered whether the actual substrate of ABH1 is an AP site. To purposely enhance the content of abasic sites, the MNNG-treated samples were incubated at 50 °C for 6 h before being incubated with ABH1. DNA containing AP sites undergoes a slow rate of spontaneous cleavage under these conditions due to β-elimination of the open-ring aldehyde species [23]. Nevertheless, this treatment was shown to significantly increase the rate of DNA degradation by ABH1 (data not shown) suggesting that abasic sites (as opposed to methylated bases) may be the actual target of ABH1’s DNA cleavage activity.
Fig. 1.

Binding and cleavage of DNA by ABH1. (A) ABH1 binds to DNA and RNA. Linearized pUC18 (0.3 – 0.5 μg) was used directly, treated with MNNG to produce predominantly 7-meG and 3-meA, or subjected to SssI methyltransferase and S-adenosyl methionine to generate 5-meC-containing DNA. ABH1 was mixed with these samples, electrophoresed on 1% agarose, and stained with EtBr. (B) Degradation of MNNG-treated DNA by ABH1. Unmodified, SssI-methylated, and MNNG-treated linearized pUC18 (appr. 0.5 μg) were incubated with ABH1 (10 μM) for 3 h at 37 °C, electrophoresed, and stained. (C) ABH1 binding to dU- and AP-containing oligonucleotides. ABH1 was mixed with 100 ng of (i) oligo1 (dU at position 28) annealed to the complementary oligo1c, (ii) oligo1+oligo1c treated with UDG (30 min at 37 °C) to generate an abasic site (depicted as a star), and (iii) oligo2+oligo2c (both with dU) treated with UDG. The samples were electrophoretically analyzed. (D) ABH1 cleavage of oligonucleotides containing an abasic site. Oligo1 labeled with 32P at its 5′ end was annealed to oligo1c and treated with UDG. The effect of ABH1 (1 μM, 1 h at 37 °C) on DNA cleavage was examined by gel electrophoresis. (E) ABH1 cleavage of ds-DNA. ABH1 (5 μM) was incubated at 37 °C with oligo2 annealed to oligo2c (0.3 μg) containing either dUs or abasic sites in close proximity on opposed strands. After 90 min, samples were incubated with proteinase K at 65 °C for 1 h and analyzed by electrophoresis.
To more stringently investigate the specificity of the unexpected ABH1 activity, complementary 60-mer oligonucleotides were synthesized (Table 1) with some samples containing a single internal dU base. The ds-oligomer was treated with UDG to create an abasic site. DNA binding assays with non-labeled oligonucleotides revealed that ABH1 binds with higher affinity to DNA containing an AP-site compared to the DNA containing dU (Fig. 1C). Cleavage assays were performed with 32P end-labeled substrates containing abasic sites. As can be seen (Fig. 1D), cleavage of the labeled substrate is dependent on the presence of ABH1. To determine whether ABH1 can generate DSB by cleaving closely spaced abasic sites on opposite strands, a ds 60-mer substrate (oligo2 annealed to oligo2c) that contains one dU on each strand in close proximity (Table 1) was prepared. Incubation of the substrate with UDG and ABH1 resulted in ds cleavage to produce two fragments, with cleavage dependent on both enzymes (Fig. 1E). We conclude that ABH1 cleaves DNA at AP sites.
To address the possibility that a contaminating DNA-cleaving enzyme copurified with ABH1, three different experimental approaches were used. First, the ABH1 purified with the Ni-NTA Sepharose column was further purified by chromatography on Q-Sepharose. The peak ABH1-containing fractions coincided with AP-specific DNA cleavage activity (Fig. 2A and 2B). Second, the His-tagged ABH1 purified on the Ni-NTA resin was subjected to Superdex-75 gel filtration chromatography. The ABH1 eluted as a monomer (MW = 53 ± 6 kDa, in close agreement with the theoretical mass of 46.8 kDa) and these fractions showed AP-specific cleavage activity (Fig. 2C and 2D). Activity coincided with ABH1 in the fractions, and was not associated with larger complexes or with contaminating ~30-kDa species (possible degradation products of ABH1). The absence of activity for fractions containing larger species excludes the possibility of a contaminating DNA-cleaving enzyme tightly bound to ABH1. Finally, to completely rule out a contaminating prokaryotic AP-specific DNA cleaving enzyme, a different expression system was chosen and ABH1 was produced as a His-tagged fusion protein in Sf9 insect cells. The protein was purified by using Ni-NTA agarose beads (Fig. 3A) and AP-specific DNA cleavage was assayed with ABH1 still bound to the Ni-NTA agarose. As expected, this sample was active whereas a control sample purified from cells expressing wild type virus showed no DNA cleaving activity (Fig. 3B). Significantly, the activity of the purified enzyme from insect cells approximated that of the His-tagged ABH1 produced in E. coli as judged by comparing the amounts of product formed from equivalent amounts of proteins. Both samples purified from Sf9 cells contained small amounts of contaminating exonuclease activity, but it did not obscure the AP-specific DNA cleavage activity and the ABH1 product can still be visualized. Taken together, these results demonstrate that AP-specific DNA cleavage activity is inherent to ABH1.
Fig. 2.

AP-specific DNA cleavage activity of ABH1 after further purification. (A) SDS-PAGE of ABH1 eluted from Q-Sepharose. The fractions eluting from the Q-Sepharose column were pooled as indicated and aliquots were denatured in SDS, run on a 12% SDS-polyacrylamide gel, and visualized by Coomassie-blue staining. (B) Oligo2+oligo2C was treated with UDG, incubated with His6-ABH1 (8.6 μM for the Ni-NTA pool and 10 μM for the Q-Sepharose pool) for 2 h, digested with proteinase K at 65 °C for 15 min, analyzed by electrophoresis using a 13.5% polyacrylamide gel, and visualized by EtBr staining. No ABH1 added in the negative control. (C) SDS-PAGE of ABH1 fractions eluted from Sephadex-75 analyzed by using 12% SDS-PAGE with visualization by Coomassie-blue staining. (D) Cleavage of AP-containing oligonucleotides by His6-ABH1 eluted from the Sephadex-75 gel filtration (GF) column. Fractions (9 μl) were incubated with UDG-treated oligo2+2c (containing AP sites on both strands) for 2 h at 37 °C, subjected to proteinase K treatment, separated on a 13.5% PAGE gel, and visualized by EtBr staining. No ABH1 was added to nc; Ni-NTA purified ABH1 is analyzed in Ni-NTA.
Fig. 3.
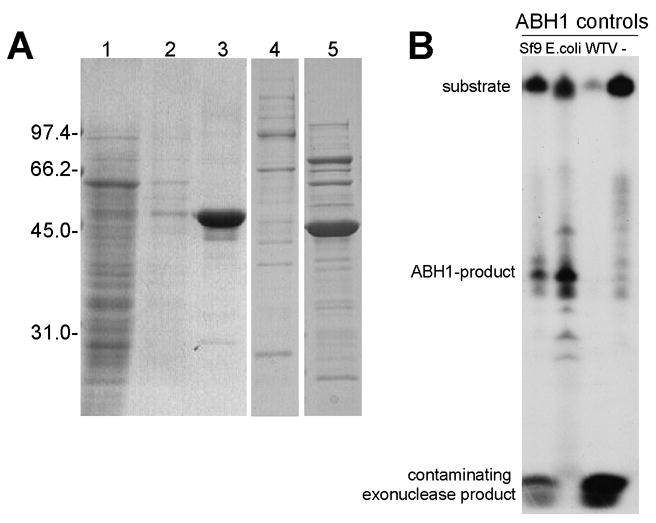
ABH1 purified from Sf9 cells exhibits AP-specific DNA cleavage activity. (A) Analysis of fractions obtained during purification of ABH1 from Sf9 cells. Insect cell extracts were incubated with Ni-NTA beads and fractions were examined by SDS-PAGE: lane 1, supernatant after removing beads; 2, wash of beads; 3, material bound to Ni-NTA beads; 4 and 5, material bound to Ni-NTA beads (after wash) from Sf9 cells expressing wild-type virus and from E. coli cells expressing His-tagged ABH1, respectively. Proteins were visualized by staining with Coomassie blue. The slight difference in sizes of the two His-tagged proteins is due to different linkers between the His-tag and the start residue of ABH1. (B) Activity assays of ABH1 purified from Sf9-cells. ABH1 purified from Sf9 cells or E. coli cells, along with Ni-NTA-enriched sample from insect cells containing wild type virus or no added ABH1, were incubated with end-labeled oligo3+3c and UDG for one hour at 37 °C. The reaction was stopped by addition of proteinase K and incubation at 65 °C for 15 min. The samples were loaded on a 20% denaturing polyacrylamide gel and the labeled products visualized by exposing to HyBlot film.
3.2 Cofactors and cosubstrate requirements
The Fe2+, 2-oxoglutarate, and ascorbate requirements of ABH1’s DNA cleavage activity were examined. These cofactors are needed for the reactivity of Fe2+/2-oxoglutarate dioxygenases that coordinate Fe2+ via a 2-His-1-carboxylate motif [17,24], a feature that is conserved in both human and murine ABH1. The addition of EDTA had no effect on the ability of ABH1 to cleave the oligonucleotide substrate, in contrast to the inhibitory effect of EDTA for the dioxygenase activities of many members of this enzyme family. Moreover, the exclusion of Fe2+, 2-oxoglutarate, and ascorbate from the reaction did not hinder activity; indeed, the presence of these additives appeared somewhat inhibitory to UDG or ABH1 (Supplemental Fig. 3A). Similarly, three point variants of potential Fe2+ ligands (H231A, D233A, and H287A), shown to be inactive for the demethylase activity [8], were unaffected in their ability to cleave abasic sites (Supplemental Fig. 3B). Furthermore, metal analysis by inductively-coupled plasma emission-mass spectrometry confirmed that Fe2+ was absent in purified protein. We conclude that, in contrast to the AlkB, ABH1, ABH2, or ABH3 demethylase activities, the AP-specific DNA cleaving activity of ABH1 does not require Fe2+, 2-oxoglutarate, or ascorbate as cofactors. Moreover, the ABH1 activity was unaffected by added Mg2+, Mn2+, or Zn2+.
3.3 ABH1 is an AP-lyase that catalyzes a β-elimination reaction
Enzymes that cleave at abasic sites can operate by either of two mechanistically distinct processes (Fig. 4A). Hydrolases, such as the human AP endonuclease 1 (Ape1), cleave to the 5′ side of the lesion while lyases, such as E. coli endonuclease III (EndoIII), employ a β-elimination reaction to yield the 3′-α,β-unsaturated aldehyde, trans-4-hydroxy-2-pentenal 5-phosphate [23,25]. We sought to distinguish which of the two mechanisms is utilized by ABH1 using two approaches.
Fig. 4.

ABH1’s DNA-cleaving reaction mechanism. (A) Comparison of AP-hydrolase and AP-lyase mechanisms. (B) Activity of ABH1 and other AP-cleavage enzymes with modified abasic sites. The oligo2 and AP-oligo2c were synthesized with THF on both strands at the position of the dU and annealed. The activity of ABH1, EndoIII, and Ape1 with these substrates (0.4 μg) was compared to the unmodified AP-oligonucleotide (5 μM ABH1, 90 min at 37 °C). (C) Analysis of ABH1 product size. Oligo1 with 5′-32P was annealed to the complementary strand, treated with UDG, and cleavage products were compared for ABH1, ApeI, and EndoIII. (D) Trapping of intermediates with NaBH4. ABH1 (5 μM), EndoIII (1 unit), and a non-enzyme control were incubated with 0.1 μM 5′-32P substrates (lanes 1 and 2, oligo3 annealed to either oligo3c or oligo3c2; lane 3, ss-oligo3) in the presence of UDG and NaBH4, and analyzed by SDS-PAGE. DNA examined without added protein migrates off the bottom of the gel.
First, the reactivity of ABH1 was examined with oligonucleotides containing THF, a modified AP-site. The β-elimination reaction, but not the hydrolase activity, is prevented by replacing the abasic site with this reduced analog [26]. The ABH1 activity was abolished in the THF-containing oligonucleotide, similar to that for EndoIII and unlike Ape1, providing evidence that the cleavage reaction proceeds according to lyase-type chemistry (Fig. 4B).
As an independent approach to assess the cleavage mechanism, the size of the product resulting from ABH1 activity was examined by using 32P-end-labeled samples and PAGE. 5′-[32P]-labeled ds-oligonucleotides containing the AP site were incubated with ABH1, Ape1, or EndoIII and the 5′ products were compared (Fig. 4C). The ABH1 product is clearly larger than that of Ape1, but also is slightly larger than that of EndoIII. Additional studies revealed the ABH1-derived 5′ product to be distinct from that obtained with EndoVIII, which catalyzes a β-elimination followed by a δ-elimination to leave a 3′ phosphate (Supplemental Fig. S4). Furthermore, treatment of these samples with polynucleotide kinase resulted in a shift in position of the EndoVIII product, consistent with removal of a 3′-terminal phosphate by this phosphatase, whereas the migration behavior of products from ABH1 and other enzymes were unchanged (Supplemental Fig. S4). These results corroborate the reaction of ABH1 as a lyase that catalyzes β-elimination, but not δ-elimination, and produces a product lacking a 3′ phosphate. We attribute the slight increases in apparent sizes of the substrate and products for the ABH1 samples, compared to those of Ape1 and EndoIII, to tight binding of ABH1 fragments to the oligonucleotides (see 4. Discussion).
Having shown that ABH1 cleaves DNA at abasic sites by a lyase mechanism, we tested whether a covalent enzyme-DNA complex could be trapped by chemical reduction as is typical for other enzymes catalyzing β-elimination reactions [27]. When ABH1 was incubated with 5′-32P labeled substrate containing an AP-site in the presence of NaBH4, a complex was observed migrating at a larger apparent molecular weight than ABH1 alone during SDS-PAGE (Fig. 4D). Enzyme-DNA complexes of similar sizes were observed when ABH1 was trapped with ds-DNA containing one or two AP-sites, and a distinct species was found with ss-DNA containing one AP-site. The appearance of a second complex in each of the lyase-containing samples may arise from incomplete denaturation of the samples and has been observed by others [25,27]. This result probably can be attributed to a complex consisting of two proteins bound to the DNA, as seen in the band at a higher molecular weight in the ABH1 samples, or to a protein-product complex as seen for EndoIII with a slower migrating band than the protein-substrate complex. When ABH1 was treated with the reducing agent in the presence of DNA lacking an AP-site, no complexes were formed. Analogous results were obtained using enzyme obtained from Sf9 cells (data not shown). These findings confirm that ABH1 is an AP-lyase that uses a covalent catalytic mechanism to cleave at AP-sites according to a β-elimination mechanism.
3.4 Turnover, stoichiometry, and oligonucleotide specificity of ABH1’s AP-lyase activity
The DNA cleavage assay was used to examine the kinetics of the lyase activity of ABH1. When ABH1 was present in five-fold molar excess over the ds-DNA substrate, product formation was approximately linear for forty min before starting to level off (Fig. 5A). Essentially all of the substrate was converted to product by 3 h if one accounts for the 0.2 μM AP-site cleavages arising from the slow rate of spontaneous chemical cleavage under these conditions [23]. The specific activity calculated for ABH1 under these conditions, calculated on the basis of the initial rate, was 0.070 ± 0.004 nmol of AP-sites cleaved per min per mg protein.
Fig. 5.
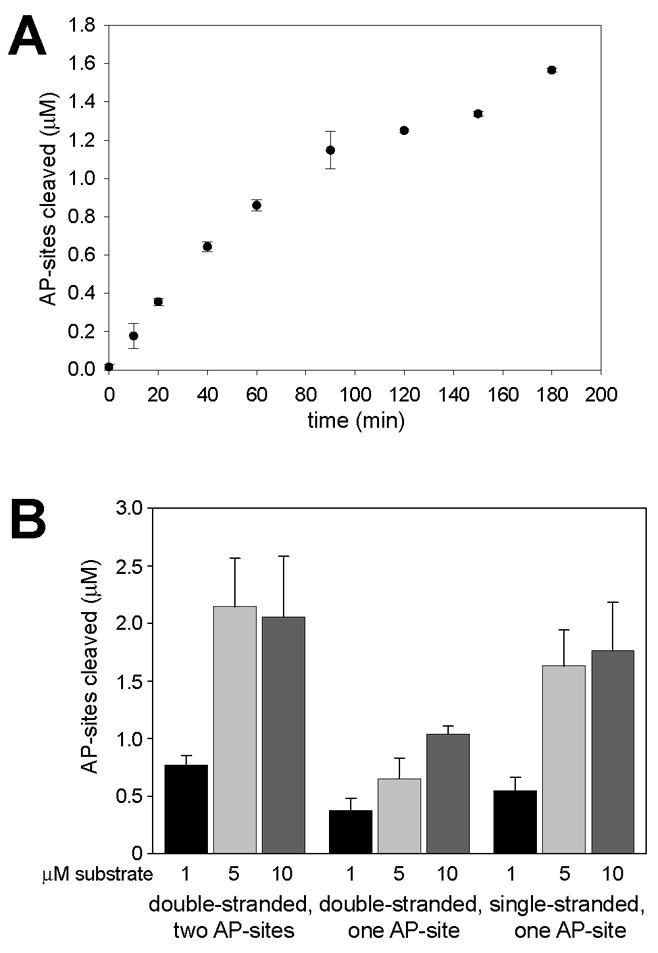
Time dependence and oligonucleotide specificity of ABH1 AP lyase activity. (A) Time dependence. ABH1 (5 μM) was incubated with oligo3+3c (1 μM) plus UDG, and aliquots were stopped at the indicated time points by addition of proteinase K and incubation at 65 °C. The experiment was performed with two independently expressed and purified ABH1 batches and activities were measured with each batch twice. (B) Substrate specificity and concentration dependence. ABH1 (5 μM) was incubated with UDG and ds-DNA (oligo3 annealed to either oligo3c or oligo3c2) or ss-DNA (oligo3) at the indicated concentrations (black bars, 1 μM; light grey, 5 μM; dark grey 10 μM) for one h at 37 °C. All activities were assayed in duplicate with at least three different preparations of ABH1. The measurement of AP-sites cleaved for ds-DNA with two AP-sites is multiplied by two to account for the two cleavages required to give rise to the product. The error bars represent standard deviations.
The lyase activity of ABH1 was examined at greater substrate-to-enzyme ratio, using a one h time point (Fig. 5B, left). Product formation increased nearly three-fold (corresponding to an initial rate of at least 0.16 ± 0.031 nmol of AP-sites cleaved per min per mg protein when enzyme and substrate were in equimolar amounts (i.e., 5-fold over the initial ratio), but turnover did not further increase when the substrate concentration was again doubled to become 2-fold over the enzyme concentration. Indeed, the concentration of product formed was always substoichiometric to the concentration of enzyme. We attribute these results to the formation of a tight complex between enzyme and product (see 4. Discussion).
The demethylase activity of ABH1 was reported to act exclusively on ss-DNA [8], in contrast to the AP-lyase activity described above. To further explore the AP lyase oliognucleotide specificity, we incubated ABH1 with ds-DNA containing an AP-site on one strand and with AP-containing ss-DNA to compare with the standard (ds-DNA with dual AP-sites) activity (Fig. 5B). ABH1 catalyzed strand cleavage with all three substrates, but showed a preference for ds-DNA with two AP-sites in close proximity on opposite strands over ss-DNA or ds-DNA with only one AP-site. The substrate preference did not change when the DNA concentration was varied.
4 Discussion
Our studies show the unexpected finding that human ABH1 exhibits DNA cleaving activity at abasic sites. ABH1 cleaves 3′ of the AP sites using a lyase mechanism as shown by the inability of enzyme to utilize THF-containing DNA as a substrate, the size of the product formed, and the ability to trap a covalent crosslink between enzyme and substrate/product with NaBH4. Several lines of evidence demonstrate the lyase activity is inherent to ABH1 and not due to a contaminating endonuclease. The lyase activity was coincident with ABH1 when chromatographed on two different resins beyond the initial Ni-NTA purification. In particular, the lyase activity matches the size of monomeric ABH1 and cannot accommodate a contaminating protein that forms a complex with ABH1. Furthermore, similar levels of lyase activity were associated with ABH1 whether produced in bacterial or Sf9 insect cells. Thus, ABH1 exhibits AP-lyase activity in addition to its previously documented demethylase activity [8].
In contrast to the 3-meC demethylase activity reported for ABH1 [8], its AP lyase activity is independent of Fe2+ or 2-oxoglutarate. Both activities require DNA binding, and our assays confirm that ABH1 binds to ds-DNA and show that it prefers binding to DNA containing an abasic site over native or dU-containing oligonucleotides. Contrary to the demethylase activity that was limited to ss-DNA the lyase activity of ABH1 cleaves both ss-DNA and ds-DNA at abasic sites, with the greatest activity found using ds-DNA containing two AP-sites. These findings suggest that the AP lyase and demethylase activities of ABH1 utilize distinct active sites.
The low level of AP lyase activity exhibited by ABH1 under the conditions tested here mirrors the low level of 3-meC demethylase activity previously measured for this protein [8]. For example, Westbye et al. reported that approximately half of a fluorescently-labeled 49-mer containing a single 3-meC was cleaved after a 15 min incubation with ABH1 in 20-fold excess over substrate. We considered various explanations to account for these low turnover numbers. One possibility is that the low activities reflect improper folding or incorrect post-translational processing due to heterologous expression of a mammalian enzyme in E. coli. For example, a recent study of activation-induced cytidine deaminase reported that only 1 in 5000 copies of the protein was active when synthesized in E. coli [28]. We showed, however, that the AP-lyase activity of ABH1 isolated from E. coli cells approximated that of the enzyme purified from Sf9 insect cells, decreasing the likelihood of this explanation. An alternative possibility is that the low activity arises due to the absence of a critical protein-protein interaction. Many DNA repair enzymes, such as those involved in base excision repair (BER) or DSB repair, require interactions with other proteins for activation, DNA binding, or dissociation from the DNA [29–34]. As described below, we have supporting evidence for the slow dissociation of ABH1 from its product.
Several findings from our in vitro investigations suggest that ABH1, once bound to the DNA, is not easily removed from its product. Only after proteinase K digestions at elevated temperatures could the product be visualized by gel electrophoresis and even after this treatment, small peptides appear to be associated with the DNA as indicated by the slower migration of the oligonucleotides in the ABH1 samples. Furthermore, ABH1 activity does not increase when the substrate-to-enzyme ratio increases from one to two. In addition, the presence of ABH1 protects the DNA product from exonuclease digestion in enriched samples from insect cells. Taken together, these findings suggest that isolated ABH1 catalyzes a single turnover reaction under the conditions examined, with the product being tightly bound to the protein. In this context, it is of interest that ABH1 prefers ds-DNA containing two AP-sites over ds-DNA containing one AP-site allowing ABH1 to introduce a DSB. Stoichiometric turnover and tight binding of ABH1 to the product of the DSB in vivo may be important for protecting the cleaved DNA ends from degradation until the DNA repair machinery can guarantee proper repair. Binding of DNA-modifying enzymes to DNA products has been reported for several mammalian glycosylases [29,35,36,37]. Thus the tight binding of ABH1 to the DNA cleavage products, and the consequently low(er) turnover rate, might be intrinsic to the enzyme and crucial for cell survival.
To our knowledge, human ABH1 is the first mammalian AlkB-homologue shown to exhibit an activity other than demethylation. Possibly related to this finding is the observation that an ABH1 isoform exists which only consists of the N-terminal 185 amino acid residues of the protein, thus removing the putative Fe2+ ligands and parts of the 2-oxoglutarate dioxygenase region. Furthermore, while the first 72 residues of ABH1 are conserved among ABH1 homologues, they exhibit no significant homology to other mammalian AlkB homologues. This result could indicate an important, demethylase-independent role for this region of the protein. We attempted to test whether the AP lyase activity is localized within a terminal domain by creating mutants of ABH1 truncated at each end. Unfortunately, these variants (missing 71 amino acids at the N-terminus with Val-72 changed to Met or missing 53 amino acids at the C-terminus) were produced as inclusion bodies in E. coli. Of interest, an isoform of ABH2 also is known to exist [20]. In that case, a splice variant lacking exon 3 results in a product of 157 amino acids missing the C-terminal 2-oxoglutarate dioxygenase domain. ABH8 also is thought to have different isoforms because the initially deposited sequence of 717 bp appears to be a splice variant of a longer form (~2 kbp) that encodes at least three distinct domains; namely, an RNA recognition motif, a methyltransferase domain as well as the 2-oxoglutarate dioxygenase domain [20]. These findings suggest that several AlkB homologues exhibit activities different from, or in addition to, the demethylase activity and which are independent of the dioxygenase domain.
Our findings lead us to consider hypotheses about the in vivo roles of ABH1. AP-endonuclease activity is important in all DNA repair pathways. In BER, a monofunctional glycosylase removes the damaged base leading to the AP-site which is then cleaved [38–41]. In mammalian cells, Ape1 is the main enzyme responsible for this activity. It is possible that ABH1 is involved in an alternative BER pathway, but no evidence has been found for an additional activity and the ubiquitous expression of Ape1 makes this an unlikely hypothesis. Alternatively, ABH1 could play a role in mitochondrial DNA repair. This is in agreement with recent studies that demonstrated a mitochondrial localization of ABH1. Mitochondria are exposed to oxidative stress and several mitochondrial DNA repair pathways have been described [42,43]. Due to the high susceptibility to oxidative damage, BER is an important repair pathway and several nuclear enzymes such as UDG, 8-oxoguanine DNA glycosylase/lyase OGG1 or Ape1 have been shown to have a mitochondrial form and to play a role in mitochondrial DNA repair. This raises the possibility that ABH1 also functions in mitochondrial BER in addition to any role in direct DNA repair by oxidative demethylation. In contrast, other authors had noted that ABH1 strongly interacts with Mrj, an essential placental gene product that mediates gene repression by recruitment of class II histone deacetylase, indicating a possible role in gene regulation [18]. It is interesting to point out that Ape1 also is a multifunctional enzyme and acts as a transcriptional activator in addition to its AP-endonuclease and 3′-diesterase activity [44,45]. To conclude, ABH1 has two different catalytic activities, an AP-lyase and a demethylase activity. Our current efforts are focused on investigating ABH1’s in vivo functions.
Supplementary Material
Acknowledgments
We thank Megan Andrzejak, Andrea Silva, Larissa Reifur, and Dr. Donna Koslowsky for assistance with selected experiments, Dr. Barbara Sedgwick for plasmid pBAR67, and Dr. Kefei Yu for comments on the manuscript. This work was supported by the National Institutes of Health (GM063582 to R.P.H. and AI048758 to K.M.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wood RD, Mitchell M, Lindahl T. Human DNA repair genes. Mut Res. 2005;577:275–283. doi: 10.1016/j.mrfmmm.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 2.Wood RD, Mitchell M, Sgouros J, Lindahl T. Human DNA repair genes. Science. 2001;291:1284–1289. doi: 10.1126/science.1056154. [DOI] [PubMed] [Google Scholar]
- 3.Duncan T, Trewick SC, Koivisto P, Bates PA, Lindahl T, Sedgwick B. Reversal of DNA alkylation damage by two human dioxygenases. Proc Natl Acad Sci. 2002;99:16660–16665. doi: 10.1073/pnas.262589799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Falnes PØ, Johansen RF, Seeberg E. AlkB-mediated oxidative demethylation reverses DNA damage in Escherichia coli. Nature. 2002;419:178–182. doi: 10.1038/nature01048. [DOI] [PubMed] [Google Scholar]
- 5.Trewick SC, Henshaw TF, Hausinger RP, Lindahl T, Sedgwick B. Oxidative demethylation by Escherichia coli AlkB directly reverts DNA base damage. Nature. 2002;419:174–178. doi: 10.1038/nature00908. [DOI] [PubMed] [Google Scholar]
- 6.Kurowski MA, Bhagwat AS, Papaj G, Bujnicki JM. Phylogenomic identification of five new human homologs of the DNA repair enzyme AlkB. BMC Genomics. 2003;4:48. doi: 10.1186/1471-2164-4-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sanchez-Pulido L, Andrade-Navarro MA. The FTO (fat mass and obesity associated) gene codes for a novel member of the non-heme dioxygenase superfamily. BMC Biochem. 2007;8:23. doi: 10.1186/1471-2091-8-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Westbye MP, Feyzi E, Aas PA, Vagbo CB, Talstad VA, Kavli B, Hagen L, Sundheim O, Akbari M, Liabakk NB, Slupphaug G, Otterlei M, Krokan HE. Human AlkB homolog 1 is a mitochondrial protein that demethylates 3-methylcytosine in DNA and RNA. J Biol Chem. 2008;283:25046–25056. doi: 10.1074/jbc.M803776200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ougland R, Zhang CM, Liiv A, Johansen RF, Seeberg E, Hou YM, Remme J, Falnes PØ. AlkB restores the biological function of mRNA and tRNA inactivated by chemical methylation. Molec Cell. 2004;16:107–116. doi: 10.1016/j.molcel.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 10.Shimada K, Nakamura M, Anai S, De Velasco M, Tanaka M, Tsujikawa K, Ouji Y, Konishi N. A novel human AlkB homologue, ALKBH8, contributes to human bladder cancer progression. Cancer Res. 2009;69:3157–3164. doi: 10.1158/0008-5472.CAN-08-3530. [DOI] [PubMed] [Google Scholar]
- 11.Gerken T, Girard CA, Tung YC, Webby CJ, Saudek V, Hewitson KS, Yeo GS, McDonough MA, Cunliffe S, McNeill LA, Galvanovskis J, Rorsman P, Robins P, Prieur X, Coll AP, Ma M, Jovanovic Z, Farooqi IS, Sedgwick B, Barroso I, Lindahl T, Ponting CP, Ashcroft FM, O’Rahilly S, Schofield CJ. The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science. 2007;318:1469–1472. doi: 10.1126/science.1151710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jia G, Yang CG, Yang S, Jian X, Yi C, Zhou Z, He C. Oxidative demethylation of 3-methylthymine and 3-methyluracil in single-stranded DNA and RNA by mouse and human FTO. FEBS Lett. 2008 doi: 10.1016/j.febslet.2008.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fischer J, Koch L, Emmerling C, Vierkotten J, Peters T, Bruning JC, Ruther U. Inactivation of the Fto gene protects from obesity. Nature. 2009;458:894–898. doi: 10.1038/nature07848. [DOI] [PubMed] [Google Scholar]
- 14.Sundheim O, Vågbo CB, Bjorås M, Sousa MML, Talstad V, Aas PA, Drablos F, Krokan HE, Tainer JA, Slupphaug G. Human ABH3 structure and key residues for oxidative demethylation to reverse DNA/RNA damage. EMBO J. 2006;25:3389–3397. doi: 10.1038/sj.emboj.7601219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang CG, Yi C, Duguid EM, Sullivan CT, Jian X, Rice PA, He C. Crystal structures of DNA/RNA repair enzymes AlkB and ABH2 bound to dsDNA. Nature. 2008;452:961–965. doi: 10.1038/nature06889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu B, Edstrom WC, Hamuro Y, Weber PC, Gibney BR, Hunt JF. Crystal structures of catalytic complexes of the oxidative DNA/RNA repair enzyme AlkB. Nature. 2006;439:879–884. doi: 10.1038/nature04561. [DOI] [PubMed] [Google Scholar]
- 17.Simmons JM, Müller TA, Hausinger RP. FeII/α-ketoglutarate hydroxylases involved in nucleobase, nucleoside, nucleotide, and chromatin metabolism. Dalton Trans. 2008:5132–5142. doi: 10.1039/b803512a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pan Z, Sikandar S, Witherspoon M, Dizon D, Nguyen T, Benirschke K, Wiley C, Vrana P, Lipkin SM. Impaired placental trophoblast lineage differentiation in Alkbh1(−/−) mice. Dev Dynamics. 2008;237:316–327. doi: 10.1002/dvdy.21418. [DOI] [PubMed] [Google Scholar]
- 19.Su AI, Cooke MP, Ching KA, Hakak Y, Walker JR, Wiltshire T, Orth AP, Vega RG, Sapinoso LM, Moqrich A, Patapoutian A, Hampton GM, Schultz PG, Hogenesch JB. Large-scale analysis of the human and mouse transcriptomes. Proc Natl Acad Sci. 2002;99:4465–4470. doi: 10.1073/pnas.012025199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsujikawa K, Koike K, Kitae K, Shinkawa A, Arima H, Suzuki T, Tsuchiya M, Makino Y, Furukawa T, Konishi N, Yamamoto H. Expression and sub-cellular localization of human ABH family molecules. J Cell Molec Med. 2007;11:1105–1116. doi: 10.1111/j.1582-4934.2007.00094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rasband WS. ImageJ. U. S. National Institutes of Health; Bethesda, Maryland, USA: 1997–2008. http://rsb.info.nih.gov/ij/ [Google Scholar]
- 22.Wyatt MD, Pittman DL. Methylating agents and DNA repair responses: Methylated bases and sources of strand breaks. Chem Res Toxicol. 2006;19:1580–1594. doi: 10.1021/tx060164e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lhomme J, Constant JF, Demeunynck M. Abasic DNA structure, reactivity, and recognition. Biopolymers. 1999;52:65–83. doi: 10.1002/1097-0282(1999)52:2<65::AID-BIP1>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 24.Hausinger RP. Fe(II)/α-ketoglutarate-dependent hydroxylases and related enzymes. Crit Rev Biochem Mol Biol. 2004;39:21–68. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 25.Piersen CE, McCullough AK, Lloyd RS. AP lyases and dRPases: commonality of mechanism. Mut Res. 2000;459:43–53. doi: 10.1016/s0921-8777(99)00054-3. [DOI] [PubMed] [Google Scholar]
- 26.Takeshita M, Chang CN, Johnson F, Will S, Grollman AP. Oligodeoxynucleotides containing synthetic abasic sites. Model substrates for DNA polymerases and apurinic/apyrimidinic endonucleases. J Biol Chem. 1987;262:10171–10179. [PubMed] [Google Scholar]
- 27.Piersen CE, Prasad R, Wilson SH, Lloyd RS. Evidence for an imino intermediate in the DNA polymerase beta deoxyribose phosphate excision reaction. J Biol Chem. 1996;271:17811–17815. doi: 10.1074/jbc.271.30.17811. [DOI] [PubMed] [Google Scholar]
- 28.Larijani M, Petrov AP, Kolenchenko O, Berru M, Krylov SN, Martin A. AID associates with single-stranded DNA with high affinity and a long complex half-life in a sequence-independent manner. Mol Cell Biol. 2007;27:20–30. doi: 10.1128/MCB.00824-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baldwin MR, O’Brien PJ. Human AP endonuclease I stimulates multiple-turnover base excision by alkyladenine DNA glycosylase. Biochemistry. 2009 doi: 10.1021/bi900517y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ilina ES, Lavrik OI, Khodyreva SN. Ku antigen interacts with abasic sites. Biochim Biophys Acta. 2008;1784:1777–1785. doi: 10.1016/j.bbapap.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 31.Waters TR, Gallinari P, Jiricny J, Swann PF. Human thymine DNA glycosylase binds to apurinic sites in DNA but is displaced by human apurinic endonuclease 1. J Biol Chem. 1999;274:67–74. doi: 10.1074/jbc.274.1.67. [DOI] [PubMed] [Google Scholar]
- 32.Weterings E, Chen DJ. The endless tale of non-homologous end-joining. Cell Res. 2008;18:114–124. doi: 10.1038/cr.2008.3. [DOI] [PubMed] [Google Scholar]
- 33.Wyman C, Kanaar R. DNA double-strand break repair: all’s well that ends well. Annu Rev Genet. 2006;40:363–383. doi: 10.1146/annurev.genet.40.110405.090451. [DOI] [PubMed] [Google Scholar]
- 34.Zharkov DO, Rosenquist TA, Gerchman SE, Grollman AP. Substrate specificity and reaction mechanism of murine 8-oxoguanine-DNA glycosylase. J Biol Chem. 2000;275:28607–28617. doi: 10.1074/jbc.M002441200. [DOI] [PubMed] [Google Scholar]
- 35.Waters TR, Gallinari P, Jiricny J, Swann PF. Human thymine DNA glycosylase binds to apurinic sites in DNA but is displaced by human apurinic endonuclease 1. J Biol Chem. 1999;274:67–74. doi: 10.1074/jbc.274.1.67. [DOI] [PubMed] [Google Scholar]
- 36.Zharkov DO, Rosenquist TA, Gerchman SE, Grollman AP. Substrate specificity and reaction mechanism of murine 8-oxoguanine-DNA glycosylase. J Biol Chem. 2000;275:28607–28617. doi: 10.1074/jbc.M002441200. [DOI] [PubMed] [Google Scholar]
- 37.Fitzgerald ME, Drohat AC. Coordinating the initial steps of base excision repair. Apurinic/apyrimidinic endonuclease 1 actively stimulates thymine DNA glycosylase by disrupting the product complex. J Biol Chem. 2008;283:32680–32690. doi: 10.1074/jbc.M805504200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.David SS, O’Shea VL, Kundu S. Base-excision repair of oxidative DNA damage. Nature. 2007;447:941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dianov GL, Sleeth KM, Dianova, Allinson SL. Repair of abasic sites in DNA. Mutat Res. 2003;531:157–163. doi: 10.1016/j.mrfmmm.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 40.Hazra TK, Das A, Das S, Choudhury S, Kow YW, Roy R. Oxidative DNA damage repair in mammalian cells: a new perspective. DNA Repair (Amst) 2007;6:470–480. doi: 10.1016/j.dnarep.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krokan HE, Nilsen H, Skorpen F, Otterlei M, Slupphaug G. Base excision repair of DNA in mammalian cells. FEBS Lett. 2000;476:73–77. doi: 10.1016/s0014-5793(00)01674-4. [DOI] [PubMed] [Google Scholar]
- 42.Larsen NB, Rasmussen M, Rasmussen LJ. Nuclear and mitochondrial DNA repair: similar pathways? Mitochondrion. 2005;5:89–108. doi: 10.1016/j.mito.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 43.Stuart JA, Brown MF. Mitochondrial DNA maintenance and bioenergetics. Biochim Biophys Acta. 2006;1757:79–89. doi: 10.1016/j.bbabio.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 44.Fritz G. Human APE/Ref-1 protein. Int J Biochem Cell Biol. 2000;32:925–929. doi: 10.1016/s1357-2725(00)00045-5. [DOI] [PubMed] [Google Scholar]
- 45.Tell G, Quadrifoglio F, Tiribelli C, Kelley MR. The Many Functions of APE1/Ref-1: Not Only a DNA Repair Enzyme. Antioxid Redox Signal. 2009;11:1–19. doi: 10.1089/ars.2008.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.