

Abstract
Hippocampal CA1 pyramidal neurons are selectively vulnerable to ischemia, while adjacent CA3 neurons are relatively resistant. Although glutamate receptor-mediated mitochondrial Ca2+ overload and dysfunction is a major component of ischemia-induced neuronal death, no direct relationship between selective neuronal vulnerability and mitochondrial dysfunction has been demonstrated in intact brain preparations. Here, we show that in organotypic slice cultures NMDA induces much larger Ca2+ elevations in vulnerable CA1 neurons than in resistant CA3. Consequently, CA1 mitochondria exhibit stronger calcium accumulation, more extensive swelling and damage, stronger depolarization of their membrane potential, and a significant increase in ROS generation. NMDA-induced Ca2+ and ROS elevations were abolished in Ca2+-free medium or by NMDAR antagonists, but not by zinc chelation. We conclude that Ca2+-overload-dependent mitochondrial dysfunction is a determining factor in the selective vulnerability of CA1 neurons.
Keywords: calcium, excitotoxicity, hippocampal slice cultures, reactive oxygen species, zinc, electron probe microanalysis
INTRODUCTION
CA1 hippocampal pyramidal neurons, but not adjacent CA3 neurons, are highly vulnerable to in vivo ischemic injury (Kirino, 2000). The selective vulnerability of CA1 has also been observed following in vitro ischemia/hypoxia or oxygen-glucose deprivation (OGD) in rodent organotypic slice cultures (Pringle et al., 1997; Perez Velazquez et al., 1997; Rytter et al., 2003). Glutamate excitotoxicity is thought to play a pivotal role in CA1 neuronal death because, among other reasons, NMDA receptor (NMDAR) and AMPA receptor (AMPAR) antagonists are neuroprotective following OGD (Pringle et al., 1997; Rytter et al., 2003). Independent avenues of research indicate that excessive NMDAR-dependent cytosolic Ca2+ elevation is a key event in excitotoxic death (Choi, 1995; Gillessen et al., 2002), although this is firmly established only in cultured neurons. However, several observations suggest the general importance of excessive Ca2+. For example, OGD-induced Ca2+ elevations have been implicated in CA1 vulnerability (Perez Velazquez et al., 1997; Bickler and Hansen, 1998; Cronberg et al., 2005) in hippocampal slices. Additionally, increasing external Ca2+ potentiates NMDA-evoked currents in CA1, but depresses them in CA3 neurons (Grishin et al., 2004); these differences were enhanced during OGD (Gee et al., 2006).
Large cytosolic Ca2+ elevations lead to mitochondrial calcium overload — a crucial step in excitotoxicity — which results in mitochondrial damage, activation of the mitochondrial permeability transition (MPT), the release of pro-apoptotic proteins, and the production of reactive oxygen species (ROS) (Starkov et al., 2004; Nicholls, 2004, 2009). This sequence is well established for isolated brain mitochondria and for neurons in culture, but information directly addressing the role of Ca2+ in ischemia-evoked mitochondrial dysfunction in in situ neurons is limited. Interestingly, MPT activation and ROS formation were found to be higher in mitochondria from CA1 than in those from CA3 (Mattiasson et al., 2003), suggesting — although there is no direct evidence — that there may be a relationship between the disparate vulnerability of CA1 vs. CA3 neurons and NMDA-induced, Ca2+ overload-mediated mitochondrial dysfunction in intact brain preparations.
This narrative has so far focused on Ca2+, on the presumption that it is the main signaling ion in excitotoxic injury cascades, but certain recent reports have questioned the fact and the role of Ca2+ elevations in CA1 neurons during OGD in acute slices, instead ascribing a pivotal role to Zn2+ (Stork and Li, 2006; Medvedeva et al., 2009). Zn2+ is known to be toxic, although its source and mechanism(s) of toxicity are largely obscure (Frederickson et al., 2005; Paoletti et al., 2009). In particular, it is not clear how Zn2+ toxicity does or does not relate to mitochondrial dysfunction in excitotoxicity, and whether mitochondria can appreciably buffer Zn2+ (Dineley et al., 2008).
In previous studies, we used, among other approaches, direct measurements of intracellular calcium concentrations to show that excitotoxic elevation of free cytosolic Ca2+ was paralleled by mitochondrial calcium overload (Pivovarova et al., 2004, 2008). Here, we report that CA1 hippocampal neurons in organotypic slice cultures are selectively vulnerable to NMDAR-dependent excitotoxicity because NMDAR over-activation induces Ca2+ elevations that are much larger in these vulnerable neurons than in resistant CA3 neurons. These results establish the previously missing link between CA1-selective neurodegeneration and Ca2+ overload-dependent mitochondrial dysfunction.
MATERIALS AND METHODS
Slice cultures and survival assays
Hippocampal organotypic slice cultures were prepared from day 6 postnatal Sprague-Dawley rats as previously described (Pozzo-Miller et al., 1997) and in accordance with the NINDS, NIH Animal Care and Use Committee Protocol No. ASP-1159. Slices display normal anatomical structure and electrophysiological properties by 7 days in vitro (DIV) and until at least 14 DIV. Prior to experiments, slices were subjected the propidium iodide (PI) viability staining (see below) and only those culture sets with no detectable dead cells were used. For experiments, 8–10 DIV slices were transferred into modified artificial cerebrospinal fluid (ACSF) containing in mM: 137 NaCl, 2.0 CaCl2, 2.0 MgSO4, 5.4 KCl, 0.3 Na2HPO4, 0.22 KH2PO4, 10 HEPES, 10 glucose, 26 sucrose (~310 mOsM, pH 7.4). Viability assays were also replicated in a 25 mM NaHCO3/CO2 buffered medium. To induce excitotoxicity, slices were exposed to 500 μM glutamate or 200 μM NMDA supplemented with 10 μM glycine in a Mg2+-free ACSF for 30 min at 37 °C. Chemical ischemia (CI) was simulated by substituting sucrose for glucose and KCl (70 mM) for NaCl in ACSF, and adding 10 mM cyanide and 2 mM 2-deoxyglucose. After exposure, cultures were washed in ACSF and returned to the incubator in growth medium for 20–24 h. As needed, inhibitors were added to the ACSF 10 min before excitotoxic or ischemic exposure. Cell death was assayed in defined regions of entire slices with 3.3 μg/ml PI staining according to a quantification method described previously (Pringle et al., 1997), modified to take into account the density of neurons, which was assayed by immunostaining for the neuron-specific marker NeuN (Chemicon, Temecula, CA). For normalization, the average relative fluorescence ratio of PI to NeuN per unit area in CA1 and CA3 regions was estimated using ImageJ software (http://rsb.info.nih.gov/ij/index.html).
Fluorescence microscopy
The Ca2+-sensitive low-affinity fluorescent probe fluo-4FF (excitation 488 nm, emission 515 nm; Invitrogen, Carlsbad, CA) was used to estimate the level of intracellular free Ca2+ in neurons. Cells in slices were loaded by incubation in 5 μM fluo-4FF AM ester in ACSF for 30 min at 37 °C. The membrane-permeant, cationic dye rhodamine-123 (Rh123, excitation 514 nm, emission 530 nm, Invitrogen) was used to assay changes in mitochondrial membrane potential (MMP). Slices were exposed for 30 min at 37 °C to 25 μM Rh123 in ACSF and washed with ACSF for additional 30 min before imaging. ROS levels were estimated using the superoxide-sensitive fluorescent indicator dihydroethidium (DHE, excitation 488 nm, emission 530 nm, Invitrogen). Slices were incubated with 4 μg/ml DHE for 15 min at 37°C and washed with ACSF.
For imaging, slices were placed in glass-bottom chambers at 25 °C on the stage of Zeiss LSM 510 confocal microscope (Carl Zeiss, Thornwood, NY; 5x objective). Central optical slices through neuronal cell layers were recorded every 30s for 20–40 min. Data are given as background-corrected ratio of fluorescence intensity to baseline fluorescence (F/F0).
Electron microscopy and electron probe x-ray microanalysis (EPMA)
For conventional electron microscopy, slice cultures were fixed, stained, Epon-embedded and sectioned using standard procedures. Sections were viewed in a JEOL 1200 transmission electron microscope (JEOL, Peabody, MA). Images were recorded digitally by means of an XR-100 CCD camera (AMT, Danvers, MA).
For EPMA, slices were frozen and cryo-sectioned as previously described (Pozzo-Miller et al., 1997; Pivovarova et al., 2002). Briefly, cryosections (~80 nm thick) were cut en face from previously marked CA1 or CA3 cell body layers of slam-frozen slice cultures by means of a Leica UC6 cryoultramicrotome (Leica, Bannockburn, IL) at − 160 °C. Pyramidal cell somatic regions were located in frozen specimens with the aid of fiducial marks identifying the strata pyramidale. Cryosections were transferred to an energy-filtering analytical cryo-electron microscope (Zeiss EM 912, Peabody, MA) and after freeze-drying were imaged using a ProScan (TRS) 2kx2k slow-scan CCD camera (Munster, Germany) controlled by AnalySIS image processing software (Olympus Soft Imaging Solutions, Lakewood, CO). X-ray spectra were recorded and processed by established procedures (Pivovarova et al., 1999). EPMA-derived total concentrations of Na, Mg, P, Cl, K and Ca measured in cytoplasm and mitochondria are given in mol/kg dry weight.
Statistics
For statistical comparisons, one-way ANOVA followed by post-hoc Dunnett multiple comparisons test was used for normally distributed data. For non-normally distributed data the nonparametric Kruskal-Wallis rank ANOVA with post-hoc Dunn’s test was used. Data are given as mean ± SEM. Analysis was performed using InStat software (GraphPad Software, San Diego, CA). In general, experiments were performed on at least three slice cultures prepared from different animals.
Reagents
Fluo-4FF, rhodamine-123 and dihydroethidium were purchased from Invitrogen (Carlsbad, CA). All other reagents were from Sigma-Aldrich (St. Louis, MO).
RESULTS
CA1-selective vulnerability is mediated by NMDAR-dependent Ca2+ elevation
We first established that in our rat organotypic slice cultures CA1 neurons were selectively vulnerable to excitotoxic insults. Chemical ischemia (CI) for 30 minutes resulted in delayed neuronal death in CA1, whereas neurons in CA3 were generally unaffected (Fig. 1). Cell death, estimated as the ratio of PI staining 24 hours after stimulus to NeuN immunofluorescence — the latter reflecting the total population of neurons — in standardized areas, was approximately twofold higher in CA1 compared to CA3 after CI, whereas there was no detectable cell death in either region before stimulation or in control slices subjected to the entire incubation protocol but without CI (not shown). This result is consistent with the previous observation that CA1 neurons of organotypic hippocampal slice cultures are selectively vulnerable to oxygen/glucose deprivation (OGD) (Gee et al., 2006). Since NMDA receptor-dependent excitotoxicity is a major component of ischemic cell death, we also tested the effects of slice exposure to toxic NMDA. As with CI, NMDA induced selective death in CA1 (PI/NeuN fluorescence ratio 2.4±0.4 times higher than in CA3, p<0.05), which was prevented by the NMDA antagonist MK-801 (Fig. 1), confirming that CA1-selective neuronal death was dependent on NMDAR activation.
Figure 1. Chemical ischemia or NMDA exposure induces selective death of CA1 hippocampal neurons.
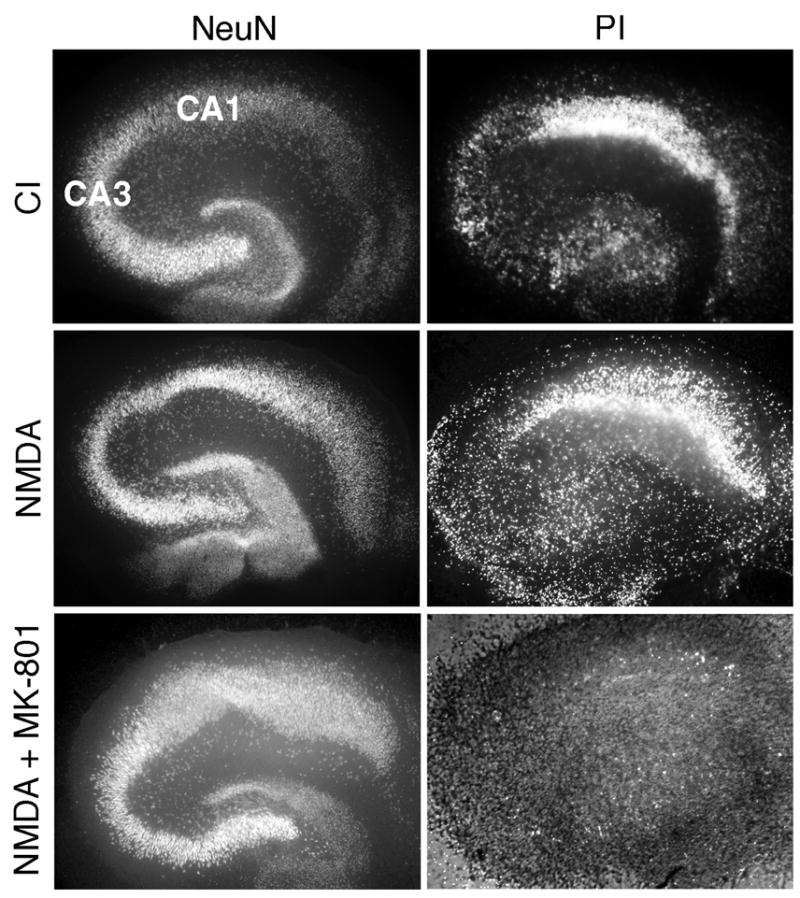
Propidium iodide (PI) staining 24 h after chemical ischemia (CI, see Methods for details) or NMDA (200 μM) exposure for 30–40 min reveals much higher PI fluorescence in CA1 compared to CA3, while staining for NeuN shows a similar density of neurons in these regions. NMDAR antagonist MK-801 completely prevents NMDA-induced cell death.
Having confirmed the expected NMDA vulnerability of CA1 neurons in hippocampal slice cultures, we next asked whether the Ca2+ overload that characterizes excitotoxic death in cultured neurons (Choi, 1995; Gillessen et al., 2002) might also distinguish vulnerable CA1 neurons from less vulnerable CA3 cells. The Ca2+-sensitive low-affinity fluorescent probe fluo-4FF revealed during 30 min of NMDA exposure a rapid, biphasic cytosolic Ca2+ increase, followed by a sustained plateau, in CA1 (Figs. 2 and 3E; Supplemental Video). Higher-magnification images demonstrate that fluorescence originates from individual CA1 neurons (Fig. 2, bottom panel). In contrast, no Ca2+ elevations were detected in CA3 neurons (Figs. 2 and 3E). Dramatic differences in CA1 vs. CA3 Ca2+ elevations were also observed during CI and glutamate exposure (Fig. 3A, C). MK-801 strongly reduced CI-induced Ca2+ elevations (Fig. 3B) and completely abolished those induced by glutamate (Fig. 3D) or NMDA (not shown), indicating that Ca2+ entry through NMDARs is a predominant component of the response. Since it was confirmed that Ca2+ responses to CI and glutamate are mediated by NMDARs, NMDA was used to induce excitotoxicity in remaining experiments.
Figure 2. NMDA induces Ca2+ elevations selectively in CA1 neurons.
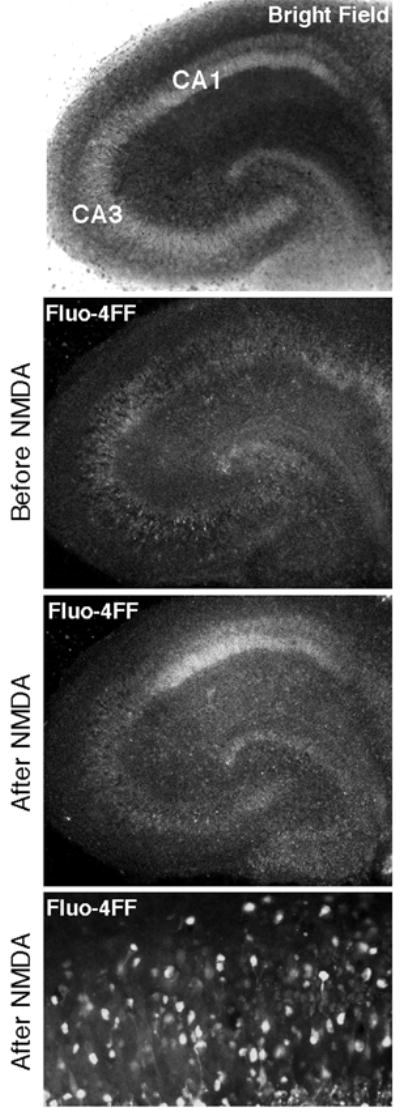
Confocal fluorescence images of fluo-4FF-loaded slices reveal NMDA-induced (200 μM, 30 min) Ca2+ changes in a representative hippocampal slice culture. Intracellular fluorescence was equally low across the entire slice area before stimulation, but increased almost exclusively in CA1 after NMDA application. Bottom panel illustrates elevated free Ca2+ in individual CA1 pyramidal neurons. Images in top three panels were taken using a 5x objective; bottom panel used a 20x objective.
Figure 3. Large Ca2+ elevations in CA1 neurons are mediated by NMDAR activation.

Ca2+ traces, presented as normalized fluo-4FF fluorescence ±SEM in the CA1 (red) and CA3 (blue) regions of hippocampal slices, demonstrate large, persistent Ca2+ elevations in CA1, but not CA3, during CI (A) or exposure to 500 μM glutamate (C) or 200 μM NMDA (E). The NMDAR antagonist MK-801 largely reduces CI-induced (B) while eliminating glutamate-induced (D) Ca2+ elevations. The Zn2+ chelator TPEN (50 μM) has no effect on Fluo-4FF fluorescence increase, while Ca2+ chelator EGTA (2 mM) completely abolished any fluorescence increase (F). Experiments were performed on slices from at least three different slices prepared from three different animals.
Certain recent studies have questioned the role of Ca2+ overload in OGD-induced CA1 degeneration in acute hippocampal slices, suggesting instead that excess Zn2+ accumulates in neurons and precedes Ca2+ deregulation (Stork and Li, 2006; Medvedeva et al., 2009). To examine this possibility in the context of CA1 excitotoxicity in slice cultures, we tested the effects of removing extracellular Ca2+ (by chelation with EGTA) or, alternatively, removing intra- and extracellular Zn2+ (by chelation with TPEN). EGTA completely eliminated NMDA-induced fluo-4FF fluorescence increases in CA1, while TPEN had no effect on fluorescence (Fig. 3F) nor on cell viability (not shown), indicating that fluo-4FF is sensitive to Ca2+, not Zn2+, and that in organotypic slice cultures elevation of Ca2 is the primary early step that initiates the selective, NMDAR-dependent death of CA1 neurons.
NMDA-induced mitochondrial calcium accumulation and damage is higher in CA1 than in CA3 neurons
There is a convincing evidence that high cytosolic Ca2+ elevations lead to mitochondrial Ca2+ overload and dysfunction, a crucial step in the excitotoxic death of cultured CNS neurons (Nicholls, 2004, 2009). Therefore, we used electron probe x-ray microanalysis (EPMA) to measure NMDA-induced changes of total Ca concentrations in neuronal cytoplasm, as well as within individual neuronal mitochondria, in CA1 and CA3 regions. Consistent with the response of free cytosolic Ca2+, excitotoxic NMDA exposure led to increases in both cytoplasmic and mitochondrial total Ca concentrations that were much higher in CA1 than in CA3 neurons (Fig. 4A, B; Table 1). Similar to previous observations for delayed excitotoxic death in dispersed hippocampal neurons (Pivovarova et al., 2004), 30 min NMDA exposure resulted in robust Ca accumulation in a subpopulation of mitochondria in both CA1 and CA3 neurons such that Ca was heterogeneously distributed both between and within mitochondria. The avidly Ca-sequestering subpopulation contained numerous Ca-rich precipitates with exceedingly high Ca concentrations (>1000 kg/kg dry weight; Table 1, footnote d). Both the mean Ca concentration (Fig. 4B, Table 1) and the fraction of strongly Ca-accumulating mitochondria (Table 1, footnote d) were larger in CA1 than in CA3 neurons. No significant changes in astrocyte Ca concentrations were detected (Fig. 4A, B).
Figure 4. NMDA-induced total calcium accumulation is higher in CA1 neurons than in CA3 neurons.

NMDA exposure (200 μM, 30 min) induced large increases in total Ca concentrations in cytoplasm (A) and mitochondria (B) of CA1 neurons, but much smaller increases in CA3; glial cell concentrations were unchanged (*, p<0.05 relative to CA1 exposed to NMDA). NMDA-induced redistribution of cytoplasmic Na and K was similar in neurons of both regions (C), but less extensive in mitochondria of CA3 neurons (D), (p<0.05). MK-801 generally prevented NMDA-induced changes in Ca, Na and K concentrations. Data are given as mean ±SEM, as measured by electron probe microanalysis (EPMA) on cryosections from at least three different slices prepared from three different animals.
Table 1.
Effect of excitotoxic stimulation on elemental concentrations in cytoplasm and mitochondria of CA1 and CA3 hippocampal neurons
| n | Na | Mg | Cl | P | K | Ca | |
|---|---|---|---|---|---|---|---|
| (kg/kg dry weight) | |||||||
| Cytoplasm | |||||||
| CA1 | |||||||
| Controla | 24 | 29±5 | 34±3 | 74±4 | 510±26 | 549±38 | 0.7±0.8b |
| NMDA | 58 | 666±20 | 31±2 | 510±20 | 356±13 | 58±4 | 38±1.9 |
| Recovery 2 h | 25 | 247±32* | 37±5 | 183±14* | 440±39 | 382±24* | 3.8±0.4* |
| NMDA+MK-801 | 34 | 132±22* | 34±3 | 123±15* | 431±22 | 362±20* | 3.2±2.0* |
| CA3 | |||||||
| NMDAc | 35 | 642±26 | 41±3** | 443±25** | 551±42** | 131±11** | 10±1.7** |
| Mitochondria | |||||||
| CA1 | |||||||
| Controla | 20 | 16±5 | 33±4 | 35±4 | 402±15 | 389±15 | −1.7±1.4b |
| NMDAd | 63 | 406±28 | 23±3 | 330±23 | 526±53 | 46±6 | 421±86 |
| Recovery 2 h | 28 | 96±14* | 36±3* | 80±7* | 345±13 | 283±14* | 3.4±2.0* |
| NMDA+MK-801 | 30 | 49±12* | 33±2* | 44±7* | 392±17 | 297±16* | 1.7±0.8* |
| CA3 | |||||||
| NMDAc,d | 47 | 346±42 | 27±2** | 286±38 | 401±27 | 112±13** | 121±51** |
Cultures were frozen immediately after exposure to NMDA (100 μM) or to NMDA with MK-801 (20μM) for 30 min. Data are given as mean ± SEM in kg/kg dry weight. The number of neurons analyzed was 10–12 for each condition, taken from slices of three different animals; n is the number of analyses.
For both cytoplasm and mitochondria, Mg, P and Ca concentrations in ‘Control’ were not different from ‘Recovery’ or ‘NMDA+MK-801’. ‘Control’ concentrations of Na, Cl are lower, while K is higher (p<0.05).
Below detection limit.
A small fraction of cells, ~15%, was not different from CA1 ‘Control’.
Average concentrations from a random sample of mitochondria that are heterogeneous with respect to the presence or absence of Ca-rich precipitates. In CA1 neurons Ca concentrations were 1324±139 (~30%) and 31±3 (~70%) kg/kg for mitochondria with and without precipitates, respectively. In CA3 comparable mitochondrial Ca concentrations were 1036±71 (~10%) and 12±3 (~90%) kg/kg.
Different from CA1 NMDA stimulation by ANOVA with post-hoc Dunnett multiple comparisons test for cytoplasm and nonparametric Kruskal-Wallis rank ANOVA with post-hoc Dunn’s test for unbalanced data for mitochondria, p < 0.05.
Different from CA1 NMDA stimulation by two-tailed unpaired t test for cytoplasm and nonparametric Mann-Whitney test for mitochondria, p < 0.05.
Ca accumulation by CA1 neurons was effectively abolished by MK-801 (Fig. 4A, B). Elevated Ca in CA1 neurons returned to pre-stimulus levels after a 2 h recovery (Table 1), indicating that in the majority of cells death was delayed relative to Ca overload. NMDAR over-stimulation also reversed the normal cytoplasmic and mitochondrial concentrations of Na and K; CA1 and CA3 neurons were similar in this respect (Fig. 4C, D). As with Ca concentration changes, Na/K reversal was prevented by MK-801 (Fig 4C, D).
In concert with total Ca overload, mitochondria of neurons (which are readily distinguished from glia at the electron microscopic level; see Supplemental Fig. 1), were characterized by massive swelling, but only in a subset of mitochondria. In unstimulated CA1 neurons, mitochondria have a normal filamentous morphology (Fig. 5, top panel). After NMDA exposure, a large fraction of mitochondria (Fig. 5, bar graph) became round, with lucent matrices and greatly increased volume (Fig. 5, middle panel). The majority of swollen mitochondria retained their high-Ca precipitates, which are preserved in frozen and freeze-substituted, but not in fixed, preparations (Fig. 5, middle panel, inset). The fraction of swollen, damaged mitochondria in CA3 neurons after NMDA exposure (Fig. 5, bottom panel) was much smaller than in CA1 neurons (Fig. 5, bar graph).
Figure 5. Fraction of damaged mitochondria after NMDA exposure is higher in CA1 than in CA3 neurons.

Electron micrographs of representative neuronal cell bodies of CA1 and CA3 regions of hippocampal slice culture conventionally fixed after exposure to NMDA (200 μM, 30 min). NMDA-treated CA1 neurons exhibit a large number of swollen mitochondria with lucent, low density matrices, while the majority of mitochondria in CA3 neurons are structurally similar to those in CA1 controls (arrows). Neurons display characteristic round, lucent nuclei (pieces of which are included in each panel for purposes of identification) and are readily distinguished from glial cells in slice preparations (see Supplemental Fig. 1). Inset: Representative field from a freeze-dried cryosection of the CA1 region of a slice culture illustrates at higher magnification a swollen mitochondrion with calcium-rich inclusions. Bars for all panels and inset, 2 μm. Bar graph compares the fraction of swollen mitochondria in CA1 (64.4±3.0%) and CA3 neurons (30.3±3.1%) after NMDA exposure. Data are means from two experiments, n=31 (CA1) and n=25 (CA3), p<0.05.
Mitochondrial Ca2+ overload and structural changes in cultured neurons are invariably accompanied by depolarization of the mitochondrial membrane potential (MMP) (Pivovarova et al., 2004; Ward et al., 2007). The fluorescent probe rhodamine 123 (Rh-123) was used to estimate and compare MMP depolarization in CA1 and CA3 neurons during excitotoxic challenge. NMDA application resulted in a larger increase in Rh-123 fluorescence in CA1 relative to CA3 (Fig. 6A). MMP depolarization was apparently only partial, however, since the protonophore FCCP, which is expected to completely depolarize all mitochondria, induced a further, strong and similar fluorescence increase in both CA1 and CA3 (Fig. 6A).
Figure 6. NMDA induces selective MMP depolarization and ROS generation in CA1 neurons.

(A) Representative traces of baseline-normalized rhodamine 123 (Rh-123) fluorescence intensities show partial MMP depolarization induced by NMDA (200 μM), which is stronger in CA1 compared to CA3. FCCP (1μM) application produced equivalent MMP depolarization in both regions. (B) Traces of normalized fluorescence of dihydroethidium (DHE) report large, persistent increases in ROS generation following NMDA (200 μM) application in CA1 but not in CA3 region. (C) Blocking mitochondrial Ca2+ uptake with FCCP (1μM) or exposure to NMDA in Ca2+-free medium (EGTA, 2mM) prevents ROS generation, but Zn2+ chelation (TPEN, 50 μM) was without effect.
CA1-selective ROS generation is mediated by mitochondrial Ca2+ overload
The excessive ROS generation associated with ischemic or excitotoxic neuronal death is also thought to be mediated by mitochondrial Ca2+ overload (Brookes et al., 2004; Starkov et al., 2004). We estimated ROS generation in hippocampal slice cultures during excitotoxic NMDA by measuring superoxide production with the fluorescent dye dihydroethidium (DHE). NMDA induced a significant increase in DHE fluorescence in CA1, but not in CA3 (Fig. 6B). ROS generation was prevented by blocking mitochondrial Ca2+ uptake, either with FCCP or by NMDA exposure in a nominally Ca2+-free medium (Fig. 6C). The Zn2+ chelator TPEN had no effect on ROS production, nor on cell viability (not shown) indicating that CA1-selective ROS generation and cell death are predominantly mediated by strong accumulation of Ca2+ in neuronal mitochondria.
DISCUSSION
This study provides evidence for the previously missing link between CA1-selective neurodegeneration and Ca2+ overload-dependent mitochondrial dysfunction, and indicates that over-activation of NMDARs in organotypic hippocampal slice cultures induces much stronger Ca2+ accumulation in ischemia-vulnerable CA1 neurons than in adjacent, resistant CA3 neurons. This difference in Ca2+ load between CA1 and CA3 neurons was paralleled by differences in mitochondrial Ca2+ overload, dysfunction and ROS generation. All of these are known key downstream events in the established pathway for glutamate excitotoxicity in isolated neurons in culture. Although glutamate excitotoxicity also appears to be a major factor in neuronal death following ischemia, the mechanistic details are largely unknown. In particular, the relative importance of Ca2+-dependent vs. Ca2+-independent mechanisms is not clear. Since we show here that selective, NMDAR-dependent Ca2+ accumulation in CA1, but not CA3, is induced by glutamate, and to a large extent by chemical ischemia, it seems likely that difference in Ca2+ loading could be a major reason for the differential susceptibility of CA1 and CA3 neurons to ischemia. These observations are consistent with reports that NMDA is more toxic to CA1 than to CA3 neurons (Aguirre and Baudry, 2009), and that OGD-induced Ca2+ elevations in CA1 neurons of rat acute hippocampal slices (Bickler and Hansen, 1998) and organotypic slice cultures (Frantseva et al., 2001) are NMDAR-dependent.
NMDAR-dependent Ca2+ overload in the mitochondria of CA1 neurons (Fig. 4, Table 1) is the trigger for CA1-selective mitochondrial damage and dysfunction, as manifested by mitochondrial swelling (Fig. 5), MMP depolarization (Fig. 6A) and ROS production (Fig. 6C). The first two of these are characteristics of Ca2+-overload-induced MPT (Bernardi et al., 2006), which implies that MPT is involved in the selective vulnerability of CA1 neurons. There is strong evidence that Ca2+-induced MPT plays a role in excitotoxic death of cultured isolated neurons (Jemmerson et al., 2005; Li et al., 2009; Nicholls, 2009), and MPT has also been implicated in delayed neuronal death following transient ischemia (Baines et al., 2005; Schinzel et al., 2005). The stronger Ca2+ accumulation and larger fraction of damaged, swollen mitochondria observed here in CA1 as compared to CA3 suggests that stronger MPT activation accounts for the selective excitotoxicity susceptibility of CA1. This potential mechanism is reinforced if, as previously reported, CA1 neuronal mitochondria are inherently more susceptibility to MPT than CA3 mitochondria (Mattiasson et al., 2003). As we previously showed in dispersed hippocampal cultures (Pivovarova et al., 2004) and confirmed here in slices, neurons mainly recover pre-stimulus Ca2+ levels within a few hours of stimulus removal, and only show signs of death much later. This delay is accounted for by the heterogeneity of mitochondrial damage, whereby damaged mitochondria initiate apoptosis-like death signals while undamaged, healthy mitochondria continue to produce the ATP required by downstream energy-dependent pathways (Pivovarova et al., 2004). We should note that mitochondrial damage has been recognized as a hallmark of neuronal degeneration, both in vitro and in vivo, for over four decades (Olney and Sharpe, 1969).
Mitochondria are a primary source of ROS, which contributes to oxidative stress and cell injury (Feissner et al., 2009). Blocking ROS generation during OGD in hippocampal slice cultures was neuroprotective (Zhou and Baudry, 2009). Mitochondrial Ca2+ accumulation is also thought to be responsible for the production of ROS during excitotoxicity (Chinopoulos and Adam-Vizi, 2006; Starkov et al., 2004). Here we observed NMDA-induced, selective generation of ROS in CA1, but not in CA3. CA1 ROS production appears to be mediated by mitochondrial Ca2+ uptake, since chelating extracellular Ca2+ with EGTA or eliminating mitochondrial Ca2+ uptake with the protonophore FCCP completely abolished ROS generation (Fig. 6C), an observation consistent with recent findings in cultured striatal neurons (Duan et al., 2007).
Recent reports suggest that the role ascribed to Ca2+ during OGD in acute hippocampal slices could in part be attributable to Zn2+ (Stork and Li, 2006; Medvedeva et al., 2009). Here, we found no effect of the membrane-permeant Zn2+ chelator TPEN on either ROS production, excitotoxic cell death, or NMDA-induced increases in fluorescence from the low-affinity Ca2+ probe fluo-4FF (Fig. 3F). In contrast, chelating external Ca2+ with EGTA was very effective, completely abolishing cytosolic Ca2+ increases. It seems unlikely that these observations are compromised by crosstalk between the cations, since in our hands fluo-4FF is Zn2+-insensitive. Therefore, these observations provide evidence that, in apparent contrast to the role of Zn2+ in OGD of acute slices, fast Ca2+ elevation following NMDA stimulation is a primary and early event in vulnerable neurons of slice cultures. Moreover, independent measurements of total Ca concentrations and distributions by EPMA indicate that exposure of hippocampal slices to NMDA results in intracellular Ca, not Zn, accumulation in vulnerable neurons. We also found only a slight and insignificant effect of the Zn2+ chelator TPEN on the production of ROS (Fig. 6C). The possibility that NMDA-induced release of Zn2+ from intracellular stores could trigger MPT and ROS generation was suggested earlier (Bossy-Wetzel et al., 2004), although other studies argue that Ca2+-dependent mitochondrial ROS generation occurs before, and is responsible for, the mobilization of toxic Zn2+ (Dineley et al., 2008). There is convincing evidence that Zn2+ chelation is protective against ischemic neuronal death (Calderone et al., 2004), but whether and how Zn2+ contributes to excitotoxicity, as well as the source of Zn2+ ions, remains to be determined (Paoletti et al., 2009).
Beyond the role of small molecules and ions, there remain interesting questions about the glutamate receptors responsible for toxic Ca2+ entry. Particularly relevant are questions about post-translational modifications and receptor trafficking. It has been reported that ischemia leads to CA1-specific, Cdk5-mediated phosphorylation of NMDARs, and that this step is directly responsible for selective death of CA1 neurons (Wang et al., 2003). These observations may link to Ca2+ overload in that prolonged activation of Cdk5 is dependent on calpain activity, which is in turn up-regulated by cytosolic, and possibly mitochondrial, Ca2+ elevations (Badugu et al., 2008). Concerning the subtype composition of GluRs, evidence suggests that ischemia induces CA1-selective downregulation of GluR2 mRNA expression and internalization of GluR2-containing AMPARs, resulting in increased Ca2+ influx via GluR2-lacking, Ca2+-permeable AMPARs (Kwak and Weiss, 2006; Liu and Zukin, 2007). These processes have been implicated in delayed neuronal death. The reasons for selective expression of Ca2+-permeable AMPARs in CA1 neurons are unknown, but NMDAR activity and Ca2+ signaling are thought to be involved in GluR2 trafficking and internalization (Lin and Huganir, 2007; Dixon et al., 2009). Thus, the CA1-selective, NMDAR-dependent Ca2+ elevations described here might serve as an upstream signal for switching AMPAR subunit composition and/or location. Many mechanistic details remain controversial, however. For example, in cortical neurons both in vitro NMDA exposure and transient ischemia were reported to reduce, in a calpain-dependent manner, the level of surface-expressed and total GluR1, but not GluR2 as might have been expected (Yuen et al., 2007).
The important question, especially for successful therapeutic interventions, is why NMDA induces selective Ca2+ elevation in CA1 neurons. Several scenarios can be envisioned. As the expression of NR1 and NR2B subunits is higher in CA1 as compared to CA3 (Coultrap et al., 2005), perhaps there are simply more surface-expressed NMDARs in CA1, and consequently a larger calcium load. Another possibility considers differential Ca2+ buffering capacities. Calbindin-D28k immunoreactivity is higher in rat CA1 than in CA3 pyramidal cells (Sloviter, 1989; Muller et al., 2005). This is consistent with our EPMA data revealing a higher total Ca content in activated CA1 neurons in the sense that both observations indicate a higher level of Ca2+ buffering in CA1. However, more Ca2+ buffering power does not seem to translate to excitotoxicity resistance. To the contrary, calbindin knockout mice showed generally reduced susceptibility to ischemic damage (Klapstein et al., 1998; Schwaller et al., 2002), while the loss of calbindin diminished VGCC-dependent Ca2+ influx during repetitive neuronal firing in the dentate gyrus and was neuroprotective (Nagerl et al., 2000). Both observations imply that enhanced Ca2+ buffering would not be neuroprotective. The evidence to this point suggests that ischemia-vulnerable and -resistant neurons are characterized by differences in cytosolic Ca2+ buffering, suggesting that this is yet another of the many factors that can play a role in ischemic vulnerability. Complexity notwithstanding, one primary excitotoxic injury event appears to be NMDAR-dependent excessive Ca2+ elevation leading to mitochondrial dysfunction and activation of downstream death pathways, mechanisms that are quite similar to those well established for dispersed cultures.
Supplementary Material
Supplemental Figure 1. Conventional electron micrographs illustrating the anatomical organization of CA1 and CA3 pyramidal neurons in organotypic slice cultures of hippocampus. Both CA1 (top and middle panels) and CA3 (bottom panel) pyramidal neurons are characterized by large, adjacent cell bodies containing round, lucent nuclei (N). Neuronal cell bodies are tightly packed in rows, an arrangement that clearly demarcates neurons. Glial somata are rarely seen between neuronal cell bodies in the stratum pyramidale, but when present are readily distinguished by a characteristic irregular nucleus with a chromatin border (GN). Bar for all panels, 2 μm.
NMDA-evoked selective Ca2+ elevations in CA1 neurons. A 30-min time-lapse record of Ca2+ dynamics during NMDA exposure (200 μM) from neurons of a representative hippocampal slice culture loaded with the low-affinity fluorescent probe fluo-4FF. NMDA induces a rapid, transient fluorescence increase followed by a sustained plateau (beginning about halfway through the video) in area CA1; there is no detectable response in CA3. Images were recorded at the neuronal cell body focal plane using a 5x objective. Images were collected every 30 sec for the first 5 min and every 60 sec for the remaining 25 min. Frame rate, 5 frames/s.
Acknowledgments
The authors are indebted to Dr. C.G. Thomas for initial electrophysiological characterization of our slice culture preparations, as well as for many helpful discussions, and to Dr. C.A. Brantner and the staff of the NINDS EM facility, Dr. Jung-Hwa Tao-Cheng, Director, for excellent technical assistance. This research was supported by the Intramural Research Program of the NIH, NINDS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguirre CC, Baudry M. Progesterone reverses 17-β-estradiol-mediated neuroprotection and BDNF induction in cultured hippocampal slices. Eur J Neurosci. 2009;29:447–454. doi: 10.1111/j.1460-9568.2008.06591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badugu R, Garcia M, Bondada V, Joshi A, Geddes JW. N terminus of calpain 1 is a mitochondrial targeting sequence. J Biol Chem. 2008;283:3409–3417. doi: 10.1074/jbc.M706851200. [DOI] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- Bernardi P, Krauskopf A, Basso E, Petronilli V, Blachly-Dyson E, Di Lisa F, Forte MA. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 2006;273:2077–2099. doi: 10.1111/j.1742-4658.2006.05213.x. [DOI] [PubMed] [Google Scholar]
- Bickler PE, Hansen BM. Hypoxia-tolerant neonatal CA1 neurons: relationship of survival to evoked glutamate release and glutamate receptor-mediated calcium changes in hippocampal slices. Brain Res Dev Brain Res. 1998;106:57–69. doi: 10.1016/s0165-3806(97)00189-2. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Talantova MV, Lee WD, Scholzke MN, Harrop A, Mathews E, Gotz T, Han J, Ellisman MH, Perkins GA, Lipton SA. Crosstalk between nitric oxide and zinc pathways to neuronal cell death involving mitochondrial dysfunction and p38-activated K+ channels. Neuron. 2004;41:351–365. doi: 10.1016/s0896-6273(04)00015-7. [DOI] [PubMed] [Google Scholar]
- Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. 2004;287:C817–833. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- Calderone A, Jover T, Mashiko T, Noh KM, Tanaka H, Bennett MV, Zukin RS. Late calcium EDTA rescues hippocampal CA1 neurons from global ischemia-induced death. J Neurosci. 2004;24:9903–9913. doi: 10.1523/JNEUROSCI.1713-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinopoulos C, Adam-Vizi V. Calcium, mitochondria and oxidative stress in neuronal pathology. Novel aspects of an enduring theme. FEBS J. 2006;273:433–450. doi: 10.1111/j.1742-4658.2005.05103.x. [DOI] [PubMed] [Google Scholar]
- Choi DW. Calcium: still center-stage in hypoxic-ischemic neuronal death. Trends Neurosci. 1995;18:58–60. [PubMed] [Google Scholar]
- Coultrap SJ, Nixon KM, Alvestad RM, Valenzuela CF, Browning MD. Differential expression of NMDA receptor subunits and splice variants among the CA1, CA3 and dentate gyrus of the adult rat. Brain Res Mol Brain Res. 2005;135:104–111. doi: 10.1016/j.molbrainres.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Cronberg T, Rytter A, Wieloch T. Chelation of intracellular calcium reduces cell death after hyperglycemic in vitro ischemia in murine hippocampal slice cultures. Brain Res. 2005;1049:120–127. doi: 10.1016/j.brainres.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Dineley KE, Devinney MJ, 2nd, Zeak JA, Rintoul GL, Reynolds IJ. Glutamate mobilizes [Zn2+] through Ca2+ -dependent reactive oxygen species accumulation. J Neurochem. 2008;106:2184–2193. doi: 10.1111/j.1471-4159.2008.05536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon RM, Mellor JR, Hanley JG. Pick1-mediated glutamate receptor subunit 2 (GLUR2) trafficking contributes to cell death in oxygen/glucose deprived hippocampal neurons. J Biol Chem. 2009;284:14230–14235. doi: 10.1074/jbc.M901203200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Y, Gross RA, Sheu SS. Ca2+-dependent generation of mitochondrial reactive oxygen species serves as a signal for poly(ADP-ribose) polymerase-1 activation during glutamate excitotoxicity. J Physiol. 2007;585:741–758. doi: 10.1113/jphysiol.2007.145409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feissner RF, Skalska J, Gaum WE, Sheu SS. Crosstalk signaling between mitochondrial Ca2+ and ROS. Front Biosci. 2009;14:1197–1218. doi: 10.2741/3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantseva MV, Carlen PL, Perez Velazquez JL. Dynamics of intracellular calcium and free radical production during ischemia in pyramidal neurons. Free Radic Biol Med. 2001;31:1216–1227. doi: 10.1016/s0891-5849(01)00705-5. [DOI] [PubMed] [Google Scholar]
- Frederickson CJ, Koh JY, Bush AI. The neurobiology of zinc in health and disease. Nat Rev Neurosci. 2005;6:449–462. doi: 10.1038/nrn1671. [DOI] [PubMed] [Google Scholar]
- Gee CE, Benquet P, Raineteau O, Rietschin L, Kirbach SW, Gerber U. NMDA receptors and the differential ischemic vulnerability of hippocampal neurons. Eur J Neurosci. 2006;23:2595–2603. doi: 10.1111/j.1460-9568.2006.04786.x. [DOI] [PubMed] [Google Scholar]
- Gillessen T, Budd SL, Lipton SA. Excitatory amino acid neurotoxicity. Adv Exp Med Biol. 2002;513:3–40. doi: 10.1007/978-1-4615-0123-7_1. [DOI] [PubMed] [Google Scholar]
- Grishin AA, Gee CE, Gerber U, Benquet P. Differential calcium-dependent modulation of NMDA currents in CA1 and CA3 hippocampal pyramidal cells. J Neurosci. 2004;24:350–355. doi: 10.1523/JNEUROSCI.4933-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemmerson R, Dubinsky JM, Brustovetsky N. Cytochrome C release from CNS mitochondria and potential for clinical intervention in apoptosis-mediated CNS diseases. Antioxid Redox Signal. 2005;7:1158–1172. doi: 10.1089/ars.2005.7.1158. [DOI] [PubMed] [Google Scholar]
- Kirino T. Delayed neuronal death. Neuropathology. 2000;20(Suppl):S95–97. doi: 10.1046/j.1440-1789.2000.00306.x. [DOI] [PubMed] [Google Scholar]
- Klapstein GJ, Vietla S, Lieberman DN, Gray PA, Airaksinen MS, Thoenen H, Meyer M, Mody I. Calbindin-D28k fails to protect hippocampal neurons against ischemia in spite of its cytoplasmic calcium buffering properties: evidence from calbindin-D28k knockout mice. Neuroscience. 1998;85:361–373. doi: 10.1016/s0306-4522(97)00632-5. [DOI] [PubMed] [Google Scholar]
- Kwak S, Weiss JH. Calcium-permeable AMPA channels in neurodegenerative disease and ischemia. Curr Opin Neurobiol. 2006;16:281–287. doi: 10.1016/j.conb.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Li V, Brustovetsky T, Brustovetsky N. Role of cyclophilin D-dependent mitochondrial permeability transition in glutamate-induced calcium deregulation and excitotoxic neuronal death. Exp Neurol. 2009;218:171–182. doi: 10.1016/j.expneurol.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DT, Huganir RL. PICK1 and phosphorylation of the glutamate receptor 2 (GluR2) AMPA receptor subunit regulates GluR2 recycling after NMDA receptor-induced internalization. J Neurosci. 2007;27:13903–13908. doi: 10.1523/JNEUROSCI.1750-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SJ, Zukin RS. Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 2007;30:126–134. doi: 10.1016/j.tins.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Mattiasson G, Friberg H, Hansson M, Elmer E, Wieloch T. Flow cytometric analysis of mitochondria from CA1 and CA3 regions of rat hippocampus reveals differences in permeability transition pore activation. J Neurochem. 2003;87:532–544. doi: 10.1046/j.1471-4159.2003.02026.x. [DOI] [PubMed] [Google Scholar]
- Medvedeva YV, Lin B, Shuttleworth CW, Weiss JH. Intracellular Zn2+ accumulation contributes to synaptic failure, mitochondrial depolarization, and cell death in an acute slice oxygen-glucose deprivation model of ischemia. J Neurosci. 2009;29:1105–1114. doi: 10.1523/JNEUROSCI.4604-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller A, Kukley M, Stausberg P, Beck H, Muller W, Dietrich D. Endogenous Ca2+ buffer concentration and Ca2+ microdomains in hippocampal neurons. J Neurosci. 2005;25:558–565. doi: 10.1523/JNEUROSCI.3799-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagerl UV, Mody I, Jeub M, Lie AA, Elger CE, Beck H. Surviving granule cells of the sclerotic human hippocampus have reduced Ca2+ influx because of a loss of calbindin-D28k in temporal lobe epilepsy. J Neurosci. 2000;20:1831–1836. doi: 10.1523/JNEUROSCI.20-05-01831.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG. Mitochondrial dysfunction and glutamate excitotoxicity studied in primary neuronal cultures. Curr Mol Med. 2004;4:149–177. doi: 10.2174/1566524043479239. [DOI] [PubMed] [Google Scholar]
- Nicholls DG. Mitochondrial calcium function and dysfunction in the central nervous system. Biochim Biophys Acta. 2009;1787:1416–1424. doi: 10.1016/j.bbabio.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olney JW, Sharpe LG. Brain lesions in an infant rhesus monkey treated with monsodium glutamate. Science. 1969;166:386–388. doi: 10.1126/science.166.3903.386. [DOI] [PubMed] [Google Scholar]
- Paoletti P, Vergnano AM, Barbour B, Casado M. Zinc at glutamatergic synapses. Neuroscience. 2009;158:126–136. doi: 10.1016/j.neuroscience.2008.01.061. [DOI] [PubMed] [Google Scholar]
- Perez Velazquez JL, Frantseva MV, Carlen PL. In vitro ischemia promotes glutamate-mediated free radical generation and intracellular calcium accumulation in hippocampal pyramidal neurons. J Neurosci. 1997;17:9085–9094. doi: 10.1523/JNEUROSCI.17-23-09085.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pivovarova NB, Hongpaisan J, Andrews SB, Friel DD. Depolarization-induced mitochondrial Ca accumulation in sympathetic neurons: spatial and temporal characteristics. J Neurosci. 1999;19:6372–6384. doi: 10.1523/JNEUROSCI.19-15-06372.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pivovarova NB, Nguyen HV, Winters CA, Brantner CA, Smith CL, Andrews SB. Excitotoxic calcium overload in a subpopulation of mitochondria triggers delayed death in hippocampal neurons. J Neurosci. 2004;24:5611–5622. doi: 10.1523/JNEUROSCI.0531-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pivovarova NB, Pozzo-Miller LD, Hongpaisan J, Andrews SB. Correlated calcium uptake and release by mitochondria and endoplasmic reticulum of CA3 hippocampal dendrites after afferent synaptic stimulation. J Neurosci. 2002;22:10653–10661. doi: 10.1523/JNEUROSCI.22-24-10653.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pivovarova NB, Stanika RI, Watts CA, Brantner CA, Smith CL, Andrews SB. Reduced calcium-dependent mitochondrial damage underlies the reduced vulnerability of excitotoxicity-tolerant hippocampal neurons. J Neurochem. 2008;104:1686–1699. doi: 10.1111/j.1471-4159.2007.05080.x. [DOI] [PubMed] [Google Scholar]
- Pozzo-Miller LD, Pivovarova NB, Leapman RD, Buchanan RA, Reese TS, Andrews SB. Activity-dependent calcium sequestration in dendrites of hippocampal neurons in brain slices. J Neurosci. 1997;17:8729–8738. doi: 10.1523/JNEUROSCI.17-22-08729.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pringle AK, Iannotti F, Wilde GJ, Chad JE, Seeley PJ, Sundstrom LE. Neuroprotection by both NMDA and non-NMDA receptor antagonists in in vitro ischemia. Brain Res. 1997;755:36–46. doi: 10.1016/s0006-8993(97)00089-9. [DOI] [PubMed] [Google Scholar]
- Rytter A, Cronberg T, Asztely F, Nemali S, Wieloch T. Mouse hippocampal organotypic tissue cultures exposed to in vitro “ischemia” show selective and delayed CA1 damage that is aggravated by glucose. J Cereb Blood Flow Metab. 2003;23:23–33. doi: 10.1097/01.WCB.0000034361.37277.1B. [DOI] [PubMed] [Google Scholar]
- Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci USA. 2005;102:12005–12010. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaller B, Meyer M, Schiffmann S. ‘New’ functions for ‘old’ proteins: the role of the calcium-binding proteins calbindin-D28k, calretinin and parvalbumin, in cerebellar physiology. Studies with knockout mice. Cerebellum. 2002;1:241–258. doi: 10.1080/147342202320883551. [DOI] [PubMed] [Google Scholar]
- Sloviter RS. Calcium-binding protein (calbindin-D28k) and parvalbumin immunocytochemistry: localization in the rat hippocampus with specific reference to the selective vulnerability of hippocampal neurons to seizure activity. J Comp Neurol. 1989;280:183–196. doi: 10.1002/cne.902800203. [DOI] [PubMed] [Google Scholar]
- Starkov AA, Chinopoulos C, Fiskum G. Mitochondrial calcium and oxidative stress as mediators of ischemic brain injury. Cell Calcium. 2004;36:257–264. doi: 10.1016/j.ceca.2004.02.012. [DOI] [PubMed] [Google Scholar]
- Stork CJ, Li YV. Intracellular zinc elevation measured with a “calcium-specific” indicator during ischemia and reperfusion in rat hippocampus: a question on calcium overload. J Neurosci. 2006;26:10430–10437. doi: 10.1523/JNEUROSCI.1588-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Liu S, Fu Y, Wang JH, Lu Y. Cdk5 activation induces hippocampal CA1 cell death by directly phosphorylating NMDA receptors. Nat Neurosci. 2003;6:1039–1047. doi: 10.1038/nn1119. [DOI] [PubMed] [Google Scholar]
- Ward MW, Huber HJ, Weisova P, Dussmann H, Nicholls DG, Prehn JH. Mitochondrial and plasma membrane potential of cultured cerebellar neurons during glutamate-induced necrosis, apoptosis, and tolerance. J Neurosci. 2007;27:8238–8249. doi: 10.1523/JNEUROSCI.1984-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen EY, Gu Z, Yan Z. Calpain regulation of AMPA receptor channels in cortical pyramidal neurons. J Physiol. 2007;580:241–254. doi: 10.1113/jphysiol.2006.122754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Baudry M. EUK-207, a superoxide dismutase/catalase mimetic, is neuroprotective against oxygen/glucose deprivation-induced neuronal death in cultured hippocampal slices. Brain Res. 2009;1247:28–37. doi: 10.1016/j.brainres.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Conventional electron micrographs illustrating the anatomical organization of CA1 and CA3 pyramidal neurons in organotypic slice cultures of hippocampus. Both CA1 (top and middle panels) and CA3 (bottom panel) pyramidal neurons are characterized by large, adjacent cell bodies containing round, lucent nuclei (N). Neuronal cell bodies are tightly packed in rows, an arrangement that clearly demarcates neurons. Glial somata are rarely seen between neuronal cell bodies in the stratum pyramidale, but when present are readily distinguished by a characteristic irregular nucleus with a chromatin border (GN). Bar for all panels, 2 μm.
NMDA-evoked selective Ca2+ elevations in CA1 neurons. A 30-min time-lapse record of Ca2+ dynamics during NMDA exposure (200 μM) from neurons of a representative hippocampal slice culture loaded with the low-affinity fluorescent probe fluo-4FF. NMDA induces a rapid, transient fluorescence increase followed by a sustained plateau (beginning about halfway through the video) in area CA1; there is no detectable response in CA3. Images were recorded at the neuronal cell body focal plane using a 5x objective. Images were collected every 30 sec for the first 5 min and every 60 sec for the remaining 25 min. Frame rate, 5 frames/s.