

To the Editor:
We describe a boy without genital ambiguities, with mild dysmorphic features and developmental delay, carrying a Y-negative de novo 46,XX,t (11;22)(q23;q11.2) karyotype. Three prior cases with XX-male sex reversal with involvement of chromosome 22 and total absence of SRY have been reported. The first patient was a true hermaphrodite with a duplication of 22q13.1-qtel and a deletion of 22p13-ptel originating from a pericentric inversion in the mother [Aleck et al., 1999]. The second patient had a deletion 22q11.2 with clear symptoms of Velocardiofacial/Shprintzen (VCFS) syndrome [Phelan et al., 2003]. Extensive molecular analysis of this case did not reveal additional cytogenetic aberrations beside the deletion of the VCFS region [Erickson et al., 2003]. The third patient carried a duplication of SOX10, an SRY-homologue located at 22q13.1, but appeared to be an isolated case among other cases of XX-males [Seeherunvong et al., 2004]. This is the fourth case involving chromosome 22, the second involving 22q11.2, but the first to occur with a balanced translocation. Although it was concluded in the three former cases that the chromosome 22q aberration was not the cause of the XX-male sex reversal, but rather a chance coincidence, it is conceivable that an undefined gene involved in sexual differentiation may reside in the 22q11.2–q13.1 region.
The propositus’ female karyotype was initially determined in amnion cells at 16 weeks of gestation. Subsequent chromosome analysis of the parents showed a normal karyotype implying that the fetus carried a de novo t(11;22)(q23;q11.2). Based on data from the literature, a risk of 1–5% for a congenital abnormality in the child was estimated. The parents decided to continue the pregnancy. At week 20, an ultrasound examination unexpectedly showed male sex organs with no signs of congenital adrenal hyperplasia (CAH) or other congenital malformations observed. Re-evaluation of the fetal karyotype, patient records and laboratory data files did not indicate irregularities. A possible sample processing error was excluded by confirming parent-child kinship. Using five forensic short tandem repeat loci with known allele frequency distribution in the Dutch population (VWA, D18S51, THO1, D3S1358, and D16S539), a combined match probability of <10−6 was calculated after haplotype analysis. To further exclude a sample processing error, a second amniocentesis was performed at 22 weeks of gestation that confirmed the 46,XX,t(11;22)(q23; q11.2) karyotype. The first and second amniotic fluid samples contained respectively 1.2 and 0.9 nmol/L testosterone and 0.89 and 0.31 mU/L follicle-stimulating hormone (FSH) levels consistent with male sex (testosterone >0.35 nmol/L, FSH level <3.0 mU/L). Presence of Y-chromosomal material was excluded after the negative test results of multiplex PCR with SRY and DAZ polymorphic markers and FISH with Y-whole chromosome painting probe (WCP-Y; Vysis, Downers Grove, IL) and SRY-specific probe pDP1335 (generously provided by David Page, Whitehead Institute, Cambridge, MA). Analysis of the PCR products was performed on an ABI Prism 3100 Genetic Analyzer (Applied Biosystems, Forest City, CA) and fragment size analysis was done with GeneScan v3.7.1. All FISH experiments were essentially performed according to standard procedures [Wiegant and Dauwerse, 1995]. Adrenogenital syndrome was excluded by mutation analysis of CYP11B1 that encodes 11-beta-hydroxylase, an important enzyme in the sex-steroid synthesis pathway. Based on the results of all prenatal tests (Table I), the parents decided to continue the pregnancy.
TABLE I.
Summary of the Cytogenetic, Biochemical and Molecular Investigations on Prenatal Material From the Carrier Patient
| Material | Karyotype GTG-band |
Testosterone nmol/L |
FSH mU/L | Parental markers |
CYP11B1 mutation |
SRY, DAZ markers |
SRY FISH | WCP-Y FISH |
|---|---|---|---|---|---|---|---|---|
| Amnion 1 | 46,XX,t(11;22) | 1.2 | 0.89 | Match | Absent | Absent | Absent | Absent |
| Amnion 2 | 46,XX,t(11;22) | 0.9 | 0.31 | n.d. | n.d. | Absent | Absent | Absent |
nmol/L, nanomol per liter; FSH, follicle stimulating hormone; mU/ml, milli-units per milliliter; FSH level is <3.0 mU/L inmales; CYP11B1, cytochrome P450 subfamily XIB polypeptide 1, 11-beta-hydroxylase; SRY, sex determining region Y; DAZ, deleted in azoospermia; WCP-Y, whole chromosome painting probe for chromosome Y; n.d., not determined.
The male baby was delivered at full-term gestation without complications. His Apgar score was 9 and 10 after, respectively, 1 and 5 minutes. He weighed 3,200 g, his testicles were descended, and there were no signs of external or internal female sex organs by ultrasound examination. Umbilical cord blood and fibroblasts were collected for postnatal cytogenetic testing. The 46,XX,t(11;22)(q23;q11.2) karyo-type was confirmed by GTG-banding and FISH analysis (Fig. 1) using whole chromosome painting probes WCP-11 and WCP-22 (Vysis). Multiplex-FISH experiments using a 24-color probe kit (24XCyte; MetaSystems, Altlussheim, Germany) and dedicated digital imaging (Cytovision v3.1.1; Applied Imaging, Newcastle upon Tyne, UK) excluded the presence of additional chromosome translocations (data not shown). At clinical follow-up after 6 months, the propositus had a normal male phenotype. After 16 months, however, delays in mental and physical development were observed. At 2 years of age, he showed the psychomotor development of a 1-year-old, and had mild dysmorphic facial features including epicanthic folds, low nasal bridge, and thin upper lip.
Fig. 1.
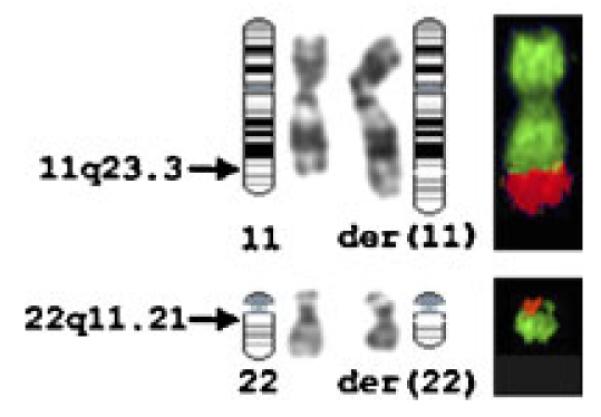
Analysis of the t(11;22) breakpoint by GTG-banding and FISH with whole chromosome painting probes WCP-11 (green) and WCP-22 (red). Normal and translocation chromosomes der(11) and der(22) were part of a metaphase cell derived from the propositus. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Male sex determination normally occurs in the presence of the SRY gene located on the Y-chromosome. Over 90% of male patients with an 46,XX karyotype contain Y-chromosomal material in their genome, most frequently because of a cryptic X;Y translocation within the Xp/Yp pseudoautosomal region. The remaining 10% of XX-males lack Y-chromosomal material. This group can be classified in three distinct phenotypic groups: normal males, males with genital ambiguity, or true hermaphrodites
To exclude classic 22q11.2 deletion syndrome, locus-specific FISH probes for HIRA (LSI 22q11.2, Vysis) and TBX1 (XX-91C, 1Mb-collection, Sanger Institute, Cambridge, UK) were used [Chieffo et al., 1997]. Both genes were present on the normal chromosome 22 and der(22) (Fig. 2A,B). Furthermore, probes RP11-189M8, RP11-566K1, RP11-251M9, and RP11-401F2 (BACPAC Resources, CHORI, Oakland, CA) were used to exclude a deletion of the second DiGeorge syndrome (DGS2) locus on 10p14 [Berend et al., 2000]. The DGS2 locus was present in two copies (Fig. 2C). In an attempt to explain the XX-sex reversal, we investigated 22q13.1 for a SOX10 duplication using fosmid probes WI2-1157M7, WI2-1323A3, and WI2-1412L4 (BACPAC Resources). Enhanced FISH signals, indicative for locus-duplication, were not observed (Fig. 2D).
Fig. 2.
FISH analysis of (A–C) DiGeorge/Velocardiofacial (DG/VCF) Syndrome and (D) SOX10 duplication. Metaphase chromosomes derived from the balanced t(11;22) carrier patient hybridized to different fluorescently labeled probes. A: LSI 22q11.2-deletion probe (red) specific for HIRA is present on the normal 22 and der(22), while the control probe specific for ARSA (green) localizes to 22q13.3 on the normal 22 and der(11). B: BAC XX-91C (green) specific for the DG/VCF-associated gene TBX1 is present on the normal 22 and der(22) and not on the normal 11 or der(11) (red). C: BAC RP11-251M9 (green) specific for the DG/VCF-II region is present in 10p14 of both chromosome 10 (red) homologues. The results of A–C indicate that none of the common DG/VCF regions were deleted in the propositus. D: Single copy signals of fosmid probe WI2-1412L4 (green) are on the normal 22 and the der(11) as indicated by centromere 11-specific probe (red). Similar results were obtained with fosmids WI2-1157M7 and WI2-1323A3. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Balanced translocation t(11;22)(q23;q11.2) is the most common human non-Robertsonian translocation [Fraccaro et al., 1980; Zackai and Emanuel, 1980]. Carriers are frequently found in families without phenotypic abnormalities. They are, however, at increased risk for spontaneous abortions due to chromosomal unbalanced offspring. Unbalanced karyotypes originating from 3:1 meiotic segregation lead to individuals carrying a supernumerary der(22) translocation chromosome and a specific phenotype for partial trisomy 22 [Emanuel Syndrome, OMIM #609029]. Male patients with Emanuel syndrome may have abnormal sex organs ranging from undescended testes to cryptorchidism/hypospadias [Schinzel et al., 1981]. Extensive molecular studies of the t(11;22) breakpoint region demonstrate long AT-rich palindromes residing at 22q11.2 and 11q23 that give rise to frequent non-allelic homologous recombination and subsequent double strand breaks [Kurahashi and Emanuel, 2001]. The low-copy repeat (LCR) regions also represent a genomic architecture that confers a high risk for de novo chromosomal rearrangements such as microdeletions, microduplications, or translocations involving 22q11.21 breakage [Spiteri et al., 2003].
We hypothesized that the formation of a de novo t(11;22) might have caused a cryptic duplication or deletion in the breakpoint region that might explain the sex reversal and the developmental delay. The 11;22 translocation breakpoints were therefore analyzed in detail by FISH, using twenty-seven neighboring probes spanning a 3.85 Mb interval at 22q11.21–q11.22 (Fig. 3 and see Online Supplement 1 at http://www.interscience.wiley.com/jpages/1552-4825/suppmat/index.html), and six probes for band 11q23.3 (Fig. 4 and see Online Supplement 2 at http://www.interscience.wiley.com/jpages/1552-4825/suppmat/index.html). FISH probes were obtained from the 1 Mb- and Tile Path-collection (kind gift of Nigel Carter, Sanger Institute, Cambridge, UK), and the RP-11 library (BACPAC Resources). All chromosome 22-specific probes gave FISH signals on the normal chromosome 22. Twenty-one chromosome 22-specific probes gave FISH signals on either the der(22) or the der(11), indicating loci out-side the breakpoint region. Six FISH probes positioned between 18.5 and 20.0 Mb of chromosome 22 showed a FISH signal on both the der(22) and the der(11), suggestive for spanning the breakpoint or a possible microduplication. However, five of these FISH probes (RP11-432P21, cHK89, RP11-585I18, p52f6, and RP11-750K14) contain repetitive portions of LCR22-B and one (RP11-892O8) shows overlap with LCR22-D (Fig. 3). The presence of multiple LCRs in the breakpoint region may have caused cross-hybridization of FISH clones to both sides of the breakpoint. Furthermore, small gaps between neighboring probes were not interrogated by FISH. Therefore, FISH analysis was not able to precisely localize the breakpoint on chromosome 22 or to undoubtedly conclude on the presence or absence of a microduplication or microdeletion.
Fig. 3.

Graphic overview of the positions of the chromosome 22-specific FISH probes with respect to low-copy repeats (LCR) B to E. Clones hybridizing to both the der(11) and the der(22) are depicted as speckled bars.
Fig. 4.

Graphic overview of the positions of the chromosome 11-specific FISH probes. Clones hybridizing to both the der(11) and the der(22) are depicted as speckled bars.
In classic familial t(11;22), the breakpoint in chromosome 22 is located in LCR22-B, which also contains a gap in the genome sequence assembly available at the time of manuscript preparation (NCBI genome build 35, May 2004 freeze). To delineate the translocation breakpoint in chromosome 22, FISH analysis was performed with cosmid probes c68a1 and c87f9 specific for, respectively, N41 and ZNF74 and known to hybridize to unique breakpoint-flanking sequences in classic t(11;22) patients [Shaikh et al., 1999]. Cosmid c68a1 exclusively hybridized to der(22) and c87f9 to der(11) (Fig. 5A). Probes RP11-87O6 and RP11-442E11 were used for the detection of the typical familial t(11;22) breakpoint in chromosome 11 [Shaikh et al., 1999; Tapia-Páez et al., 2000] and were indeed found to span the breakpoint in chromosome band 11q23.3 (Fig. 5B). These results strongly suggest the presence of the classic 11;22 translocation, which is frequently found in healthy individuals.
Fig. 5.

FISH analysis indicating that the propositus carries the classic t(11;22) breakpoint. A, B: Metaphase chromosomes derived from the propositus hybridized to different fluorescently labeled probes. A: Cosmids c68a1 (green) and c87f9 (red) flanking the common t(11;22) breakpoint LCR-22B on 22q11.21 are both on the normal 22 showing as yellow signals. Signals for c68a1 are on the der(22) and signals for c87f9 are on the der(11). B: BAC RP11-442E11 bridging the common t(11;22) breakpoint on chromosome 11 shows signals (red) on the normal chromosome 11, der(11) and der(22). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
With conventional FISH methods, we were not able to detect any concurrent chromosomal aberration beside the classic 11;22 translocation that might explain the sex reversal or developmental delay. Alternatively, high-resolution genome-wide interrogation by tiling-path array-CGH was performed to possibly reveal copy number changes in parts of the genome other than the 11;22 breakpoint region that may underlie the phenotype. Array-CGH was performed using 32,448 BAC clones [Krzywinski et al., 2004] on a single micro-array slide with an average genomic tiling-resolution of 100 bp on a custom-designed analysis platform essentially described elsewhere [De Vries et al., 2005]. Using differentially labeled DNA from the propositus and a female normal reference pool in a duplicate dye-swap experiment, no genomic gains or losses were observed throughout chromosomes 1–22, X and Y (see Online Supplement 3 at http://www.interscience.wiley.com/jpages/1552-4825/suppmat/index.html).
Consistent with earlier reports on XX sex reversal and chromosome 22 aberrations, we were unable to determine a possible underlying genetic cause. More research is mandatory at the molecular level to identify unknown factors in the sex determination pathway downstream of SRY, which may also influence facial morphology and psychomotor development.
Supplementary Material
ACKNOWLEDGMENTS
We thank A.J.H. Hamers (emeritus-Head of Cytogenetics Unit, azM) for intellectual input and supervision of prenatal cytogenetic diagnoses, K. Mebis-Verhees and N. Jonker-Houben from the Cytogenetics Unit for cytogenetic analyses, Dr. J. Herbergs from the Molecular Diagnostics Unit for genomic marker analyses, Professors J. Nijhuis and J. Offermans from the Department of Obstetry-Gynecology for ultrasound data and valuable discussion, Dr. P. Menheere from the Department of Clinical Chemistry for biochemical analysis, Dr. H. Scheffer from the Molecular Diagnostic Laboratory, Dr. R. Pfundt and I. Neefs from the Laboratory of Molecular Medicine, Human Genetics Department, Radboud University Nijmegen Medical Center, Nijmegen, for respectively CYP11B1 mutation and array-CGH analysis; and the propositus and his parents for participation in this study. This study was supported in part by CA39926 from the National Institutes of Health, USA (BSE).
Grant sponsor: National Institutes of Health, USA; Grant number: CA39926.
Footnotes
This article contains supplementary material, which may be viewed at the American Journal of Medical Genetics website at http://www.interscience.wiley.com/jpages/1552-4825/suppmat/index.html.
REFERENCES
- Aleck KA, Argueso L, Stone J, Hackel JG, Erickson RP. True hermaphroditism with partial duplication of chromosome 22 and without SRY. Am J Med Genet. 1999;85:2–4. doi: 10.1002/(sici)1096-8628(19990702)85:1<2::aid-ajmg2>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Berend SA, Spikes AS, Kashork CD, Wu JM, Daw SC, Scambler PJ, Shaffer LG. Dual-probe fluorescence in situ hybridization assay for detecting deletions associated with VCFS/DiGeorge syndrome I and DiGeorge syndrome II loci. Am J Med Genet. 2000;91:313–317. [PubMed] [Google Scholar]
- Chieffo C, Garvey N, Gong W, Roe B, Zhang G, Silver L, Emanuel BS, Budarf ML. Isolation and characterization of a gene from the DiGeorge chromosomal region homologous to the mouse Tbx1 gene. Genomics. 1997;43:267–277. doi: 10.1006/geno.1997.4829. [DOI] [PubMed] [Google Scholar]
- De Vries BB, Pfundt R, Leisink M, Koolen DA, Vissers LE, Janssen IM, van Reijmersdal S, Nillesen WM, Huys EH, de Leeuw N, Smeets D, Sistermans EA, Feuth T, van Ravenswaaij-Arts CM, van Kessel A Geurts, Schoenmakers EF, Brunner HG, Veltman JA. Diagnostic genome profiling in mental retardation. Am J Hum Genet. 2005;77:606–616. doi: 10.1086/491719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuel Syndrome On-line Mendelian Inheritance in Man #609029. http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=609029.
- Erickson RP, Skinner S, Jacquet H, Campion D, Buckley PG, Mantripragada KK, Dumanski JP. Does chromosome 22 have anything to do with sex determination: Further studies on a 46,XX,22q11.2 del male. Am J Med Genet Part A. 2003;123A:64–67. doi: 10.1002/ajmg.a.20489. [DOI] [PubMed] [Google Scholar]
- Fraccaro M, Lindsten J, Ford CE, Iselius L. The 11q;22q translocation: A European collaborative analysis of 43 cases. Hum Genet. 1980;56:21–51. doi: 10.1007/BF00281567. [DOI] [PubMed] [Google Scholar]
- Krzywinski M, Bosdet I, Smailus D, Chiu R, Mathewson C, Wye N, Barber S, Brown-John M, Chan S, Chand S, Cloutier A, Girn N, Lee D, Masson A, Mayo M, Olson T, Pandoh P, Prabhu AL, Schoenmakers E, Tsai M, Albertson D, Lam W, Choy CO, Osoegawa K, Zhao S, de Jong PJ, Schein J, Jones S, Marra MA. A set of BAC clones spanning the human genome. Nucleic Acids Res. 2004;32:3651–3660. doi: 10.1093/nar/gkh700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurahashi H, Emanuel BS. Long AT-rich palindromes and the constitutional t(11;22) breakpoint. Hum Mol Genet. 2001;10:2605–2617. doi: 10.1093/hmg/10.23.2605. [DOI] [PubMed] [Google Scholar]
- Phelan MC, Rogers RC, Crawford EC, Brown LG, Page DC. Velocardiofacial syndrome in an unexplained XX male. Am J Med Genet Part A. 2003;116A:77–79. doi: 10.1002/ajmg.a.10833. [DOI] [PubMed] [Google Scholar]
- Schinzel A, Schmid W, der Maur P Auf, Moser H, Degenhardt KH, Geisler M, Grubisic A. Incomplete trisomy 22. I. Familial 11/22 translocation with 3: 1 meiotic disjunction. Delineation of a common clinical picture and report of nine new cases from six families. Hum Genet. 1981;56:249–262. doi: 10.1007/BF00274675. [DOI] [PubMed] [Google Scholar]
- Seeherunvong T, Perera EM, Bao Y, Benke PJ, Benigno A, Donahue RP, Berkovitz GD. 46,XX sex reversal with partial duplication of chromosome arm 22q. Am J Med Genet Part A. 2004;127A:149–151. doi: 10.1002/ajmg.a.20630. [DOI] [PubMed] [Google Scholar]
- Shaikh TH, Budarf ML, Celle L, Zackai EH, Emanuel BS. Clustered 11q23 and 22q11 breakpoints and 3:1 meiotic malsegregation in multiple unrelated t(11;22) families. Am J Hum Genet. 1999;65:1595–1607. doi: 10.1086/302666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiteri E, Babcock M, Kashork CD, Wakui K, Gogineni S, Lewis DA, Williams KM, Minoshima S, Sasaki T, Shimizu N, Potocki L, Pulijaal V, Shanske A, Shaffer LG, Morrow BE. Frequent translocations occur between low copy repeats on chromosome 22q11.2 (LCR22s) and telomeric bands of partner chromosomes. Hum Mol Genet. 2003;12:1823–1837. doi: 10.1093/hmg/ddg203. [DOI] [PubMed] [Google Scholar]
- Tapia-Páez I, O’Brien KP, Kost-Alimova M, Sahlen S, Kedra D, Bruder CE, Andersson B, Roe BA, Hu P, Imreh S, Blennow E, Dumanski JP. Fine mapping of the constitutional translocation t(11;22)(q23;q11) Hum Genet. 2000;106:506–516. doi: 10.1007/s004390000287. [DOI] [PubMed] [Google Scholar]
- Wiegant J, Dauwerse JG. Multi-colored chromosomes by fluorescence in situ hybridization. In: Verma RS, Babu A, editors. Human chromosomes principles and techniques. McGraw-Hill; New York: 1995. pp. 210–218. [Google Scholar]
- Zackai EH, Emanuel BS. Site-specific reciprocal translocation, t(11;22) (q23;q11), in several unrelated families with 3:1 meiotic disjunction. Am J Med Genet. 1980;7:507–521. doi: 10.1002/ajmg.1320070412. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.