

Abstract
A new series of N-carboxyphenylpyrrole ligands were designed using GeometryFit based on an x-ray crystal structure of gp41. The synthesized ligands showed significant inhibitory activities against HIV gp41 6-helix bundle formation, HIV-1 mediated cell-cell fusion and HIV-1 replication.
AIDS causes very serious public health problem and economic burden. Globally, an estimated 33 million people are living with HIV, with nearly 7,500 new infections each day. 1 So far, 33 anti-HIV drugs (including 5 fixed-dose combinations) have been approved by FDA with 30 of them belonging to reverse transcriptase inhibitors (RTI) and protease inhibitors (PI).2 Combination therapies have shown significant synergistic effects.3 However, there is still no cure or effective vaccine for AIDS, and current anti-HIV drugs are facing increasing prevalence of viral resistance and side effects.4,5 Therefore, it is in urgent need to develop novel anti-HIV drugs addressing new targets other than RTI and PI.
HIV infects cells through endocytosis and envelope glycoprotein- and dynamin- dependent fusion.6 Gp41, a transmembrane subunit of HIV-1 envelope glycoprotein, plays a crucial role in HIV fusion and entry.7 Gp41 exists as a trimer and the ectodomain of each monomer contains an N-terminal peptide (N-peptide) and a C-terminal peptide (C-peptide). HIV fusion involves the insertion of the gp41 N-peptides into host cell membrane and the subsequent binding of the C-peptides (anchored to viral membrane) to N-peptides, which forms a 6-helical bundle bringing the viral membrane and host cell membrane to proximity for fusion. Gp41 is a proven drug target - the peptides derived from the gp41 C-peptides were found as potent HIV fusion inhibitors able to block the N-peptides from binding to the C-peptides.8, 9, 10, 11, 12, 13 One of the peptides, T-20 (Fuzeon/Enfuvirtide, a 36-amino acid synthetic peptide)14,15,16 was approved by FDA in 2003 for treating patients with multi-drug resistant HIV. However, T-20 is not orally available and has high production cost. 17 Therefore, developing orally available nonpeptide small-molecule fusion inhibitors targeting gp41 is highly desirable.
So far, no small-molecule anti-HIV drug targeting gp41 has been successfully developed, and gp41 has long been questioned for its druggability for: 1) It is difficult for small molecules to block the very strong protein-protein interactions between gp41 C-peptides (red, Fig. 1) and N-peptides (blue, Fig.1); 2) gp41 N-peptide bundle is highly hydrophobic and lack of deep pockets to allow strong binding with small molecules, which makes it an even more difficult drug target.
Figure 1.
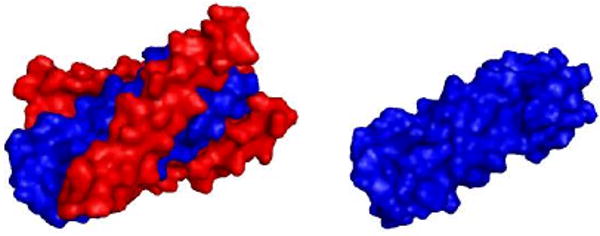
gp41 C- and N-peptide bundle (left); gp41 N-peptide bundle (right). Protein source: 1AIK (PDB code).8
Despite the formidable challenges, a few small-molecule gp41 inhibitors with modest activities at micromolar concentrations were discovered during the past 10 years mostly via high-throughput screening and diverse compound library synthesis.18, 19, 20, 21, 22, 23, 24, 25 A12 (Fig. 2) is one of the most promising compounds discovered by Xie and Jiang's labs recently.26 Notably, A12 is not only able to inhibit gp41 6-helical bundle formation (EC50 37.36 μM) but also druglike.
Figure 2.
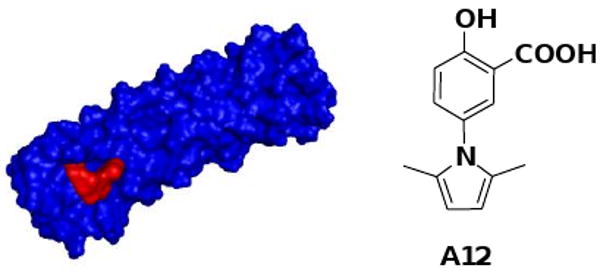
A12 (red) docked into gp41 N-peptide bundle (blue). Protein source: 2R5D (PDB code).28
Until Aug. 2009, over 130 x-ray/NMR 3D structures related to gp41 have been released from the Protein Data Bank (PDB),8, 27, 28, 29 which provide invaluable information to understand gp41's function at the atomic level as well as lay a solid foundation for structure-based design of small-molecule gp41 inhibitors.
However, structure-based de novo design of small-molecule gp41 inhibitors remains extremely challenging largely due to the lack of effective de novo drug design methodologies.30 Hamilton's pioneer work31 in de novo design of gp41 surface antagonists led to a novel small-molecule gp41 inhibitor (EC50 ∼15 μM), however, its further optimization is difficult partly because its design was based on general α-helix mimicry of gp41 rather than a specific binding pocket on gp41. Most recently, Liu et al successfully designed a novel small-molecule gp41 inhibitor (IC50 31 μM, inhibition of gp41 helical bundle formation) using their fragment-based de novo ligand design technology, unfortunately, this compound failed to show activity in a HIV mediated cell-cell fusion inhibitory assay in the real life.32
We believe that A12 (Fig. 1) could serve as a valuable starting point for further drug design targeting gp41 by modifying this structure using GeometryFit, a proprietary knowledge-oriented, computer-aided and structure-based de novo drug design methodology developed by GeometryLifeSci.33
Based on our docking study (gp41 N-peptide source:28PDB ID: 2r5d) using AutoDock 4.0,34 we identified a binding mode of A12 (Fig. 3, A12: pink sticks) quite different from that proposed previously by Xie and Jiang.26 In our proposed binding mode, the calculated binding free energy of ΔG is - 6.5 kcal/mol and the three key contact motifs identified within the gp41 binding pocket are: 1) the flexible and positively-charged Lys38 (cyan sticks), which could form a strong salt bridge with the negatively charged carboxylic group of A12; 2) the hydrophilic Gln41 (green), which could form two H-bonds with the same carboxylic group of A12; 3) the highly hydrophobic pocket (yellow) composed of Leu29, Leu32, Thr33, Val34 and Ile37, which happens to nicely complement the shape of the hydrophobic pyrrole ring of A12 to form very favorable hydrophobic interactions. These three key contact motifs may account for the binding of A12 to gp41, leading to the inhibition of gp41 6-helix bundle formation.
Figure 3.
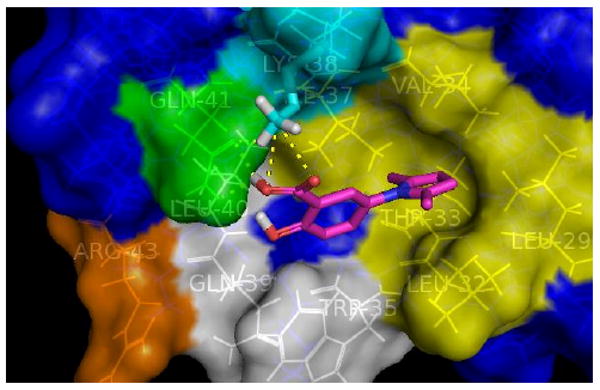
Calculated most favorable binding mode of A12
In addition to the three key contact motifs, we then explored additional potential contact motifs within the binding pocket in order to find new ligands with improved binding affinities. The most favorable binding mode of A12 suggests that its phenolic group do not contribute significantly to its overall binding affinity but do have plenty of room to extend into additional contact motifs within the binding pocket, such as Arg43 (orange, Fig 3.) and the binding pocket composed of Trp35, Gln39, Leu40 (white, Fig. 3).
Using GeometryFit, we designed a new ligand GLS-12 (Fig. 4) based on the A12 structure, in which the phenolic group of A12 was replaced by a phenyl group to not only fit into the shape of the pocket composed of Trp35, Gln39, Leu40 (white, Fig. 4) and Gln41 (green, Fig. 4) but also generate favorable hydrophobic interactions with Trp35, Gln39, Leu40 (white, Fig. 4). The subsequent docking study confirmed that, in the most favorable binding mode of GLS-12 (Fig. 4), the new ligand well maintains the interactions with the previous three key contact motifs while docking nicely into the new binding pocket with its phenyl group. The calculated binding free energy of ΔG was increased to -7.0 kcal/mol.
Figure 4.
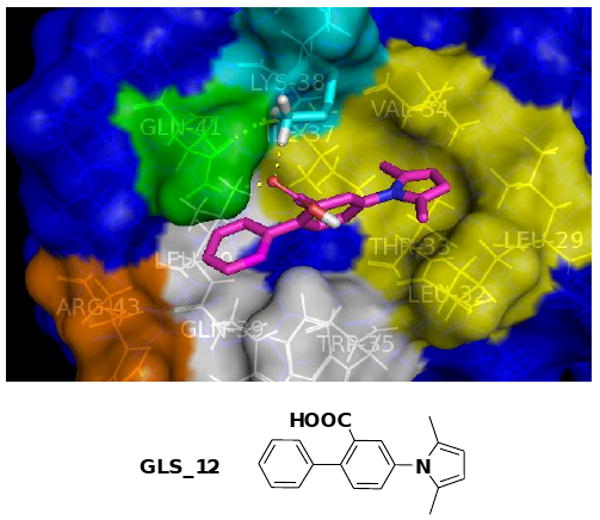
Calculated most favorable binding mode of GLS_12
To further increase the binding affinity, we decided to recruit the positively charged Arg43 (orange, Fig. 4) as an additional contact motif within the binding pocket. Using GeometryFit, we designed another new ligand GLS-22 (Fig. 5) based on the structure of GLS-12, in which a negatively charged carboxylic acid handle was added at the para position of the phenyl ring in order to form a strong electrostatic interaction with Arg43. The subsequent docking study confirmed that, in the most favorable binding mode of GLS-22 (Fig. 5), the new ligand's carboxylic group could form a salt bridge with Arg43 and at the same time well maintain the interactions with the previous four key binding sites. As a result, the calculated binding free energy of ΔG was increased to - 8.9 kcal/mol.
Figure 5.
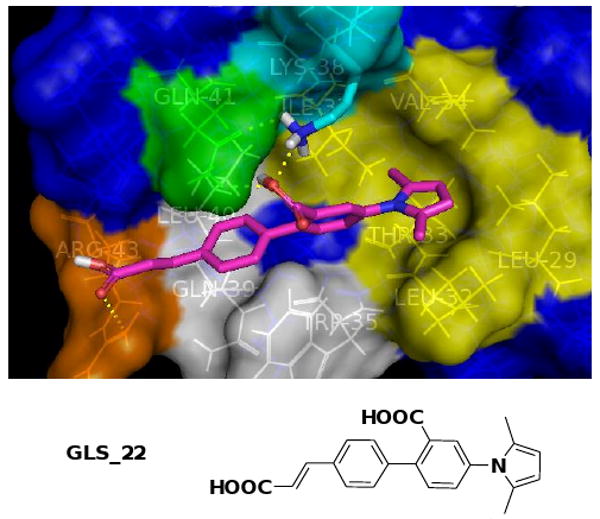
Calculated most favorable binding mode of GLS_22
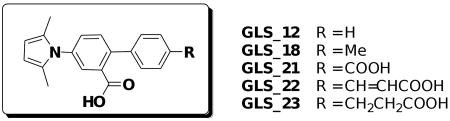
In this preliminary study, total of five new ligands were designed, synthesized and screened for their inhibitory activities against 1) HIV gp41 six-helix bundle (6-HB) formation, 2) HIV-1-mediated cell-cell fusion (CF) and 3) HIV-1 replication (p24 and CPE), as previously described,21,26 using A12 as a control. The synthesis is illustrated in Scheme 1 and the bio-assay results are summarized in Table 1.
Scheme 1.

Reagents and conditions: (i) H2SO4, MeOH, reflux; (ii) 2,5-Hexanedione, p-TsOH, Toluene, reflux; (iii) Tf2O, Et3N, DCM, 0 °C; (iv) PdCl2(PPh3)2, K2CO3, THF:H2O (2:1), reflux; (v) Malonic acid, Piperidine, Pyridine, reflux; (vi) 1) NaOH, EtOH, 2) HCl; (vii) Ethyl (triphenylphosphoranyliden)acetate, DCM; (viii) Pd/C, H2, THF:MeOH (4:1).
Table 1.
Anti-HIV bioassay results vs calculated binding ΔG
 | |||||||
|---|---|---|---|---|---|---|---|
| Ligand35 | ΔG(calcu.) (Kcal/mol) |
IC50 (μM) | CC50a (μM) | SIb | |||
| p24 | CPE | CF | 6-HB | CC50 / IC50 (p24) | |||
| A12 | -6.5 | 28.19±3.79 | 55.45±16.59 | 43.24±2.28 | 29.39±5.47 | 333.84±22.88 | 11.84 |
| GLS-12 | -7.0 | 17.76±2.74 | 37.92±7.55 | 59.17±2.00 | 28.96±2.27 | 289.80±13.18 | 16.31 |
| GLS-18 | -6.9 | 18.19±2.04 | 79.54±8.16 | 51.61±3.38 | 21.26±2.88 | 280.04±38.11 | 15.39 |
| GLS-21 | -8.7 | 25.63±1.92 | 33.93±6.66 | 26.01±1.31 | 20.50±1.11 | 355.23±24.47 | 13.85 |
| GLS-22 | -8.9 | 4.91±0.69 | 7.71±1.64 | 3.60±0.27 | 20.73±2.11 | 255.28±3.73 | 51.99 |
| GLS-23 | -8.6 | 8.30±1.34 | 13.08±3.19 | 8.30±0.11 | 21.75±0.75 | 227.27±22.86 | 27.38 |
CC50: 50% cytotoxicity concentration
SI: Selectivity Index
All 5 designed new ligands showed better activities than A12 in all assays except GLS_18 in the CPE assay and GLS_12 & 18 in the CF assay. The assay results are in accordance with our predictions using GeometryFit in most cases. The designed new ligands (GLS_21,22,23) targeting two more contact motifs within the binding pocket than those of A12 all showed better activities across all four assays, whereas the ligands (GLS_12,18) targeting only one more hydrophobic contact motif than those of A12 mostly showed comparable activities, which indicates that the second additional contact motif, Arg43, may contribute more significantly to the increased binding affinity. The best ligand, GLS_22, showed ∼ 6-7 folds better inhibitory activity than A12 in the HIV replication assays, ∼12 folds in the HIV cell-cell fusion assay and ∼ 1.4 folds in the gp41 six-helix bundle formation assay. All five new ligands had low cytotoxicity (CC50: 227-355 μM), and the most potent ligand, GLS_22, had the highest Selectivity Index (SI) (CC50/IC50 (p24): 51.99).
Summary
In this preliminary study, we identified a new binding mode of A12 involving three key contact motifs within the binding pocket, based on which we explored two additional potential contact motifs within the binding pocket and designed five new derivatives of A12 using GeometryFit. All five new ligands showed improved anti-HIV activities in almost all assays, which suggests that our design model and methodology are reliable, paving the way for our de novo design of novel small-molecule HIV inhibitors targeting gp41 in the future.
Acknowledgments
This work was supported by GeometryLifeSci, LLC, an NIH grant (AI46221) and grant funding from National Science Foundation of China (20942001) and Start-up Foundation for New Investigators from Guangzhou Institute of Biomedicine and Health (GIBH).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Qiang Zhu, Email: zhu_qiang@gibh.ac.cn.
Shibo Jiang, Email: SJiang@NYBloodCenter.org.
Yun Liao, Email: leonard_liao@geometrylifesci.com.
References and notes
- 1.World Health Statistics. WHO; 2009. p. 11. [Google Scholar]
- 2.http://www.hivandhepatitis.com/hiv_and_aids/hiv_treat.html.
- 3.De Clercq EJ. Clin Virol. 2004;30:115. doi: 10.1016/j.jcv.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 4.Richman DD, Morton SC, Wrin T, Hellmann N, Berry S, Shapiro MF, Bozzette SA. AIDS. 2004;18:1393. doi: 10.1097/01.aids.0000131310.52526.c7. [DOI] [PubMed] [Google Scholar]
- 5.1) Carr A, Cooper DA. Lancet. 2000;356:1423. doi: 10.1016/S0140-6736(00)02854-3. [DOI] [PubMed] [Google Scholar]; 2) Johnson& Johnson strengthens warning on HIV drug. 2009 Aug 26; Wed. 5:32pm EDT. http://www.reuters.com/article/marketsNews/idINN2628355520090826?rpc=44.
- 6.1) Miyauchi K, Kim Y, Latinovic O, Morozov V, Melikyan GB. Cell. 2009;137(3):433. doi: 10.1016/j.cell.2009.02.046. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]; 2) Fackler OT, Peterlin BM. Curr Biol. 2000;10:1005. doi: 10.1016/s0960-9822(00)00654-0. [DOI] [PubMed] [Google Scholar]
- 7.Moore JP, Jameson BA, Weiss RA, Sattentau QJ. In: Viral Fusion Mechanisms. Bentz J, editor. CRC Press; Boca Raton, FL: 1993. pp. 233–289. [Google Scholar]
- 8.Chan DC, Fass D, Berger JM, Kim PS. Cell. 1997;89:263. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 9.Chan DC, Kim PS. Cell. 1998;93:681. doi: 10.1016/s0092-8674(00)81430-0. [DOI] [PubMed] [Google Scholar]
- 10.Jiang S, Zhao Q, Debnath AK. Curr Pharm Des. 2002;8:563. doi: 10.2174/1381612024607180. [DOI] [PubMed] [Google Scholar]
- 11.Jiang S, Lin K, Strick N, Neurath AR. Nature. 1993;365:113. doi: 10.1038/365113a0. [DOI] [PubMed] [Google Scholar]
- 12.Wild CT, Shugars DC, Greenwell TK, McDanal CB, Matthews TJ. Proc Natl Acad Sci U S A. 1994;91:9770. doi: 10.1073/pnas.91.21.9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu M, Blacklow SC, Kim PS. Nat Struct Biol. 1995;2:1075. doi: 10.1038/nsb1295-1075. [DOI] [PubMed] [Google Scholar]
- 14.Kilby JM, Eron JJ. N Engl J Med. 2003;348:2228. doi: 10.1056/NEJMra022812. [DOI] [PubMed] [Google Scholar]
- 15.Lalezari JP, Henry K, O'Hearn M, Montaner JS, Piliero PJ, Trottier B, Walmsley S, Cohen C, Kuritzkes DR, Eron JJ, Jr, Chung J, DeMasi R, Donatacci L, Drobnes C, Delehanty J, Salgo MN. Engl J Med. 2003;348:2175. doi: 10.1056/NEJMoa035026. [DOI] [PubMed] [Google Scholar]
- 16.Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. Nature. 1997;387:426. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- 17.Liu S, Wu S, Jiang S. Curr Pharm Des. 2007;13:143. doi: 10.2174/138161207779313722. [DOI] [PubMed] [Google Scholar]
- 18.Kazmierski WM, Kenakin TP, Gudmundsson KS. Chem Biol Drug Des. 2006;67:13. doi: 10.1111/j.1747-0285.2005.00319.x. [DOI] [PubMed] [Google Scholar]
- 19.Debnath AK, Radigan L, Jiang SJ. Med Chem. 1999;42:3203. doi: 10.1021/jm990154t. [DOI] [PubMed] [Google Scholar]
- 20.Zhao Q, Ernst JT, Hamilton AD, Debnath AK, Jiang S. AIDS Res Hum Retroviruses. 2002;18:989. doi: 10.1089/08892220260235353. [DOI] [PubMed] [Google Scholar]
- 21.Jiang S, Lu H, Liu S, Zhao Q, He Y, Debnath AK. Antimicrob Agents Chemother. 2004;48:4349. doi: 10.1128/AAC.48.11.4349-4359.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jin BS, Lee WK, Ahn K, Lee MK, Yu YG. J Biomol Screen. 2005;10:13. doi: 10.1177/1087057104269726. [DOI] [PubMed] [Google Scholar]
- 23.Xu Y, Lu H, Kennedy JP, Yan X, McAllister LA, Yamamoto N, Moss JA, Boldt GE, Jiang S, Janda KD. J Comb Chem. 2006;8(4):531. doi: 10.1021/cc0600167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu S, Lu H, Zhao Q, He Y, Niu J, Debnath AK, Wu ST, Jiang S. Biochimica et Biophysica Acta. 2005;1723:270. doi: 10.1016/j.bbagen.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 25.Jiang S, Debnath AK, Lu H. U S patent 0287319 A1. 2006
- 26.Liu K, Lu H, Hou L, Qi Z, Teixeira C, Barbault F, Fan BT, Liu S, Jiang S, Xie L. J Med Chem. 2008;51(24):7843. doi: 10.1021/jm800869t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.1) Tan K, Liu J, Wang J, Shen S, Lu M. Proc Natl Acad Sci U S A. 1997;94(23):12303. doi: 10.1073/pnas.94.23.12303. [DOI] [PMC free article] [PubMed] [Google Scholar]; 2) Shu W, Ji H, Lu M. J Biol Chem. 2000;275(3):1839. doi: 10.1074/jbc.275.3.1839. [DOI] [PubMed] [Google Scholar]; 3) Shu W, Liu J, Ji H, Radigen L, Jiang S, Lu M. Biochemistry. 2000;39(7):1634. doi: 10.1021/bi9921687. [DOI] [PubMed] [Google Scholar]; 4) Wang S, York J, Shu W, Stoller MO, Nunberg JH, Lu M. Biochemistry. 2002;41(23):7283. doi: 10.1021/bi025648y. [DOI] [PubMed] [Google Scholar]
- 28.Welch BD, VanDemark AP, Heroux A, Hill CP, Kay MS. Proc Natl Acad Sci U S A. 2007 Oct 23;104(43):16828. doi: 10.1073/pnas.0708109104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.http://www.rcsb.org/pdb.
- 30.Mauser H, Guba W. Curr Opin Drug Discovery Dev. 2008;11(3):365. [PubMed] [Google Scholar]
- 31.Ernst JT, Kutzki O, Debnath AK, Jiang S, Lu H, Hamilton AD. Angew Chem Int Ed. 2002;41(2):278. doi: 10.1002/1521-3773(20020118)41:2<278::aid-anie278>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 32.Liu B, Joseph RW, Dorsey BD, Schiksnis RA, Katrina N, Bukhtiyarova M, Springman EB. Bioorg Med Chem Lett. 2009;19:5693. doi: 10.1016/j.bmcl.2009.08.018. [DOI] [PubMed] [Google Scholar]
- 33.http://geometrylifesci.com.
- 34.Morris G, Goodsell D, Halliday R, Huey R, Hart W, Belew R, Olson A. J Comput Chem. 1998;19:1639. [Google Scholar]
- 35.Spectral data of the ligands synthesized: A12: 1H NMR (CDCl3, 400 MHz) δ 10.46(br, 1H), 7.80 (s 1H), 7.38 (d, J = 8.4 Hz, 1H), 7.11 (d, J = 8.8 Hz, 1H), 5.91 (s, 2H), 2.04(s, 6H). MS: [M+1]+. 232 (ES+APCI). GLS_12: 1H NMR (CDCl3, 400 MHz) δ 7.80 (s 1H), 7.46-7.38 (m, 7H), 5.93 (s, 2H), 2.17(s, 6H). MS: [M+1]+. 292 (ES+APCI). GLS_18: 1H NMR (CDCl3, 400 MHz) δ 7.78 (s 1H), 7.46 (d, J = 8.0 Hz, 1H), 7.39 (d, J = 8.0 Hz, 1H), 7.30-7.22 (m, 4H), 5.93 (s, 2H), 2.41 (s, 3H), 2.09(s, 6H). MS: [M+1]+. 306 (ES+APCI). GLS_21: 1H NMR (DMSO-d6, 400 MHz) δ 7.9 (d, J = 8.0 Hz, 2H), 7.60-7.51 (m, 5H), 5.85 (s, 2H), 2.03(s, 6H). MS: [M+1]+. 336 (ES+APCI). GLS_22: 1H NMR (DMSO-d6, 400 MHz) δ 7.75 (d, J = 8.0 Hz, 2H), 7.64 (d, J = 16.0 Hz, 1H), 7.56-7.51 (m, 3H), 7.45 (d, J = 8.4 Hz, 2H), 6.58 (d, J = 16.0 Hz, 1H), 5.84 (s, 2H), 2.02(s, 6H). MS: [M+1]+. 362 (ES+APCI). GLS_23: 1H NMR (DMSO-d6, 400 MHz) δ 7.52-7.47 (m, 3H), 7.40-7.28 (m, 4H), 5.84 (s, 2H), 2.88 (t, 2H), 2.59 (t, 2H), 2.09(s, 6H). MS: [M+1]+. 364 (ES+APCI).