

Abstract
Palindromic sequences are dispersed in the human genome and may cause chromosomal translocations in humans. They constitute unsequenced gaps in the human genome because of their resistance to PCR amplification, cloning into vectors, and sequencing. We have overcome these difficulties by using a combination of optimized PCR conditions, cloning in a recombination-deficient E. coli strain, and RNA polymerases in sequencing. Using these methods, we analyzed a palindromic AT-rich repeat (PATRR) in the neurofibromatosis type 1 (NF1) gene on chromosome 17 (17PATRR). The 17PATRR manifests a size polymorphism due to a highly variable length of (AT)n dinucleotide repeats within the PATRR. 17PATRRs can be categorized into two types: a longer one that comprises a nearly or completely perfect palindrome, and a shorter one that represents its deleted asymmetric derivative. In vitro analysis shows that the longer 17PATRR is more likely to form a cruciform structure than the shorter one. Two reported t(17;22)(q11;q11) patients with NF1, whose breakpoints were identified within the 17PATRR, have translocations that are derived from perfect or nearly perfect palindromic alleles. This implies that the symmetric structure of a PATRR can induce a translocation. We identified conserved PATRRs within the NF1 gene in great apes and similar inverted repeats in two Old World monkeys, but not in New World monkeys or other mammals. This indicates that the palindromic region appeared approximately 25 million years ago and elongated during primate evolution. Although such palindromic regions are usually unstable and disappear rapidly due to deletion, the 17PATRR in the NF1 gene was stably conserved during evolution for reasons that are still unknown.
Keywords: palindrome, translocation, NF1
INTRODUCTION
In April 2003, the Human Genome Project was declared finished. Since that time, efforts have been directed toward sequencing the small percentage of regions that remain unsequenced. However, there are still many unclonable or unsequenceable gaps in the genome, such as duplicated segments, centromeric alpha satellites, and regions around other highly repetitive units [Eichler et al., 2004]. Palindromic repetitive sequences remain as a subset of these gaps because of difficulties in PCR amplification, cloning, and sequencing. Frequent deletions prevent cloning in E. coli or yeast [Leach, 1994; Gordenin et al., 1993], while PCR or sequencing is also difficult because of the secondary structures adopted by the template DNA.
We previously reported three types of characteristic palindromic sequences at the breakpoints of recurrent non-Robertsonian translocations in humans, termed palindromic AT-rich repeats (PATRRs). The first PATRR was identified at the breakpoint of the constitutional t(11;22)(q23;q11) [Kurahashi et al., 2000a]. During reconstruction of the original breakpoint sequences of chromosomes 11 and 22 from the der(11) and der(22) sequences of several t(11;22) carriers, we realized that the AT-rich sequences at the breakpoint were palindromic [Kurahashi et al., 2000a,b; Edelmann et al., 2001]. Since the PATRR in chromosome 11 (11PATRR) was deleted from the BAC encompassing the breakpoint, we analyzed the authentic 11PATRR with significant difficulty by using nested PCR followed by generation of a series of deletion mutants of the PCR products. The palindromic sequence was finally identified at the breakpoint region on normal chromosome 11, and the breakpoints were located at the center of the palindrome [Kurahashi et al., 2001a]. Although the evidence also indicated the existence of another PATRR at the chromosome 22 breakpoint (22PATRR), the original 22PATRR remains uncloned. The putative 22PATRR is located in one of the chromosome-specific low-copy-repeats (LCRs) on chromosome 22 [Shaikh et al., 2000] that prevents specific amplification of 22PATRR.
The third PATRR was found at the breakpoint of another constitutional translocation, t(17;22)(q11;q11), which is associated with a common autosomal-dominantly inherited disorder, neurofibromatosis type 1 (NF1; MIM# 162200). The causative gene, NF1, had been positionally cloned using several translocations [O’Connell et al., 1989] and deletions located at 17q11 [Viskochil et al., 1990; Wallace et al., 1990]. The two reported t(17;22)s disrupt the NF1 gene, which is responsible for the disease seen in the translocation carriers [Kehrer-Sawatzki et al., 1997; Kurahashi et al., 2003]. The breakpoint sequences of the two cases also display a highly AT-rich composition, and the original sequence was presumed to be palindromic. Recent findings of a non-recurrent t(4;22) and a t(1;22) [Nimmakayalu et al., 2003; Gotter et al., 2004] also demonstrate the existence of an inverted-repeat or AT-rich palindrome at the breakpoints on original chromosomes 4 and 1, respectively.
In addition, we have also demonstrated frequent de novo t(11;22)s in the sperm of healthy males at a frequency of about 1/100,000 to 1/10,000 [Kurahashi and Emanuel, 2001b]. The identification of PATRRs at all of these translocation breakpoints strongly suggests that the PATRRs mediate these gross rearrangements. The location of all breakpoints at the center of the respective palindromes implies involvement of their secondary structures, the so-called cruciform configuration. Further, the variable frequencies of the t(11;22) in the sperm of different individuals is likely to be affected by sequence polymorphism of the palindromes among individuals [Kurahashi and Emanuel, 2001b].
Taken together, palindrome-mediated chromosomal translocation appears to be one of the universal pathways for human genome rearrangements. In spite of the technical difficulties associated with the study of such rearrangements, our observations prompted us to further characterize PATRRs. In this study, we established methods to rapidly and reproducibly analyze these PATRRs by using an optimized condition for PCR, recombination-deficient E.coli for cloning, and RNA polymerases for sequencing. We applied these methods to a detailed analysis of the characteristics of 17PATRR. Our results strongly suggest that PATRRs play a significant role in predisposing to chromosomal rearrangements. Additionally, their hypervariability may indicate that specific configurations impart a differential risk for “genomic instability.”
MATERIALS AND METHODS
Subjects and Cell Lines
Peripheral blood samples were obtained from 20 unrelated individuals after they gave informed consent. Cell lines from African green monkey (Cercopithecus aethiops) COS7 and owl monkey (Aotus trivirgatus) OMK(637-69) were obtained from Riken (Wako, Japan) and ATCC (Manassas, VA), respectively. A cell line from cotton-top tamarin (Saguinus oedipus; B95-8) was a gift of Dr. Kohtaro Yamamoto. Fibroblast cell lines from gorilla (Gorilla gorilla; AG05251A), and rhesus monkey (Macaca mulatta; AG08316) were obtained from the Coriell Cell Repositories (Camden, NJ) [Shaikh et al., 2000]. Genomic DNA was purified by a standard method [Sambrook et al., 1989].
Amplification of 17PATRR
PCR amplification of 17PATRR was carried out with the use of ExTaq (Takara, Kyoto, Japan). PCR primers for 17PATRR, JF17, and JF17.1 (as described previously by Kurahashi et al. [2003]) were modified at their 5′ ends by adding the T3 or T7 promoter sequences underlined in the following: T3-JF17: 5′-ATTAACCCTCACTAAAGGG-CATGTAGACACTCACCCAGCTC-3′, T7-JF17.1: 5′-TAATACGACTCACTATAGGG-GCAGATGTCCCAAATAGCATC-3′. To confirm the amplification of orthologous regions of human 17PATRR, another pair of primers was also designed to produce longer products: hNF1-F: 5′-TTGGAATACATGACTCCATGGCT-3′ and hNF1-R: 5′-GCACTGGTTTTGATGAAACTGTC-3′. These primers were annealed to the sites where amino acid coding sequences of the NF1 gene are conserved from mammal to fish (data not shown). They amplified primate orthologous regions between human NF1 exon 40 and 41 (the exon numbers followed ENST00000358273 in Ensembl database), and it was confirmed that the identical regions were amplified by these two primer sets. The locations of the primers are shown in Figure 1A. The nucleotide sequences were deposited to the DDBJ database (Mishima, Japan): human (accession numbers AB195812, AB195813, AB195814, and AB195815), gorilla (AB195810 and AB195811), rhesus monkey (AB195808), African green monkey (AB195809), tamarin (AB195807), and owl monkey (AB195806). An alignment of these sequences is depicted in Supplementary Figure S1 (available online at www.interscience.wiley.com/jpages/1059-7794/suppmat). The PCR conditions were set according to the manufacturer’s specifications, except that the deoxyribonucleotide concentration was increased to 400 μM. The concentration of magnesium ion was set at 2 mM. About 50 ng of genomic DNA was used as a template for each amplification. The PCR cycles were as follows: heat at 94°C for 2 min, 30 cycles of at 94°C for 30 sec and at 60°C for 5 min, followed by a final incubation at 60°C for 10 min. The products were fractionated on 2% agarose gels, visualized by 0.5 μg/ml ethidium bromide staining, and purified from the gel slices by QIAquick Gel Extraction Kit (Qiagen, Tokyo, Japan) according to the manufacturer’s instructions, with the exception that the gel melting temperature was set at 25°C to avoid denaturing the AT-rich DNA fragments.
FIGURE 1.

17PATRR amplification and cloning. A: Genomic structure of the 17PATRR. Exons of the NF1 gene and the 17PATRR are indicated by open and filled boxes, respectively. The location of the PCR primers is designated by an arrowhead. B: PCR of 17PATRR. Left panel: Conventional three- and two-step PCR (lanes 2–5). Lane 2 shows three-step PCR at 94°C for 30 sec, 60°C for 30 sec, and 72°C for 1 min. Lanes 3 and 4 indicate two-step PCR at 94°C for 30 sec, followed by 60°C for 1 min or 60°C for 5 min, respectively. Middle panel: Optimization of extension temperature for amplification of the 17PATRR (lanes 6–13). Extension temperatures of 60–68°C in two-step PCR were tested in two individuals: (a) individual heterozygous for 17-L-PATRR and 17-S-PATRR, (b) individual homozygous for the 17-L-PATRRs. Right panel: PCR products of 17PATRRs from five subjects (lanes 15–19). Variable length polymorphism of the PCR products was observed. Samples 1 and 2 appeared to be homozygotes, while samples 3–5 are heterozygotes. Upper faint bands in samples 3–5 (indicated by the asterisk) represent by-products due to heteroduplexes generated during the PCR reaction. They appeared only in the samples heterozygous for 17-L-PATRR and 17-S-PATRR, which was confirmed by a PCR experiment using cloned DNA as a template (data not shown). C: Sequencing chromatograms. The upper panel shows the results from conventional cycle sequencing with Taq polymerase, whereas the lower panel indicates sequencing withT7 RNA polymerase. D: 17-L-PATRR inserts cloned in pUC19 and pBelo-BAC11 vector. Left panel: Insert length in pUC19 clones. The plasmid DNAs from seven individual clones including a full-length 17PATRR were digested by restriction endonucleases. The 3-kb band indicates the vector. In the two lower bands, the longer bands correspond to the intact fragment of the 17PATRR (arrowhead), while the shorter bands correspond to rearranged products (arrow).The clone in lane 4 carried more intact inserts than the others.The colony size at picking does not correlate with the ratio of the rearranged inserts to the intact inserts. Right panel: Comparison between pUC19 and BAC vector. There is less rearranged insert in the BAC vector compared to pUC19. m and nc indicate size markers and negative control without any template DNA, respectively.
Cloning and Sequencing of 17PATRR
The purified PCR products were ligated into pBluescript II (KS +) vector (Stratagene, La Jolla, CA), which was digested at the EcoRV site, and T-tails were added [Finney et al., 1998]. The ligated DNA was transformed into JM109, DH5α, or SURE cells (Stratagene, La Jolla, CA) by electroporation. The transformed cells were spread onto LB-agar plates containing 100 μg/ml of ampicillin. Colonies were inoculated into 2 ml of prewarmed LB media containing 100 μg/ml of ampicillin and cultured for 12 hr. The plasmid-DNAs were purified by conventional alkali-SDS methods [Sambrook et al., 1989]. The DNAs were digested with BamHI and HindIII and fractionated on a 2% agarose gel to separate full-length inserts from deleted derivatives that were generated during culture. The fragments were used for RNA sequencing or recloning into pUC19 or pBeloBAC11 at their multi-cloning sites. BAC DNA clones were isolated by alkali-SDS methods and purified by two rounds of cesium chloride-ethidium bromide density centrifugation [Sambrook et al., 1989]. The sequences of purified PCR fragments or plasmid DNAs were determined by the CUGA sequencing kit for the ABI 377 (Nippon Genetech, Tokyo, Japan) [Sasaki et al., 1998]. Sequences were determined from at least three plasmid clones to avoid misinterpretation generated by PCR errors.
The single nucleotide polymorphisms (SNPs) near the 17PATRR were genotyped by either restriction digestion or sequencing of the PCR products. For the genotyping of dbSNP964288, the PCR cycle and primers were as follows: heat at 94°C for 2 min, and 30 cycles at 94°C for 30 sec, 60°C for 30 sec, and 72°C for 30 sec, i50F1: 5′-GAAATAGGACAGCCACTTGGAAG-3′ and e51R: 5′-GTTGGCCTGAGAAGGTTGCCCCAT-3′. The amplified products from 50 ng of genomic DNA were digested with PacI (NEB, Beverly, MA).
Analysis of Cruciform Formation
For cruciform digestion, 50 ng of each DNA sample was digested by 0.2 U of T7 endonuclease (NEB, Beverly, MA) or 1 U of S1 nuclease (Takara, Kyoto, Japan) for 1 hr at 37°C. XhoI (Takara, Kyoto, Japan) was used for digestion control. The buffer conditions were as follows: T7 endonuclease, XhoI, and no enzyme were used in 50 mM NaCl, 10 mM Tris-HCl (pH7.9), 10 mM MgCl2, and 1 mM DTT; and S1 nuclease was used in 30 mM Naacetate (pH4.6), 280 mM NaCl, and 1 mM ZnSO4. To map the nuclease recognition sites, we used 17-L-PATRR of p-(AT)10/d-(AT)10 and 17-S-PATRR of p-(AT)25 cloned in pBeloBAC11 at the BamHI and HindIII sites. The BAC vector was used as a control. Then 10 ng of the DNA was incubated at 37°C for 2 hr with 3 U of T7 endonuclease or 1 U of S1 nuclease in buffers as described above. After extraction with phenol/chloroform and precipitation by ethanol with a carrier, the DNA was digested with BamHI and HindIII to cut out the insert including the PATRR. The fragments were dephosphorylated by TsAP (Invitrogen, Tokyo, Japan) at 65°C for 15 min, extracted with phenol/chloroform to inactivate the enzyme, and ethanol-precipitated. The DNA was denatured at 100°C for 10 min and then phosphorylated by T4 polynucleotide kinase (Takara, Kyoto, Japan) with [γ-32P]ATP (Wako, Tokyo, Japan). After purification by ethanol precipitation, the labeled DNA was heated in 80% formamide, 4 mM EDTA (pH8.0) for 2 min at 95°C, and loaded onto 4% acrylamide/6 M urea gel in 1 × TBE buffer [Sambrook et al., 1989]. After electrophoresis, the gel was dried and exposed to X-ray films. For a size marker, 1 ng of MspI-digested pBR322 (Wako, Tokyo, Japan) was loaded after labeling. For native gel electrophoresis, symmetric 17-L-PATRR of p-(AT)10/d-(AT)10, asymmetric 17-L-PATRR of p-(AT)10/d-(AT)20, and 17-S-PATRR of p-(AT)25 in pBluescriptII were heated at 65°C in buffer (10 mM Tris-Cl, pH8.0, 1 mM EDTA, pH 8.0 and 50 mM NaCl) for 30 min, and then quickly frozen in liquid nitrogen. Agarose gel electrophoresis was carried out on 0.8% agarose in 1 × TAE buffer [Sambrook et al., 1989] at 4°C for 16 hr at 2 V/cm.
Analysis of the Nucleotide Sequences
The nucleotide sequences of humans and mammals were aligned with the aid of the ClustalW alignment system in DDBJ (www.ddbj.nig.ac.jp/search/ex–clustalw-j.html). Secondary structures were predicted by the mfold web server (www.bioinfo.rpi.edu/applications/mfold/) [Zuker, 2003] with default parameters, except that sodium ion condition and correction type were set at 1.0 M and polymer, respectively. Repetitive sequences in the genomes were identified by RepeatMasker (www.repeatmasker.org/cgi-bin/WEBRepeatMasker). The genomic sequences of chimpanzee, dog, and rodent were obtained from the Ensembl Genome Browser (www.ensembl.org/). SNP data were obtained from dbSNP (www.ncbi.nlm.nih.gov/SNP/, Build 124).
RESULTS
Amplification, Sequencing, and Cloning of 17PATRR
PCR amplification of PATRRs has been difficult to achieve using the conventional PCR methodology [Kurahashi and Emanuel, 2001a]. We tried several PCR conditions to amplify the 17PATRRs directly from genomic DNA samples. Conventional three-step PCR with an extension temperature at 72°C failed to amplify the 17PATRRs (Fig. 1B, lane 2). The use of a polymerase with strand-displacement activity also failed (data not shown). It is reasonable to assume that optimization of the extension temperature is important, since a higher temperature dissociates newly synthesized AT-rich strands, and a lower temperature would cause intrastrand annealing of the palindromic region. Thus, we tested various extension temperatures using twostep PCR, which resulted in successful amplification of the 17PATRR. The optimal temperature for the annealing and extension step was found to be approximately 60–62.5°C (Fig. 1B, lanes 6–13). With this condition, a 1-min extension time was inadequate (Fig. 1B, lane 3) because the rate of extension was likely to be slower than that used for conventional three-step PCR with an extension temperature at 72°C. We extended the extension times (e.g., 5 min for amplification of 500 bp, and 10 min for 1.6 kb; Fig. 1B, lane 4). Using these conditions, we amplified the 17PATRRs from 20 healthy individuals. The heterogeneous size of the products indicated a highly polymorphic composition of this region (Fig. 1B, right panel).
Similarly, conventional cycle sequencing reactions with Taq polymerase were also compromised by frequent stalling at the start site of the palindrome (Fig. 1C, upper panel). Several extension temperatures were tested, but they failed. We carried out another sequencing method, RNA polymerase-based sequencing [Sasaki et al., 1998], which has been reported to allow sequencing of templates that form secondary structures. We performed PCR using primers equipped with T3/T7 promoters. The PCR products were directly sequenced using RNA polymerases, which resulted in successful sequencing of the PCR products (Fig. 1C, lower panel). In heterozygous individuals whose bands could be separated by gel-electrophoresis, we obtained sequences of high quality. The rest of the samples showed duplicated signals following the (AT)n repeat regions at the middle of the 17PATRR, indicating length polymorphisms of the (AT)n between the alleles. We determined the sequence of these samples by cloning into a plasmid vector and sequencing multiple clones.
The PCR products were ligated into a high-copy plasmid vector and transformed into E. coli. When common strains such as JM109 were transformed with 17PATRR-bearing plasmids, no intact insert was obtained, and the majority of the palindromic region was lost. When we used SURE, a recombination-deficient E. coli strain that has sbcC, recJ, umuC, and uvrC mutations, the insert for each clone produced two bands. This indicates that rearrangement had occurred and shortened the length of the insert during bacterial culture (Fig. 1D, lanes 2–8). Sequencing these bands revealed that the longer bands included intact full-length inserts. The shorter bands represent the rearranged products produced by deleting between two (AT)n repeat regions in the 17PATRR, as described below (Fig. 2A, human-L-re). Although some fraction of the plasmids harbors deletions, this recombination-deficient strain yields better results when the 17PATRR is cloned. An insert in a BAC vector seemed to be more stable than when the 17PATRR was propagated as high-copy plasmids (Fig. 1D, lanes 10 and 11).
FIGURE 2.

Nucleotide sequence of the 17PATRR. A: Alignment of the 17-L-PATRRs,17-S-PATRRs of human (human-L and human-S, respectively), and a rearranged 17-L-PATRR in E.coli (human-L-re). Proximal and distal (AT)n repeats (p-(AT)n and d-(AT)n) are shown by brackets. B: Putative hairpin formation of the 17-L-PATRR. The example shows a perfect palindrome. The (AT)n repeat region is indicated by a box. The region absent in 17-S-PATRR is underlined. C: The strong correlation between the two types of PATRR and the neighborhood SNPs is shown. An SNP (dbSNP964288) at 17 kb downstream from the 17PATRR in the individuals and cell lines link to 17-L or S-PATRR alleles (L or S) completely. Lane m is a size marker.
Nucleotide Sequence of 17PATRRs
We investigated human 17PATRRs using the strategy described above. A total of 20 individuals (17 Japanese and three non-Japanese) were analyzed. The 17PATRRs can be categorized into two groups. Twenty-two of 40 chromosomes carry longer PATRRs (17-L-PATRRs) and the other 18 chromosomes carry shorter PATRRs (17-S-PATRRs). Typical sequences of the 17-L- and 17-S-PATRRs are depicted in Figure 2A (human-L and human-S, respectively). The 17-L-PATRR comprises an almost perfect or completely perfect palindrome, while the 17-S-PATRR appeared to represent its deleted derivative. The size of the 17-L-PATRR shown in Figure 2A was 187 bp. All of the proximal and distal arms of these PATRRs harbor (AT)n dinucleotide-repeat regions (p-(AT)n and d-(AT)n), which appear to cause rearrangements when cloned into plasmids in E. coli (Fig. 2A, human-L-re).
The sequences of the 17-L-PATRRs were nearly identical to those of the putative PATRRs reconstructed from the breakpoint sequences of two constitutional t(17;22) patients [Kehrer-Sawatzki et al., 1997; Kurahashi et al., 2003], and were identical to the sequence of the corresponding NF1 genomic region in the database (GenBank accession number AC004526). In the 17-L-PATRRs, length polymorphism was observed at the (AT)n dinucleotide-repeats, which are summarized in Table 1. The majority of the p-(AT)n were n = 10, while d-(AT)n showed variable lengths (n = 10–20). The symmetrical 17-L-PATRR is shown in Figure 2B, while alleles other than d-(AT)10 comprise asymmetrical palindromes. No other nucleotide differences were observed among the 17-L-PATRR alleles.
TABLE 1.
Number of (AT)n Repeats in Human 17PATRR
| p-(AT)n | d-(AT)n | Alleles | Cell lines | |
|---|---|---|---|---|
| 17L | ||||
| 10 | 10 | 3 | ||
| 10 | 12 | 2 | HepG2, 293 | |
| 10 | 14 | 7 | HT1080 | |
| 10 | 15 | 2 | THP1 | |
| 10 | 16 | 2 | ||
| 10 | 18 | 1 | THP1 | |
| 10 | 19 | 2 | ||
| 10 | 20 | 1 | HeLa | |
| 11 | 15 | 1 | ||
| 15 | 16 | 1 | ||
| Total | 22 | |||
| 17S | ||||
| 20(c) | - | 1 | ||
| 22(c) | - | 1 | HepG2 | |
| 23(c) | - | 2 | ||
| 24(c) | - | 5 | HeLa, HT1080 | |
| 25(c) | - | 6 | 293 | |
| 26(c) | - | 2 | ||
| 24(c) | - | 1a | ||
| Total | 18 | |||
| Total | 40 |
36 nucleotide duplication at p-(AT)n.
, substitution of T to C at 3rd (AT)n.
The size of the 17-S-PATRR shown in Figure 2A was 161 bp. This appears to originate from a 46 bp deletion mostly in the distal arm of the 17-L-PATRR including the center of the palindrome. All of the 17-S-PATRRs have the same 46 bp deletion at the same region, having polymorphic p-(AT)n similar to the 17-L-PATRR (Table 1). An unusual allele demonstrated a 36 nucleotide duplication around p-(AT)n (Table 1, asterisk). The sequences of regions other than the (AT)n were identical among the 17-S-PATRRs.
In comparison with the 17-L-PATRR, the p-(AT)n of the 17-S-PATRRs appeared to be about twice as long as that of the 17-L-PATRRs, with two subtle sequence differences. Since these differences were common in all 17-S-PATRRs, the 17-S-PATRRs were most likely derived from a single original allele, which is supposed to have been generated from a 17-L-PATRR or its derivative by deletion of a part of the palindrome. It is possible that this rearrangement occurred once in the human lineage and no other rearrangements have occurred subsequently, except for the (AT)n extension.
We further investigated the extent of the polymorphism in several human cell lines. Four cell lines–293, HeLa, HT1080, and HepG2–were heterozygous for 17-L-PATRR/17-S-PATRR, while THP1 was homozygous for 17-L-PATRR (Table 1), indicating that cell lines that have been in long-term culture still carry intact 17-L-PATRRs. These results suggest that 17PATRRs are stably transmitted during mitotic cell division.
The SNPs around the 17PATRR were genotyped, and the possible linkage of 17-L/S-PATRR to these SNPs was examined. All of the SNPs investigated showed complete linkage with the PATRR genotypes among the Japanese and non-Japanese individuals and cell lines of several ethnicities (Fig. 2C, Supplementary Table S1). This means that the divergence of 17-L-PATRR and 17-S-PATRR occurred a long time ago, and they have been maintained since then throughout human history.
Analysis of In Vitro Cruciform Formation
Since we succeeded in cloning the 17PATRR, we then examined the in vitro cruciform formation of the 17PATRR plasmid. First, we digested closed circular clones of the 17PATRR by T7 endonuclease I or S1 nuclease, which cut at the base or tip of the cruciform DNA, respectively. The 17-L-PATRR plasmid was cleaved into a linear form, suggesting the cruciform structure of the plasmid (Fig. 3A, lanes 3 and 4). It is also possible that the 17-L-PATRR can be cut merely because of the (AT)n repeats within it. Indeed, a part of the 17-S-PATRR that does not comprise the palindrome was also cleaved into a linear form (Fig. 3A, lanes 7 and 8). Thus, we mapped the position of the cleavage sites of these nucleases (Fig. 3B and C). The results demonstrate that the 17-L-PATRR was cut in the vicinity of the bottom or the tip of the putative cruciform conformation that was computationally predicted using mfold software (Fig. 2B). Mapping data also demonstrated that the 17-S-PATRR forms a small cruciform, but it seemed to extrude at the p-(AT)n repeat and several of its flanking bases (Fig. 3C).
FIGURE 3.

In vitro analysis of the cruciform formation of the 17PATRR plasmid. A: Digestion of cruciform DNA withT7 endonuclease I or S1 nuclease. Symmetric 17-L-PATRR (L-sym),17-S-PATRR (S), and control clone with no insert (vector) in the pBeloBAC11 vector were digested withT7 endonuclease I or S1 nuclease. For a digestion control, XhoI was used to cut the DNA at one site. Arrowheads indicate open circular (oc), linear (linear), and closed circular (cc) form DNA, respectively. B: Mapping of cleavage sites.17-L-PATRR (L-sym), 17-S-PATRR (S), and control (vector) were incubated withT7 endonuclease I (T), S1 nuclease (S), or without enzyme (n), followed by restriction enzyme digestion, end-labeling, and electrophoresis under denaturing conditions. Each band is numbered.T-5 and T-6 indicate faint bands in laneT. C: Diagrams of cleavage sites.The 17PATRRs are indicated by thick lines. Cleavage sites are depicted by arrowheads. T7 endonuclease I cleaved the DNAs only at the 5′ end of each hairpin.T4 endonucleaseVII, an analogous enzyme forT7 endonuclease I, was previously reported to cleave the plasmid harboring the palindromic region in a similar fashion [Lilley and Kemper, 1984; Lilley and Hallam, 1984]. 17-S-PATRR was mainly digested at different sites compared to 17-L-PATRR, indicating that a distinct internal (AT)n repeat of 17-S-PATRR forms a small cruciform.The faint bands in 17-S-PATRR were similar in size to those of the major bands in 17-L-PATRR, indicating that a small fraction of 17-S-PATRR extrudes a large cruciform. D: Unwound plasmids by cruciform extrusion. Agarose gel electrophoresis of the symmetric 17-L-PATRR (L-sym, p-(AT)10,d-(AT)10), 17-L-PATRR (17L-asym, p-(AT)10, d-(AT)20), and 17-S-PATRR (17-S, p-(AT)24) was performed. DNA was electrophoresed with (H) or without (N) heat denaturation. Closed circular DNA of10 and 3 kb was used as size markers (m1and m2).The bands appearing on the upper side of the gel correspond to dimer or multimer forms of the plasmids, which were characteristic of clones replicated in SURE cells.The positions of the monomer, dimer, and trimer highly negatively supercoiled DNA are indicated by arrows.
Next, we analyzed the conformation of the 17PATRR plasmids using standard agarose gel electrophoresis. Palindromic regions form a cruciform in negatively supercoiled DNA by unwinding the negative superhelicity [Sinden, 1994]. When PATRR plasmids extrude cruciforms, they migrate as a ladder in standard agarose electrophoresis [Kurahashi et al., 2004]. When 17PATRR plasmids were examined, unwound plasmids migrating as a ladder were observed in all symmetric (17-L-PATRR (p-(AT)10; d-(AT)10)) and asymmetric (17-L-PATRR (p-(AT)10; d-(AT)20); 17-S-PATRR) plasmids at lane N (Fig. 3D). The symmetric 17-L-PATRR plasmid showed extensive laddering, while the asymmetric 17-L showed less of a ladder pattern, indicative of a more unwound state for the symmetric 17-L-PATRR plasmid. On the other hand, the 17-S-PATRR plasmid showed only a small number of unwound bands, which suggests that the 17-S-PATRR extrudes a smaller cruciform at the (AT)n repeat region compared to the 17-L-PATRR.
When these plasmids were heat-denatured, the ladder on an agarose gel electrophoresis derived from unwound plasmid disappeared for the asymmetric 17-L-PATRR and 17-S-PATRR, while the symmetric 17-L-PATRR continued to demonstrate an extensive ladder (Fig. 3D, lanes H). This suggests that once it is extruded, the cruciform conformation is preserved in the symmetric 17-L-PATRR and it either tolerates heat denaturation or quickly re-extrudes after denaturation, in contrast to the asymmetric 17-L-PATRR.
This propensity to extrude cruciform arms depends on the size and symmetry of the palindrome, and is predicted to influence susceptibility to t(17;22) generation. In the analysis of the der(17) and der(22) of this translocation, both reported t(17;22) cases originated from a 17-L-PATRR. The first reported case of constitutional t(17;22) showed p-(AT)20 and d-(AT)17 at the der(17) and der(22) breakpoints, respectively, which comprise nearly perfect palindromes [Kehrer-Sawatzki et al., 1997]. The second t(17;22) case demonstrated (AT)11 on both derivative chromosomes, which constitute a completely symmetric palindrome [Kurahashi et al., 2003]. The fact that the symmetric 17PATRR forms a stable cruciform strongly supports our hypothesis that the symmetry of the PATRRs mediates translocation through susceptibility to cruciform extrusion [Kurahashi et al., 2000a].
Conservation of Palindromic Sequences in Primates
We identified two types of 17PATRRs in humans, and no other rearranged form was observed. Thus, the 17PATRR appears to be stably transmitted and conserved in the human lineage. However, it has been shown that palindromic structures are unstable and susceptible to deletion into an asymmetric form in the eukaryotic genome [Nag and Kurst, 1997; Nasar et al., 2000; Farah et al., 2002]. To investigate the origin of the 17PATRR, we examined 17PATRRs in several cell lines derived from nonhuman primates, gorillas, and Old and New World monkeys. Using PCR conditions similar to those applied for the human 17PATRRs, we successfully amplified and sequenced the primate-orthologous regions in the NF1 gene. We also analyzed the 17PATRR region for the common chimpanzee (P. troglodytes) as deposited in the Ensembl database (May 2004 release; contig number AADA01234954). Surprisingly, the gorilla and chimpanzee have similar 17PATRRs in the intron of the NF1 gene.
The gorilla also has two alleles: one is nearly identical to the human 17-L-PATRR, and the other is its deleted derivative (as with human 17-S-PATRR). The 17-L-PATRRs of the chimpanzee and gorilla comprise nearly symmetrical palindromic structures, both of which may be susceptible to forming cruciform structures (Fig. 4A and B). The gorilla and chimpanzee also have size variation in the (AT)n, suggesting that there exists a size polymorphism of the (AT)n as seen in humans. The shorter allele in the gorilla seems to be a derivative of the 17-L-PATRR with a deletion including the center of the palindrome, but the deleted region is different from that in humans (Figs. 2B and 5). Therefore, it is likely that 17-S-PATRRs in humans and gorillas generated independently from their respective 17-L-PATRRs. Interestingly, the gorilla 17-S-PATRR still maintains its symmetry, in contrast to the human 17-S-PATRR, which was deleted into an asymmetric form.
FIGURE 4.

A: Sequence alignment of the17PATRRs in primates.The arrows indicate the range of the17PATRRs.The triangle indicates the symmetric center of human 17-L-PATRR. −L and −S designate 17-L-PATRR and 17-S-PATRR, respectively. Human: human sequence as shown in Figure 2; chimpanzee: P. troglodytes; gorilla: G. gorilla; rhesus: rhesus monkey (M. mulatta); green: African green monkey (C. aethiops); tamarin: cotton top tamarin (S. oedipus); owl: owl monkey (Aotus trivirgatus); and -: sequence gap. B: Schematic secondary structures of primate 17PATRRs. 17-L-PATRRs of humans, chimpanzees, and gorillas are shown.The symmetric gorilla 17-S-PATRR is also indicated as gorilla-S, and the putative deleted region is underlined in gorilla-L. Inverted repeats in Old World, rhesus, and African green monkeys may also form cruciform structures.
FIGURE 5.
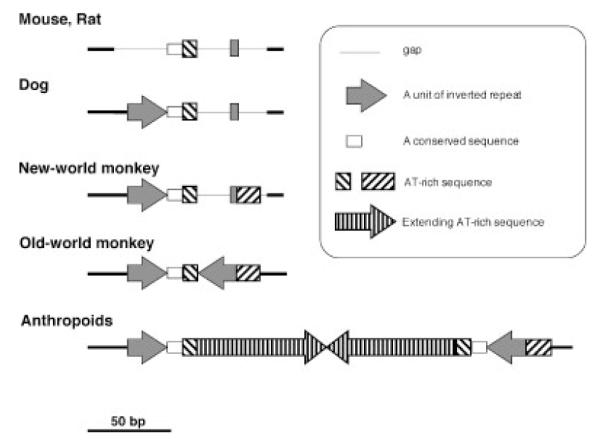
Schematic representation of17PATRR organization in mammals.The structure for the Old World monkey was based on that for the rhesus monkey, and the structure for anthropoids was based on human organization.The schematic for dogs, mice, and rats was based on data from the database.
We found that rhesus and African green monkeys had no PATRR-like sequence in the NF1 gene, but still had a part of the 17PATRR (Fig. 4A). A secondary structure prediction revealed that the short sequences of the Old World monkeys also constitute small inverted repeat sequences (Fig. 4B). The sequences of the putative cruciform base regions in monkeys are quite similar to those in humans. New World monkeys, tamarins, and owl monkeys (Fig. 4B), and other mammals (dogs, mice, and rats) in the database do not have such a palindromic region within the NF1 gene, although one arm of the short palindromic sequences observed in Old World monkeys is present. These results suggest that the 17PATRR was generated in the primate lineage as a short palindromic structure after the Old World monkeys diverged about 25 million years ago. During the evolution of the human and great ape lineages, the PATRR increased in size by lengthening of the AT-rich regions and generating symmetry, although some alleles decreased in size by partial deletion.
DISCUSSION
We determined the 17PATRR sequences of human and other primates rapidly and reproducibly using a combination of suitable PCR conditions, cloning in recombination-deficient E.coli cells, and sequencing by RNA polymerases. The conditions described in this study should aid in the analysis of various templates that are difficult to sequence. SURE cells conventionally have been used for cloning direct or inverted repeats, and they were also useful for the cloning of the 17PATRR-containing fragments. Other strains, such as DH5α and JM109, did not preserve the 17PATRR fragment stably in plasmids and led to the deletion of almost the entire palindromic sequence. This appears to explain why the putative 22PATRR is underrepresented in human BAC libraries, and the 11PATRR was completely deleted from the corresponding BAC clone [Kurahashi et al., 2000a]. Recently, the stable maintenance of long palindromes was reported in the SAE2 gene mutant of Saccharomyces cerevisiae [Rattray, 2004]. Using suitable strains for library construction and the methods for DNA amplification and sequencing described in this article, it should be possible to fill a number of the unsequenced gaps in genome sequencing projects.
Recently, considerable data concerning PATRR-mediated translocations have been accumulated [Kehrer-Sawatzki et al., 1997; Kurahashi et al., 2000a, 2003; Edelmann et al., 2001; Kurahashi and Emanuel, 2001a, 2001b; Nimmakayalu et al., 2003; Gotter et al., 2004]. We previously proposed that PATRRs adopt a cruciform structure that mediates certain translocations, and that symmetry of the palindrome is likely to influence the susceptibility to translocation [Kurahashi and Emanuel, 2001a]. In the case of the 17PATRR, the breakpoints of two cases of balanced t(17;22)(q11;q11) were both derived from the 17-L-PATRR that comprises a nearly or completely perfect palindromic structure. It is reasonable to imagine that individuals with a symmetric 17PATRR might have a higher risk for generating de novo t(17;22)s in sperm than those with asymmetric 17PATRRs. Unfortunately, no direct evidence was obtained to compare the frequency distribution of de novo t(17;22)s between the symmetric and asymmetric types, since no translocation was observed in sperm from several healthy individuals (<5 × 10−6). However, we show that the symmetric 17PATRR forms a cruciform structure more readily than the asymmetric 17PATRRs in vitro, which suggests that the symmetric 17PATRR is more likely to cause the t(17;22) translocation than the asymmetric 17PATRR. On the other hand, palindromic sequences induce meiotic and mitotic recombinations in yeast [Nag and Kurst, 1997; Nasar et al., 2000; Farah et al., 2002]. In mitotic cells, such palindromic sequences form hairpin structures in the lagging strand of the replication fork during DNA synthesis, which causes replication to stall and induces nucleolytic cleavage [Lobachev et al., 2002]. Double-strand breaks at the 17PATRR in mitosis may induce illegitimate recombination or deletion through intrachromosomal recombination between the LCRs flanking the NF1 region, causing the second hit in the normal allele of the NF1 gene in NF1 patients [Dorschner et al., 2000]. In this context, the association between the 17PATRR allele type and the prevalence of NF1-related tumors deserves further investigation to elucidate the propensity of 17PATRR to induce a double-strand break, and the role played by 17PATRRs in the predisposition to chromosomal rearrangements.
An analysis of the 17PATRR in primates, chimpanzees, and gorillas revealed that their 17PATRRs are almost identical to those in humans. Similar short, inverted repeat sequences were also found in the Old World monkeys. The sequences in mice, rats, and dogs in the database, and in New World monkeys as reported here, showed no palindromic sequence in this region. Figure 5 shows a scheme of 17PATRR organization. We speculate that the 17PATRR was generated accidentally in the primate lineage as a small inverted repeat sequence and developed into a large PATRR during anthropoid evolution.
How, then, did PATRRs expand during primate evolution? Hypervariable (AT)n in the 17PATRRs indicate that an AT-rich sequence, such as (AT)n, is susceptible to expansion by replication slippage. There is a short stretch of ATs in the middle of the 17PATRR in Old World monkeys, which might have the potential to increase in size. After the (AT)n achieved a considerable size, it may have formed a cruciform by the force of negative supercoiling in the chromosomal context. The double-strand break may have been generated by diagonal cleavage of the cruciform structure with a nuclease such as a Holliday junction resolvase [Lobachev et al., 2002]. The hairpin structure would have dissociated, generating a 3′ protruding end, which then would have annealed out of alignment and been filled in (Fig. 6). Alternatively, the protruding end may simply have been filled in, and then ligated to another end by a repair mechanism such as non-homologous end joining. The final product could have been a large palindrome with a long (AT)n at its center, similar to the PATRR in primates.
FIGURE 6.

Amodel for17PATRR development.
The conservation of the 17PATRR in both humans and great apes was an unexpected finding, since palindromic sequences are generally thought to be unstable in the genome. In bacterial and eukaryotic genomes, long palindromes are unstable and adopt secondary structures that appear to promote deletion [Leach, 1994; Collick et al., 1996; Akgün et al., 1997; Waldman et al., 1999; Cunningham et al., 2003]. In a eukaryotic genome, transgenes that form large inverted repeats are unstable in both mitosis and meiosis, leading to rearrangement or complete loss of the transgene [Collick et al., 1996]. Alu elements in primate genomes tend to be more frequently organized in tandem than in inverted configurations, whereas experimentally introduced inverted Alu elements are frequently lost from yeast genomes [Lobachev et al., 2000]. Therefore, the conservation of the 17PATRR is unusual. One possibility is that unlike an Alu inverted repeat, the 17PATRR is too short to be eliminated rapidly. Another possibility is that the palindromic sequence has been maintained because it is necessary for NF1 gene function. This function may be specific to primates, since dogs and rodents do not have such a structure. We identified an individual who is homozygous for the human 17-S-PATRR, suggesting that homozygosity for the 17-S-PATRR is not fatal and an inverted repeat structure serves no critical function for survival. In vitro studies demonstrate that the 17-S-PATRR still adopts a small cruciform configuration. The cruciform structure of the 17PATRR in the intron of the NF1 gene may have a certain function, such as alternative splicing or modification of splicing efficiency. Further studies will elucidate the cellular function of this cruciform DNA.
Supplementary Material
ACKNOWLEDGMENT
We thank Dr. Nobuhiro Hayashi for valuable discussions.
Grant sponsor: Ministry of Education, Science, Sports and Culture of Japan; Grant number:16012262;16390102;16710148; Grant sponsor: 21st Century COE Program, Ministry of Education, Science, Sports and Culture of Japan; Grant number: F33; Grant sponsor: NIH; Grant number: CA39926; HD26979; GM64725.
Footnotes
The Supplementary Material referred to in this article can be accessed at www.interscience.wiley.com/jpages/1059-7794/suppmat.
REFERENCES
- Akgün E, Zahn J, Baumes S, Brown G, Liang F, Romanienko PJ, Lewis S, Jasin M. Palindrome resolution and recombination in the mammalian germ line. Mol Cell Biol. 1997;17:5559–5570. doi: 10.1128/mcb.17.9.5559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collick A, Drew J, Penberth J, Bois P, Luckett J, Scaerou F, Jeffreys A, Reik W. Instability of long inverted repeats within mouse transgenes. EMBO J. 1996;15:1163–1171. [PMC free article] [PubMed] [Google Scholar]
- Cunningham LA, Coté AG, Cam-Ozdemir C, Lewis SM. Rapid, stabilizing palindrome rearrangements in somatic cells by the center-break mechanism. Mol Cell Biol. 2003;23:8740–8750. doi: 10.1128/MCB.23.23.8740-8750.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorschner MO, Sybert VP, Weaver M, Pletcher BA, Stephens K. NF1 microdeletion breakpoints are clustered at flanking repetitive sequences. Hum Mol Genet. 2000;9:35–46. doi: 10.1093/hmg/9.1.35. [DOI] [PubMed] [Google Scholar]
- Edelmann L, Spiteri E, Koren K, Pulijaal V, Bialer MG, Shanske A, Goldberg R, Morrow BE. AT-rich palindromes mediate the constitutional t(11;22) translocation. Am J Hum Genet. 2001;68:1–13. doi: 10.1086/316952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichler EE, Clark RA, She X. An assessment of the sequence gaps: unfinished business in a finished human genome. Nat Rev Genet. 2004;5:345–354. doi: 10.1038/nrg1322. [DOI] [PubMed] [Google Scholar]
- Farah JA, Hartsuiker E, Mizuno K, Ohta K, Smith GR. A 160-bp palindromic is a Rad50 Rad32-dependent mitotic recombination hotspot in Schizosaccharomyces pombe. Genetics. 2002;161:461–468. doi: 10.1093/genetics/161.1.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finney M, Nisson PE, Rashtchian A. Molecular cloning of PCR products. In: Ausubel FM, editor. Current protocols in molecular biology. John Wiley & Sons, Inc.; New York: 1998. Unit 15.7. [DOI] [PubMed] [Google Scholar]
- Gordenin DA, Lobachev KS, Degtyareva NP, Malkova AL, Perkins E, Resnick MA. Inverted DNA repeats: a source of eukaryotic genomic instability. Mol Cell Biol. 1993;13:5315–5322. doi: 10.1128/mcb.13.9.5315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotter AL, Shaikh TH, Budarf ML, Rhodes CH, Emanuel BS. A palindrome-mediated mechanism distinguishes translocations involving LCR-B of chromosome 22q11.2. Hum Mol Genet. 2004;13:103–115. doi: 10.1093/hmg/ddh004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehrer-Sawatzki H, Haussler J, Krone W, Bode H, Jenne DE, Mehnert KU, Tummers U, Assum G. The second case of a t(17;22) in a family with neurofibromatosis type 1: sequence analysis of the breakpoint regions. Hum Genet. 1997;99:237–247. doi: 10.1007/s004390050346. [DOI] [PubMed] [Google Scholar]
- Kurahashi H, Shaikh TH, Hu P, Roe BA, Emanuel BS, Budarf ML. Regions of genomic instability on 22q11 and 11q23 as the etiology for the recurrent constitutional t(11;22) Hum Mol Genet. 2000a;9:1665–1670. doi: 10.1093/hmg/9.11.1665. [DOI] [PubMed] [Google Scholar]
- Kurahashi H, Shaikh TH, Zackai EH, Celle L, Driscoll DA, Budarf ML, Emanuel BS. Tightly clustered 11q23 and 22q11 breakpoints permit PCR-based detection of the recurrent constitutional t(11;22) Am J Hum Genet. 2000b;67:763–768. doi: 10.1086/303054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurahashi H, Emanuel BS. Long AT-rich palindromes and the constitutional t(11;22) breakpoint. Hum Mol Genet. 2001a;10:2605–2617. doi: 10.1093/hmg/10.23.2605. [DOI] [PubMed] [Google Scholar]
- Kurahashi H, Emanuel BS. Unexpectedly high rate of de novo constitutional t(11;22) translocations in sperm from normal males. Nat Genet. 2001b;29:139–140. doi: 10.1038/ng1001-139. [DOI] [PubMed] [Google Scholar]
- Kurahashi H, Shaikh T, Takata M, Toda T, Emanuel BS. The constitutional t(17;22): another translocation mediated by palindromic AT-rich repeats. Am J Hum Genet. 2003;72:733–738. doi: 10.1086/368062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurahashi H, Inagaki H, Yamada K, Ohye T, Taniguchi M, Emanuel BS, Toda T. Cruciform DNA structure underlies the etiology for palindrome-mediated human chromosomal translocations. J Biol Chem. 2004;279:35377–35383. doi: 10.1074/jbc.M400354200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach DRF. Long DNA palindromes, cruciform structures, genetic instability and secondary structure repair. Bioessays. 1994;16:893–900. doi: 10.1002/bies.950161207. [DOI] [PubMed] [Google Scholar]
- Lilley DMJ, Hallam LR. Thermodynamics of the ColE1 cruciform. J Mol Biol. 1984;180:179–200. doi: 10.1016/0022-2836(84)90436-4. [DOI] [PubMed] [Google Scholar]
- Lilley DM, Kemper B. Cruciform-resolvase interactions in supercoiled DNA. Cell. 1984;36:413–422. doi: 10.1016/0092-8674(84)90234-4. [DOI] [PubMed] [Google Scholar]
- Lobachev KS, Stenger JE, Kozyreva OG, Jurka J, Gordenin DA, Resnick MA. Inverted Alu repeats unstable in yeast are excluded from the human genome. EMBO J. 2000;19:3822–3830. doi: 10.1093/emboj/19.14.3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobachev KS, Gordenin DA, Resnick MA. The Mre11 complex is required for repair of hairpin-capped double-strand breaks and prevention of chromosome rearrangements. Cell. 2002;108:183–193. doi: 10.1016/s0092-8674(02)00614-1. [DOI] [PubMed] [Google Scholar]
- Nag DK, Kurst A. A 140-bp-long palindromic sequence induces double-strand breaks during meiosis in the yeast Saccharomyces cerevisiae. Genetics. 1997;146:835–847. doi: 10.1093/genetics/146.3.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasar F, Jankowski C, Nag DK. Long palindromic sequences induce double-strand breaks during meiosis in yeast. Mol Cell Biol. 2000;20:3449–3458. doi: 10.1128/mcb.20.10.3449-3458.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmakayalu MA, Gotter AL, Shaikh TH, Emanuel BS. A novel sequence-based approach to localize translocation break-points indentifies the molecular basis of a t(4;22) Hum Mol Genet. 2003;12:2817–2825. doi: 10.1093/hmg/ddg301. [DOI] [PubMed] [Google Scholar]
- O’Connell P, Leach RJ, Ledbetter DH, Cawthon RM, Culver M, Eldridge JR, Frej A-K, Holm TR, Wolff E, Thayer MJ, Schafer AJ, Fountain JW, Wallace MR, Collins FS, Skolnick MH, Rich DC, Fournier REK, Baty BJ, Carey JC, Leppert MF, Lathrop GM, Lalouel J-M, White RL. Fine structure DNA mapping studies of the chromosomal region harboring the genetic defect in neurofibromatosis type I. Am J Hum Genet. 1989;44:51–57. [PMC free article] [PubMed] [Google Scholar]
- Rattray AJ. A method for cloning and sequencing long palindromic DNA junctions. Nucleic Acids Res. 2004;32:e155. doi: 10.1093/nar/gnh143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning. 2nd edition Cold Spring Harbor Laboratory; New York: 1989. [Google Scholar]
- Sasaki N, Izawa M, Watahiki M, Ozawa K, Tanaka T, Yoneda Y, Matsuura S, Carninci P, Muramatsu M, Okazaki Y, Hayashizaki Y. Transcriptional sequencing: a method for DNA sequencing using RNA polymerase. Proc Natl Acad Sci USA. 1998;95:3455–3460. doi: 10.1073/pnas.95.7.3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh TH, Kurahashi H, Saitta SC, O’Hare AM, Hu P, Roe BA, Driscoll DA, McDonald-McGinn DM, Zackai EH, Budarf ML, Emanuel BS. Chromosome 22-specific low copy repeats and the 22q11.2 deletion syndrome: genomic organization and deletion endpoint analysis. Hum Mol Genet. 2000;9:489–501. doi: 10.1093/hmg/9.4.489. [DOI] [PubMed] [Google Scholar]
- Sinden RR. DNA structure and function. Academic Press; San Diego: 1994. [Google Scholar]
- Viskochil D, Buchberg AM, Xu G, Cawthon RM, Stevens J, Wolff RK, Culver M, Carey JC, Copeland NG, Jenkins NA, White R, O’Connell P. Deletions and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locus. Cell. 1990;62:187–192. doi: 10.1016/0092-8674(90)90252-a. [DOI] [PubMed] [Google Scholar]
- Waldman AS, Tran H, Goldsmith EC, Resnick MA. Long inverted repeats are an at-risk motif for recombination in mammalian cells. Genetics. 1999;153:1873–1883. doi: 10.1093/genetics/153.4.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace MR, Marchuk DA, Andersen LB, Letcher R, Odeh HM, Saulino AM, Fountain JW, Brereton A, Nicholson J, Mitchell AL, Brownstein BH, Collins FS. Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients. Science. 1990;249:181–186. doi: 10.1126/science.2134734. [DOI] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.