

Abstract
HIV-1 viral budding involves binding of the viral Gagp6 protein to the ubiquitin E2 variant domain of the human tumor susceptibility gene 101 protein (Tsg101). Recognition of p6 by Tsg101 is mediated in part by a proline-rich motif that contains the sequence “Pro-Thr-Ala-Pro” (“PTAP”). Using the p6-derived 9-mer sequence “PEPTAPPEE”, we had previously improved peptide binding affinity by employing N-alkylglycine (“peptoid”) residues. The current study applies ring-closing metathesis macrocyclization strategies to Tsg101-binding peptide-peptoid hybrids as an approach to stabilize binding conformations and to observe the effects of such macrocyclization on Tsg101-binding affinity and bioavailability.
Viral release is a necessary component of HIV-1 replication. To be an efficient process, viral budding relies on recruitment of the ubiquitin E2 variant (UEV) domain of the human tumor susceptibility gene 101 protein (Tsg101) by major structural proteins of HIV-1.1,2 This entails the direct interaction of the Tsg101 UEV domain with a proline rich motif (PRM) within the viral Gagp6 protein that contains a conserved sequence, Pro-Thr/Ser-Ala-Pro [“P(T/S)AP”].3,4 Over-expression of the Tsg101 UEV inhibits virus release by interfering with processing of the p6 domains and this effect is abrogated by mutation within the P(T/S)AP binding site. In theory, blocking this critical Tsg101-p6 interaction could prevent viral budding and provide a basis for new targeted antiretroviral therapies.5,6 This is supported by the recent finding that inhibition of HIV budding can be achieved by cyclic peptides that interfere with Tsg101-Gag interactions.7
In an effort to develop Tsg101-binding inhibitors using the reported p6-derived 9-mer wild-type (WT) sequence “P1E2P3T4A5P6P7E8E9,” we had previously improved Tsg101-binding affinity by applying hydrazone- and hydrazide-library techniques.8 We also reported an oxime-based post solid-phase diversification approach to peptide library construction.9–11 However, the peptide mimetics resulting from this earlier work showed poor biovailability in cell-based experiments. Because poor cellular uptake was thought to contribute to the low bioavailability, peptides were conjugated with known membrane carrier antennapedia and HIV-Tat (48–60) peptides.12 Although these constructs did exhibit apparent enhanced cellular uptake, the Tsg101 binding affinity of the conjugates was seriously disrupted. Therefore, we sought an alternate approach that could improve cell bioavailability while maintaining Tsg101-binding affinity. We took note of the fact that cyclic versions of linear peptides can result in better cell-permeability. Additionally, appropriately restricting solution conformations through cyclization can also afford higher affinity.13–17 The objective of the current study was to apply RCM strategies to the WT 9-mer peptide and to observe the effects that such macrocyclization would have on Tsg101-binding affinity and cellular bioavailability.
Synthetic design
RCM macrocyclization requires the insertion into the parent peptide of two terminal alkene units that serve as ring-closing segments. Many examples of RCM macrocyclizations of peptides have been reported that rely on placement of alkenyl side chains onto the α-carbons of ring junction glycine residues. However, our recent examination of N-substituted glycines (NSGs) within the parent Tsg101 9-mer peptide8 suggested the potential utility N-alkenylglycine residues as ring-forming units. The use of NSGs is of note, because such residues constitute an important class of peptide mimetics termed “peptoids.”18 Since the overall macrocycle ring size and the insertion locations of the alkenyl chains that would most effectively provide the best combination of affinity, stability and bioavailability were not known, we envisioned placing the needed N-alkenylglycines within the N-terminal and C-terminal “P1E2” and “E8E9” sequences, respectively. This would allow RCM macrocyclization with maintenance of the critical “P3T4A5P6” recognition region. By employing a range of chain lengths in the N-alkenyl side chains, a series of macrocycles (1–4) could be constructed that would vary in ring size and location of ring-closure attachments (Figure 1). In this fashion, we could explore a variety of macrocycle structures in an attempt to arrive at optimum configurations. A similar generation of libraries of macrocyclic peptides has previously been described as an approach at interrogating bioactive conformations.19–21
Figure 1.
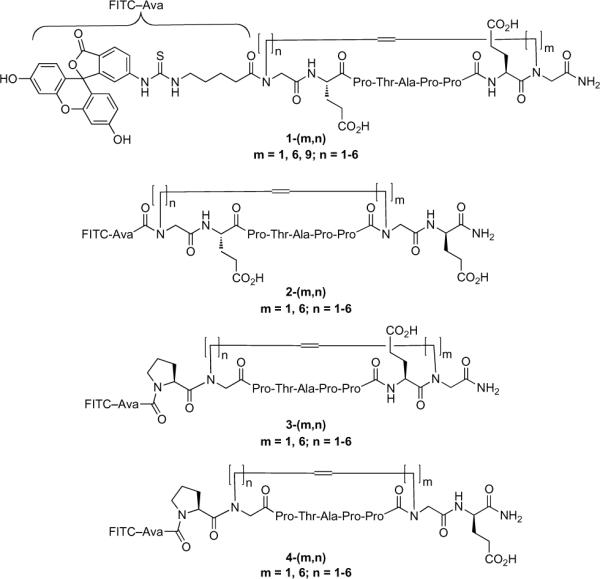
Structures of macrocycles prepared in the current study. Amino-terminal tagging with fluoroscein isothiocyanate (FITC) permitted the performance of fluorescence anisotropy-based Tsg101 binding assays.
Synthesis
The requisite ring junction-forming N-alkenylglycine residues were prepared as their N-Fmoc-protected derivatives (8a–8g, shown in Scheme 1). Treatment of methyl glycinate hydrochloride (6) with appropriate alkenyl bromides H2C=CH[CH2]nBr (5a–5g, where n = 1–6 and 9), in the presence of triethylamine in acetonitrile gave the corresponding N-alkenylglycine methyl esters (7a–7g). This reaction has previously been reported using alkali or alkali earth metal bases.22 Hydrolysis of the methyl ester (aqueous sodium hydroxide) followed by reaction with 9-fluorenylmethoxycarbonyl-N-hydroxysuccinimide (Fmoc-OSu) in the presence of NaHCO3 in aqueous dioxane gave desired reagents 8a–8g.
Scheme 1.
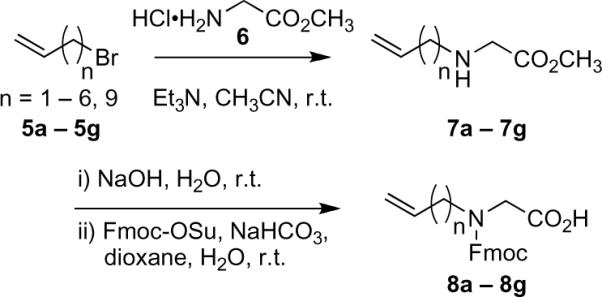
Synthesis of Fmoc-protected N-alkenylglycine residues, where a, n = 1 (over all yield 18%, based on the alkenyl bromide); b, n = 2 (13%); c, n = 3 (17%); d, n = 4 (23%); e, n = 5 (19%); f, n = 6 (21%); g, n = 9 (22%).
Synthesis of macrocycles 1–4 was by solid-phase methods using NovaSyn® TGR resin and standard Fmoc protocols. Side chain protection of glutamic acid residues was as their tert-butyl esters. The precursor open-chain peptides [9-(m,n)] (the use of “m” and “n” in compound numbering is described in Figure 1) were built up on the resin and amino-terminally acylated with an N-Fmoc 5-aminovaleric acid (N-Fmoc Ava) group. Various combinations of N-alkenylglycines were substituted for residues P1 and E9 (Scheme 2). The NFmoc Ava groups were then deprotected and the peptides were acylated using fluorescein isothiocyanate (FITC). Cleavage of the peptides from the resin (TFA) was accompanied by simultaneous side chain deprotection. HPLC purification provided the linear open-chain products 1'-(m,n). Similar protocols were followed in which N-alkenylglycines were used to replace residues P1 and E8; E2 and E9; E2 and E8 to yield linear open chain analogues 2'-(m,n), 3'-(m,n) and 4'-(m,n), respectively.
Scheme 2.
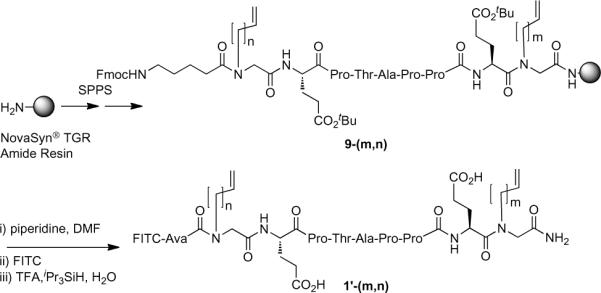
Solid-phase synthesis of open chain macrocycles 1'-(m,n).
Macrocyclizations of intermediates 9-(m,n) were conducted on the resin at room temperature in CH2Cl2 using Hoveyda-Grubbs 2nd generation RCM catalyst [(1,3-bis-(2,4,6-trimethylphenyl)-2-imidazolidinylidene)dichloro(o-isopropoxyphenylmethylene)ruthenium]23 (Scheme 3). Except for substrates containing short alkenyl side chains (the peptide-peptoid hybrids containing either two residues of 8a or one residue each of 8a and 8b), the open chain precursors were completely consumed in the RCM reaction. The efficiency of ring closure was sequence-dependent, with members from the 2-series giving the highest percentage of expected ring-closed product. Finally, amino terminal acylation with FITC as described above and cleavage of peptides from the resin provided the target macrocyclic peptides 1-(m,n), 2-(m,n), 3-(m,n) and 4-(m,n) (Figure 1). RCM is known to provide mixtures of alkenyl E/Z geometric isomers during the methathesis reaction. Previously reported examples of RCM macrocyclizations performed on large peptide substrates have highlighted the difficulties of obtaining defined E/Z geometries at the ring-forming alkenyl junction.24–28 Consistent with these prior studies, our current work does not explicitly define the geometries of RCM ring closure, nor does it extrapolate conformations of the resulting macrocycles.
Scheme 3.
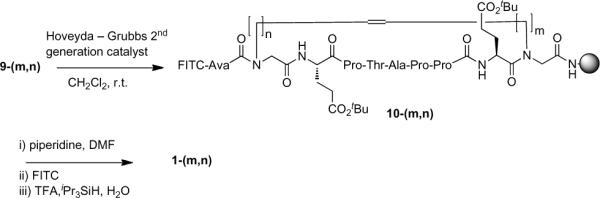
Solid-phase synthesis of target macrocycles 1-(m,n).
Tsg101 binding affinities
Determination of Tsg101 binding affinities was conducted using a fluorescence anisotropy assay.8 Open-chain peptide-peptoid hybrids 1', 2' and 3' exhibited affinities similar to the WT parent 9-mer peptide (Kd = 50–60 M).8 Macrocyclic peptide-peptoid hybrids 2 and 4 showed significantly reduced binding affinities, potentially indicating induced conformations that were unfavorable for binding. Certain members of the 1 and 3 series macrocycles showed binding affinities greater than those of the WT peptide (Table 1). The highest affinities were provided by 1-(6,6) (Kd = 19 μM), 1-(9,2) (Kd = 17 μM), and 1-(9,3) (Kd = 11 μM). The latter analogue showed 5-fold higher affinity than the WT peptide and 10-fold higher affinity than the open-chain analogue 1'-(1,1) (Kd = 120 μM).
Table 1.
Tsg 101 binding affinities of selected compounds.a
| No. | kd (μM) | No. | kd (μM) |
|---|---|---|---|
| 1-(6,4) | 38 | 1-(9,5) | 32 |
| 1-(6,5) | 24 | 1-(9,6) | 23 |
| 1-(6,6) | 19 | 3-(6,1) | 47 |
| 1-(9,2) | 17 | 3-(6,2) | 44 |
| 1-(9,3) | 11 | 3-(6,5) | 35 |
| 1(9,4) | 22 | 3-(6,6) | 31 |
Binding affinities were determined using a fluorescence anisotropy assay as described in reference 7.
Cellular uptake
An objective of the current study was to examine the effects of macrocyclization on cellular uptake. For α-helices, RCM macrocyclization can lead to more cell-permeable ligands.29–34 This effect has been attributed to several factors that include increased lipophillicity imparted by the ring-closing alkenyl hydrocarbon chain. It has also been shown that peptoids can serve as platforms for enhanced cellular uptake.35,36 This is due in part to the replacement of peptide backbone amide hydrogens by N-alkyl groups. Because macrocycles prepared within the current series contain both peptoid bonds and ring-closing alkenyl hydrocarbon chains, we anticipated an increase in cellular uptake. The inclusion of FITC tags on the synthetic peptide-peptoid hybrids permitted measurement of cellular uptake by fluorescence microscopy. Accordingly, a subset of peptide-peptoid hybrids was examined for cellular uptake. All macrocyclic analogues showed increased cellular uptake relative to control WT 9-mer peptide (Table 3). The best cellular uptake was shown by macrocycles having larger ring sizes, with 1-(6,6) showing the greatest uptake. In contrast, the corresponding ring-open macrocycle precursors did not show appreciable increased cell permeability as compared to the WT 9-mer peptide. This may indicate that increased hydrophobicity imparted by the alkenyl chains is not the sole reason for increased cell permeability of the macrocycles.
Our current report details the design, synthesis and biological evaluation of four sets of RCM-derived macrocyclic peptide-peptoid hybrids. Several distinct families were defined by different positioning of the ring-closing segments within the peptide backbones. To explore conformational effects of macrocyclization, the ring-closing alkenyl chains were varied to provide a total of 11 gradually enlarging macrocycles for each family. This gave ring sizes from 23 to 39 members. The best cellular uptakes were shown by larger ring sizes. Peptide-peptoid hybrid 1-(6,6) (total ring size of 39) exhibited the best combination of Tsg101-binding affinity (Kd = 19 μM) and cellular uptake. Work is in progress to evaluate the antiviral efficacy of macrocycles such as 1-(6,6) in whole cell systems.
Supplementary Material
Table 2.
Uptake of selected peptides into HeLa cells.a
| No. | Relative Uptakeb | No. | Relative Uptakeb |
|---|---|---|---|
| WT 9-mer | 74 ± 34 | ||
| 1'-(1,1) | 74 ± 14 | 1'-(6,6) | 35 ± 17 |
| 2'-(1,1) | 79 ± 30 | 2'-(6,6) | 40 ± 12 |
| 3'-(1,1) | 98 ± 32 | 3'-(6,6) | 42 ± 12 |
| 4'-(1,1) | 74 ± 22 | 4'-(6,6) | 45 ± 14 |
| 1-(1,3) | 175 ± 60 | 3-(1,3) | 229 ± 54 |
| 1-(6,2) | 100 ± 44 | 3-(6,2) | 77 ± 23 |
| 1-(6,6) | 328 ± 75 | 3-(6,6) | 199 ± 57 |
| 2-(1,3) | 146 ± 44 | 4-(1,3) | 272 ± 72 |
| 2-(6,2) | 100 ± 42 | 4-(6,2) | 76 ± 34 |
| 2-(6,6) | 194 ± 31 | 4-(6,6) | 200 ± 65 |
Data generated as described in the Experimental procedures.
Values calculated as the average fluorescence intensities quantified using softWoRx 3.7.0 software from cells treated with the indicated peptides.
At least 20 cells from 4–6 different fields each containing approximately 5 cells were quantified for fluorescent intensities. Data are shown as means ± average deviation.
Acknowledgements
This Work was supported in part by the Intramural Research Program of the NIH, Center for Cancer Research, NCI-Frederick and the National Cancer Institute, National Institutes of Health, under contract N01-CO-12400.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting information Supporting information associated with this article, including reaction yields and analytical data for products 8a – 8g, mass spectral data for peptides and peptide – peptoid hybrids and Tsg101 – binding affinities can be found, in the online version, at doi:10.1016/j.bmcl.2009.10.105.
References and notes
- 1.von Schwedler UK, Stuchell M, Mueller B, Ward DM, Chung H-Y, Morita E, Wang HE, Davis T, He G-P, Cimbora DM, Scott A, Kraeusslich H-G, Kaplan J, Morham SG, Sundquist WI. Cell. 2003;114:701. doi: 10.1016/s0092-8674(03)00714-1. [DOI] [PubMed] [Google Scholar]
- 2.Mazze FM, Degreve L. Acta Virologica. 2006;50:75. [PubMed] [Google Scholar]
- 3.Garrus JE, von Schwedler UK, Pornillos OW, Morham SG, Zavitz KH, Wang HE, Wettstein DA, Stray KM, Cote M, Rich RL, Myszka DG, Sundquist WI. Cell. 2001;107:55. doi: 10.1016/s0092-8674(01)00506-2. [DOI] [PubMed] [Google Scholar]
- 4.Freed EO. Trends Microbiol. 2003;11:56. doi: 10.1016/s0966-842x(02)00013-6. [DOI] [PubMed] [Google Scholar]
- 5.Turpin JA. Expert Rev. Anti-Infect. Ther. 2003;1:97. doi: 10.1586/14787210.1.1.97. [DOI] [PubMed] [Google Scholar]
- 6.Reeves JD, Piefer AJ. Drugs. 2005;65:1747. doi: 10.2165/00003495-200565130-00002. [DOI] [PubMed] [Google Scholar]
- 7.Tavassoli A, Lu Q, Gam J, Pan H, Benkovic SJ, Cohen S. ACS Chem. Biol. 2008;3:757. doi: 10.1021/cb800193n. [DOI] [PubMed] [Google Scholar]
- 8.Liu F, Stephen AG, Adamson CS, Gousset K, Aman MJ, Freed EO, Fisher RJ, Burke TR., Jr. Org. Lett. 2006;8:5165. doi: 10.1021/ol0622211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu F, Stephen AG, Waheed AA, Aman MJ, Freed EO, Fisher RJ, Burke TR., Jr. ChemBioChem. 2008;9:2000. doi: 10.1002/cbic.200800281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu F, Thomas J, Burke TR., Jr. Synthesis. 2008:2432. doi: 10.1055/s-2008-1078600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu F, Stephen AG, Fisher RJ, Burke TR., Jr. Bioorg. Med. Chem. Lett. 2008;18:1096. doi: 10.1016/j.bmcl.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.von Heijne G. J. Membrane Biol. 1990;115:195. doi: 10.1007/BF01868635. [DOI] [PubMed] [Google Scholar]
- 13.Piserchio A, Salinas GD, Li T, Marshall J, Spaller MR, Mierke DF. Chem. Biol. 2004;11:469. doi: 10.1016/j.chembiol.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 14.Horswill AR, Benkovic SJ. Cell Cycle. 2005;4:552. doi: 10.4161/cc.4.4.1585. [DOI] [PubMed] [Google Scholar]
- 15.Jiang S, Li Z, Ding K, Roller PP. Curr. Org. Chem. 2008;12:1502. [Google Scholar]
- 16.Linde Y, Ovadia O, Safrai E, Xiang Z, Portillo FP, Shalev DE, Haskell-Luevano C, Hoffman A, Gilon C. Biopolymers. 2008;90:671. doi: 10.1002/bip.21057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Antos JM, Popp MWL, Ernst R, Chew GL, Spooner E, Ploegh HL. J. Biol. Chem. 2009;284:16028. doi: 10.1074/jbc.M901752200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simon RJ, Kania RS, Zuckermann RN, Huebner VD, Jewell DA, Banville S, Ng S, Wang L, Rosenberg S, Marlow CK, Spellmeyer DC, Tan R, Frankel AD, Santi DV, Cohen FE, Bartlett PA. Proc. Nat. Acad. Sci. USA. 1992;89:9367. doi: 10.1073/pnas.89.20.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reichwein JF, Wels B, Kruijtzer JAW, Versluis C, Liskamp RMJ. Angew. Chem. Int. Ed. 1999;38:3684. doi: 10.1002/(sici)1521-3773(19991216)38:24<3684::aid-anie3684>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 20.Reichwein JF, Versluis C, Liskamp RMJ. J. Org. Chem. 2000;65:6187. doi: 10.1021/jo000759t. [DOI] [PubMed] [Google Scholar]
- 21.Davies JS. J. Pept. Sci. 2003;9:471. doi: 10.1002/psc.491. [DOI] [PubMed] [Google Scholar]
- 22.Cho JH, Kim BM. Tetrahedron Lett. 2002;43:1273. doi: 10.1016/S0040-4039(02)01113-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kingsbury JS, Harrity JPA, Bonitatebus PJ, Hoveyda AH. J. Am. Chem. Soc. 1999;121:791. [Google Scholar]
- 24.Reichwein JF, Wels B, Kruijtzer JAW, Versluis C, Liskamp RMJ. Angew. Chem. Int. Ed. 1999;38:3684. doi: 10.1002/(sici)1521-3773(19991216)38:24<3684::aid-anie3684>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 25.Chapman RN, Dimartino G, Arora PS. J. Am. Chem. Soc. 2004;126:12252. doi: 10.1021/ja0466659. [DOI] [PubMed] [Google Scholar]
- 26.Bernal F, Tyler AF, Korsmeyer SJ, Walensky LD, Verdine GL. J. Am. Chem. Soc. 2007;129:2456. doi: 10.1021/ja0693587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, Korsmeyer SJ. Science. 2004;305:1466. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chapman R, John L, Kulp I, Patgiri A, Kallenbach NR, Bracken C, Arora PS. Biochemistry. 2008;47:4189. doi: 10.1021/bi800136m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Henchey LK, Jochim AL, Arora PS. Curr. Opin. Chem. Biol. 2008;12:692–697. doi: 10.1016/j.cbpa.2008.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bird GH, Bernal F, Pitter K, Walensky LD. Methods Enzymol. (Drug-Nucleic Acid Interactions) 2008;446:369–386. doi: 10.1016/S0076-6879(08)01622-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kutchukian PS, Yang JS, Verdine GL, Shakhnovich EI. J. Am. Chem. Soc. 2009;131:4622–4627. doi: 10.1021/ja805037p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schafmeister CE, Po J, Verdine GL. J. Am. Chem. Soc. 2000;122:5891. [Google Scholar]
- 33.Wang D, Liao W, Arora PS. Angew. Chem. Int. Ed. 2005;44:6525. doi: 10.1002/anie.200501603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, Korsmeyer SJ. Science. 2004;305:1446. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Unciti-Broceta A, Diezmann F, Ou-Yang CY, Fara MA, Bradley M. Bioorg. Med. Chem. 2009;17:959. doi: 10.1016/j.bmc.2008.02.068. [DOI] [PubMed] [Google Scholar]
- 36.Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. Proc. Nat. Acad. Sci. USA. 2000;97:13003. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.