

Summary
Aims
Evaluate discs large homolog 2 (DLG2) as a positional candidate gene for disposition index (DI) in the Insulin Resistance Atherosclerosis Family Study (IRAS-FS) African-American sample.
Methods
SNPs (n=193) were selected for genotyping in 580 African-American individuals using a modified tagging algorithm. Follow-up genotyping was carried out within regions associated with DI. A subset of highly associated, uncorrelated SNPs were used as covariates in the linkage analysis to assess their contribution to linkage.
Results
Evidence of association with DI was observed at the DLG2 locus (admixture-adjusted P=0.050-8.7×10-5) with additional signals observed in follow-up genotyping of 17 SNPs (P=0.033-0.0012). Inclusion of highly associated, uncorrelated SNPs as covariates in the linkage analysis explained linkage at the DLG2 locus (90.8cM) and reduced the maximal LOD score (72.0cM) from 4.37 to 3.71.
Conclusions
Evidence of association and an observed contribution to evidence for linkage to DI was observed for SNPs in DLG2 genotyped on the African-American individuals from the IRAS-FS. Although not the only gene in the region, these results suggest that variation at the DLG2 locus contributes to maintenance of glucose homeostasis through regulation of insulin sensitivity and β-cell function as measured by DI.
Keywords: African Americans, Glucose Homeostasis, Disposition Index, SNPs, Linkage, Association
Introduction
Maintenance of glucose homeostasis encompasses both peripheral insulin sensitivity and β-cell function. It has been widely observed that insulin-resistant subjects have markedly increased insulin secretory function compared with insulin-sensitive subjects. This compensatory relationship or negative feedback loop has been quantified experimentally and demonstrated to function as a hyperbolic relationship between insulin sensitivity and β-cell function [1]. The disposition index (DI) quantifies the relationship between insulin sensitivity (SI) and pancreatic β-cell function (AIR); DI=SIxAIR [2]. Type 2 diabetes (T2D) is characterized by the failure of this compensatory relationship and reduced DI is a strong predictor of T2D [3]. Through investigation of the relationship between SI and AIR quantified by DI, we aim to identify molecular mechanism(s) that detrimentally modulate glucose homeostasis.
In a genome-wide scan based on 284 nondiabetic African Americans from 21 pedigrees recruited by the Insulin Resistance Atherosclerosis Family Study (IRAS-FS), evidence for linkage to DI on chromosome 11q was observed with a LOD score of 3.21 at 81.0cM flanked by markers D11S2371 and D11S2002 [4]. Following fine mapping with microsatellite markers in the initial family sample (Set 1) and in an independent set of 214 African-American subjects in 21 pedigrees (Set 2), the resulting linkage signal increased to a LOD score of 4.80 at 80.0cM near marker D11S937. Suggestive evidence for linkage to acute insulin response (AIR) at two separate locations flanking the DI peak (64.0cM, LOD 2.77, flanked by markers D11S4076 and D11S981; and 85.0cM, LOD 2.54, flanked by markers D11S4172 and D11S2002) was also observed, but no evidence of linkage to the insulin sensitivity index (SI) [5]. The goal of this study was to evaluate the DLG2 locus as a positional candidate gene for DI and assess its contribution to the observed linkage signal.
Subjects
Study design, recruitment and phenotyping for the IRAS-FS have been described in detail [6]. Briefly, the IRAS-FS is a multi-center study designed to identify the genetic determinants of quantitative measures of glucose homeostasis and adiposity in African Americans and Hispanic Americans. A clinical examination was performed that included an interview, a frequently sampled intravenous glucose tolerance test (FSIGT), anthropometric measurements, and blood collection. The Institutional Review Board of the clinical and analysis sites approved the study protocol and all study participants provided their written informed consent. Measures of glucose homeostasis included those from the FSIGT using the reduced sampling protocol [7-9] calculated by mathematical modeling methods (MINMOD) [10]: insulin sensitivity (SI), acute insulin response (AIR) and disposition index (DI). Individuals with a self-reported diagnosis of diabetes or fasting glucose>126 mg/dL, were excluded from this analysis. This analysis includes 580 African-American individuals from 42 pedigrees. Distributions of the primary phenotypes are listed in Table 1.
Table 1. Demographic summary of IRAS-FS African American participants.
| African Americans | |||
|---|---|---|---|
| n | Mean ± SD | Median | |
| Subjects | 580 | ||
| Demographics | |||
| Age (years) | 580 | 42.9 ± 14.0 | 41.5 |
| Female Gender (%) | 344 | 59.2 % | |
| BMI (kg/m2) | 575 | 30.0 ± 6.8 | 29.0 |
| Glucose Homeostasis | |||
| SI (×10-5min-1/[pmol/L]) | 500 | 1.63 ± 1.17 | 1.41 |
| AIR (pmol/L) | 499 | 1005.7 ± 826.2 | 771.5 |
| DI (SI × AIR; × 10-5min-1) | 499 | 1425.7 ± 1269.2 | 1151.5 |
| Fasting Glucose (mg/dL) | 512 | 94.7 ± 9.7 | 93.0 |
Materials and Methods
Genotyping
SNPs were chosen for genotyping within the DLG2 gene (longest annotated transcript; Chr11:82843701-85015962, NCBI Build 36.1 hg18) using a modified tagging algorithm. SNPs were identified for genotyping based on binning Illumina-designable SNPs according to a threshold linkage disequilibrium score (r2) [11]. This algorithm specifically tagged SNPs (as opposed to haplotypes) and was agnostic towards haplotype block structure, although larger bins were likely to encompass haplotype block regions. Genotypic data from the Yoruba (YRI) population of the International HapMap project [12] was used as the best ancestral model for the IRAS-FS African-American population. The risk with use of YRI data alone is that African-American allele frequencies will differ from the ancestral population. Caucasians could be used to estimate the Caucasian ancestry limit, but it is not certain that the simple genetic drift model of ancestral allele frequency spectrum is applicable or useful in this case. A total of 193 SNPs were selected for typing at the DLG2 locus on the Illumina BeadArray system at the Centers for Inherited Disease Research (CIDR).
Seventeen additional SNPs which captured additional variation in DLG2 were selected for follow-up using the Tagger program [19] of Haploview. This genotyping was performed on the Sequenom MassArray Genotyping System [13]. Blind duplicates were included to evaluate genotyping accuracy.
Statistical Analysis
Each SNP was examined for Mendelian inconsistencies using PedCheck [14]. Genotypes inconsistent with Mendelian inheritance were converted to missing. Maximum likelihood estimates of allele frequencies were computed using the largest set of unrelated African-American individuals (n=58), genotypes were tested for departures from Hardy-Weinberg proportions and LD structure was evaluated using Haploview 4.0 [15], using the block definition from Gabriel et al. [16]. To evaluate coverage, DLG2 genotypes (Chr11:82843701-85015962) from the HapMap YRI population of the International HapMap project were evaluated. Specifically, using the Tagger program [19] of Haploview the amount of common variation captured was assessed by forced inclusion of SNPs typed and calculation of r2 across the interval using a minor allele frequency (MAF) threshold of 0.05 and the aggressive tagging algorithm.
To test for association between individual SNPs and the quantitative phenotype DI, variance component analysis was performed as implemented in SOLAR [17]. When necessary, quantitative traits were transformed to best approximate the distributional assumptions of the test and minimize heterogeneity of the variance. Thus, the results reported represent analyses on the square root of DI. To test for association between each SNP and T2D we used a threshold parameterization of the variance component measured genotype model as implemented in SOLAR. For each phenotype, the two degree of freedom test of genotypic association was performed. In addition, a priori genetic models (dominant, additive and recessive) were computed (i.e., dominant model contrasts those with the major allele versus those without, additive model tests for a dose effect in the number of alleles, and recessive model contrasts individuals homozygous for the minor allele versus not). If the overall evidence of genotypic association was significant, the results from the genetic models were examined directly. If the overall genotypic association was not significant, the results from the genetic models were examined after adjusting for the three comparisons using a Bonferroni adjustment. This approach is consistent with the Fisher's protected least significant difference multiple comparisons procedure. To assess the influence of admixture on the tests of association, a principal components (PC) analysis was performed using 39 ancestry informative markers (AIMs; ∼2 per chromosome based on chromosome size). The 39 AIMs were available on all subjects (n=580) and these data were merged with HapMap data for CEPH (n=90) and Yoruba (n=90) populations. The total proportion of variance explained by the first PC (PC1) was 40% and was used as a covariate in the analysis to adjust for admixture. The second and third PCs did not separate from the remaining 36 PCs and explained ∼3% of the genetic variation. Only PC1 correlated with HapMap populations. Tests reported here were computed adjusting for age, gender, BMI and with/without admixture adjustment. Adjustments for multiple comparison tests were not performed due to selection of the positional candidate gene based on a priori evidence of linkage.
The results of initial and fine mapping linkage analyses in the IRAS-FS African American pedigrees have been previously reported [4, 5]. Briefly, testing for evidence of linkage to a QTL, i.e. DI, was carried out using the variance component approach implemented in the SOLAR software package [17]. To test whether a particular subset of SNPs contributed, either directly or through linkage disequilibrium with the QTL, to the evidence for linkage, a subset of highly associated, uncorrelated SNPs was entered into the QTL linkage analysis and the change in the magnitude of the LOD score calculated. If the polymorphism directly or indirectly contributes to the evidence for linkage, the initial LOD score will be reduced in a model that includes the polymorphism as a covariate. The genetic location of the DLG2 locus was calculated by extracting all markers from the deCODE Genetic Map [18] for chromosome 11 and mapping the chromosomal sex-averaged genetic positions to the latest human genome physical map positions (NCBI Build 36.1, hg 18). Using R version 2.4.0 (http://www.r-project.org/index.html) a smoothing spline was fit through these points and the spline was used to interpolate genetic map positions corresponding to physical positions of gene loci. For DLG2 we computed a single representative physical map position as the mean of the 5′ end of the most centromeric exon in any known annotated splice variant and the 3′ end as the most telomeric.
Results
This study evaluated genetic data from 580 African Americans in the IRAS-FS of whom 499 had FSIGT-derived measures of glucose homeostasis. Table 1 summarizes the descriptive statistics of the sample. In general, the sample included more women than men (59.2% women) and was overweight (BMI=30.0±6.8kg/m2).
SNP genotyping from the Illumina Custom Bead Array platform at CIDR, was >99% efficient (0.045% missing data rate) and after Mendelian error corrections using PedCheck blind duplicate samples included in genotyping (n=22) had minimal discordances (0.014% inconsistency rate). Using the Tagger program [19] of Haploview, the 192 SNPs genotyped in the initial screen (rs1483394, was not genotyped by HapMap) captured common variation (MAF>0.05; aggressive tagging algorithm) of 1833 SNPs with a mean r2=0.71 in the HapMap Yoruba population. Examination of Hardy-Weinberg equilibrium in the DLG2 gene region (n=193 SNPs) identified five SNPs (rs1432049, rs964464, rs7945737, rs6592211 and rs7116340) that deviated from expected proportions (P≤0.05). The region with the strongest evidence of association contained SNPs rs10501545 (admixture-adjusted P=1.91×10-4), rs1945309 (admixture-adjusted P=8.72×10-5; Figure 1B) which were modestly correlated (r2=0.41, Figure 2). The results of the analysis at the DLG2 locus are summarized in Figure 1 and Appendix I.
Figure 1.
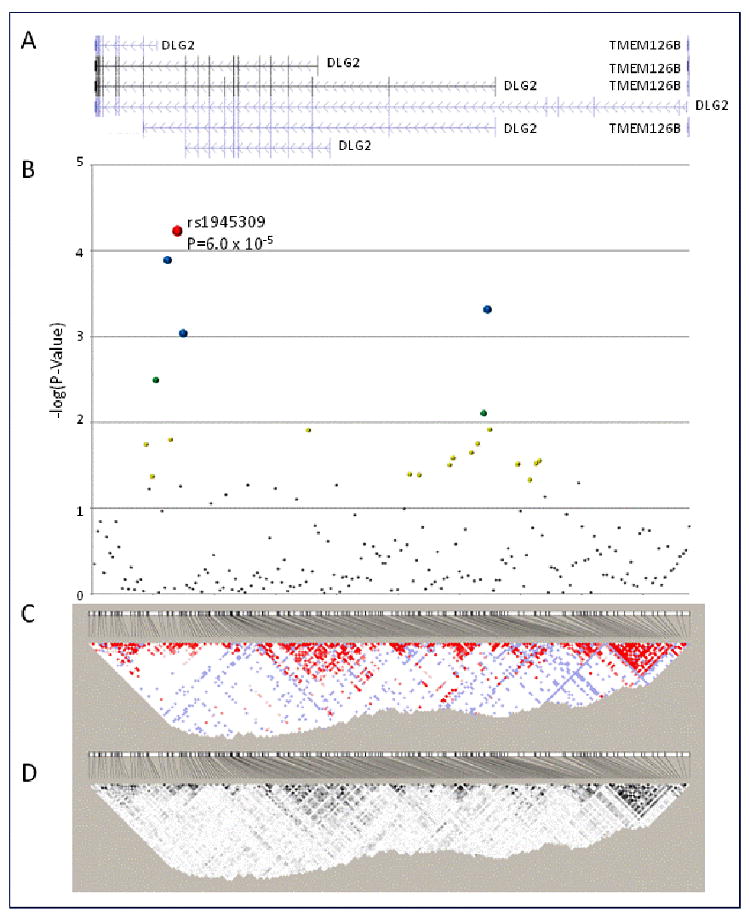
Association analysis and linkage disequilibrium structure of 193 SNPs genotyped across the DLG2 locus (NCBI Build 36.1 hg 18 chr11: 82844368-85006039). A. annotated transcripts across the DLG2 locus (NCBI Build 36.1 hg 18 chr11: 82844368-85006039). B. Association analysis of 195 DLG2 SNPs with disposition index (DI) in the combined African American population from the IRAS-FS. 2df association P-values are presented as the –log(P-Value) for SNPs along a condensed, equidistant x-axis. C and D. Haploview-generated LD map of the 27 SNPs at the DLG2 locus in unrelated African Americans (n=58) from the IRAS-FS. The number within each box indicates the D' and r2 statistic value between the corresponding two SNPs in panels C and D, respectively.
Figure 2.
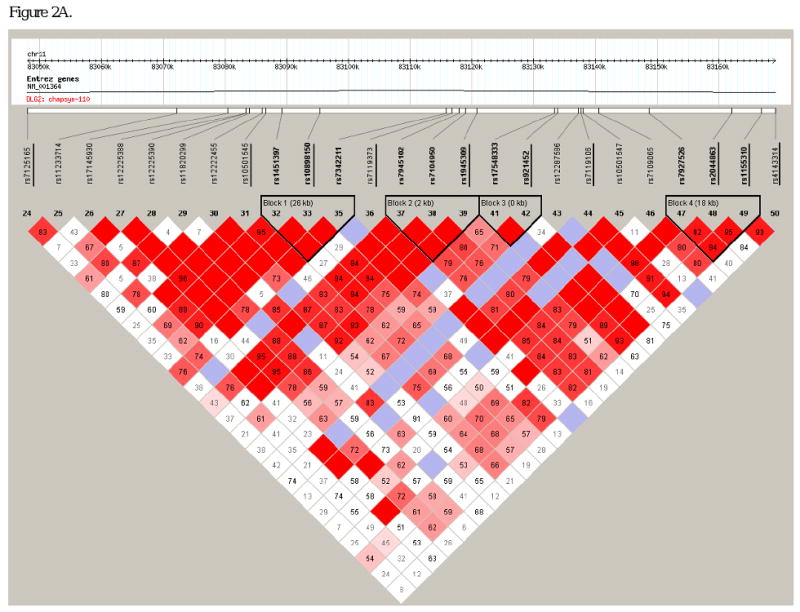

Linkage disequilibrium structure within intron 13 of DLG2. Haploview-generated LD map of the 27 SNPs at the DLG2 locus in unrelated African Americans (n=58) from the IRAS-FS. The number within each box indicates the D' and r2 statistic value in panels A and B, respectively. SNPs from the initial analysis of 193 SNPs (underlined) are intercalated with SNPs from the follow-up analysis.
In order to choose additional SNPs for follow-up analysis, the haplotype block structure of the interval harboring theses DI associated SNPs was examined. SNP rs10501545 was contained in an 8kb LD block and rs1945309 fell in an inter-block region (Figure 2A and B, SNPs underlined). In order to capture common variation across the region and interrogate block boundaries, 17 additional SNPs were typed in a follow-up study. Using the Tagger program of Haploview these 27 SNPs (10 from the initial screen and 17 follow-up SNPs) captured common variation at 88 SNPs (MAF>0.05; aggressive tagging algorithm) with a mean r2=0.85. Marker genotyping success rates were ≥90% for the additional 17 SNPs genotyped and blind duplicates were concordant. PedCheck analysis resulted in the exclusion of 12 of 9,877 genotypes. In the follow-up genotyping (n=17 SNPs) a single SNP deviated from expected Hardy-Weinberg proportions (rs7927526, P=0.023). Reexamination of the LD structure across the region (Figure 2) with a minor allele frequency threshold of 0.10 revealed three LD blocks. SNP rs10501545 fell in an inter-block region, Block 1 contained SNPs rs1451397 to rs7342211 (26kb), Block 2 contained SNPs rs7945102 to rs1945309 (2kb) and Block 3 contained SNPs rs17548333 to rs921452 (<1kb). Association analysis of the follow-up SNPs is presented in Table 2. Of the 14 additional SNPs evaluated with a MAF>0.10, nine SNPs showed evidence of association with DI (admixture-adjusted P=0.033-0.0012). Association within the context of LD block structure revealed additional SNPs within blocks 1 and 2 with evidence of association with DI (admixture-adjusted P=0.029-0.0012) and which were not highly correlated with the original signal (rs1945309; r2<0.45, respectively).
Table 2. Association analysis of 27 SNPs genotyped within the associated interval (NCBI Build 36.1, hg 18 chr11: 83048044-83169143) identified from the initial (underlined) and follow-up association analyses with disposition index (DI) across the DLG2 locus in IRAS-FS African Americans.
| Genotypic Mean ± s.d. (n)3 | 2df | 2df | |||||
|---|---|---|---|---|---|---|---|
| SNP | Alleles1 | MAF2 | 1/1 | 1/2 | 2/2 | P-value4 | P-value5 |
| rs7125165 | G/A | 0.47 | 1469 ± 1561 (139) | 1429 ± 1167 (258) | 1352 ± 1084 (96) | 0.87 | 0.86 |
| rs11233714 | G/A | 0.26 | 1361 ± 1307(287) | 1507± 1244 (161) | 1425 ± 910 (21) | 0.15 | 0.18 |
| rs17145930 | G/T | 0.21 | 1338 ± 1191 (282) | 1489 ± 1285 (166) | 1730 ± 1662 (37) | 0.12 | 0.14 |
| rs12225388 | G/A | 0.47 | 1444 ± 1307 (242) | 1448 ± 1249 (210) | 1204 ± 1206 (41) | 0.20 | 0.23 |
| rs12225390 | G/A | 0.22 | 1328 ± 1219(333) | 1623 ± 1404 (135) | 1578 ± 835 (16) | 0.013 | 0.014 |
| rs11820299 | T/A | 0.1 | 1414 ± 1236 (354) | 1470 ± 1437 (117) | 1604 ± 824 (13) | 0.34 | 0.32 |
| rs12222455 | T/A | 0.21 | 1339 ± 1230 (331) | 1626 ± 1403 (139) | 1578 ± 835 (16) | 0.030 | 0.033 |
| rs10501545 | A/G | 0.48 | 1607 ± 1341 (123) | 1477 ± 1335 (271) | 1059 ± 900 (99) | 1.30×10-4 | 1.91×10-4 |
| rs1451397 | C/T | 0.45 | 1199 ± 1217 (140) | 1464 ± 1251 (252) | 1574 ± 1321 (91) | 0.0034 | 0.0042 |
| rs10898150 | G/T | 0.42 | 1248 ± 1329 (150) | 1504 ± 1288 (258) | 1499 ± 1104 (85) | 0.016 | 0.021 |
| rs7102070 | C/T | 0.03 | 1289 ± 504 (5) | 1371 ± 1280 (123) | 1446 ± 1280 (365) | 0.45 | 0.43 |
| rs7342211 | G/A | 0.19 | 1327 ± 1208 (323) | 1672 ± 1418 (144) | 1272 ± 965 (15) | 0.018 | 0.029 |
| rs7119373 | G/A | 0.21 | 1401 ± 1192 (21) | 1454 ± 1386 (207) | 1405 ± 1190 (265) | 0.84 | 0.81 |
| rs7945102 | A/G | 0.44 | 1625 ± 1458 (145) | 1449 ± 1251 (256) | 1033 ± 892 (79) | 0.0015 | 0.0012 |
| rs7104950 | T/C | 0.46 | 1346 ± 990 (50) | 1297 ± 1214 (239) | 1596 ± 1384 (204) | 0.13 | 0.15 |
| rs1945309 | G/A | 0.34 | 1244 ± 1124 (224) | 1543 ± 1394 (226) | 1756 ± 1237 (43) | 6.00×10-5 | 8.72×10-5 |
| rs11233737 | G/T | 0.08 | 1431 ± 1310 (381) | 1401 ± 1121 (100) | 1194 ± 740 (5) | 0.83 | 0.82 |
| rs17548333 | C/T | 0.28 | 1321 ± 1082 (270) | 1565 ± 1506 (196) | 1455 ± 1150 (27) | 0.52 | 0.57 |
| rs921452 | C/T | 0.21 | 1357 ± 1235 (346) | 1616 ± 1383 (135) | 1250 ± 910 (12) | 0.060 | 0.070 |
| rs12287586 | G/C | 0.07 | 1397 ± 1284 (427) | 1629 ± 1193 (62) | 1333 ± 1436 (4) | 0.69 | 0.70 |
| rs7119106 | A/T | 0.44 | 1621 ± 1455 (145) | 1427 ± 1236 (271) | 1051 ± 924 (77) | 9.3×10-4 | 0.0013 |
| rs10501547 | A/T | 0.13 | 1396 ± 1280 (251) | 1481 ± 1322 (185) | 1375 ± 1088 (57) | 0.86 | 0.89 |
| rs7109065 | T/C | 0.47 | 1258 ± 1018 (136) | 1396 ± 1258 (261) | 1834 ± 1643 (80) | 0.02 | 0.019 |
| rs7927526 | T/C | 0.45 | 1511 ± 1320 (161) | 1409 ± 1167 (234) | 1324 ± 1320 (98) | 0.88 | 0.92 |
| rs2044863 | G/C | 0.31 | 1415 ± 1308 (255) | 1453 ± 1269 (205) | 1339 ± 1036 (33) | 0.80 | 0.79 |
| rs1155310 | G/A | 0.39 | 1439 ± 1402 (184) | 1413 ± 1210 (240) | 1436 ± 1141 (69) | 0.87 | 0.91 |
| rs4143314 | T/C | 0.35 | 1404 ± 1274 (250) | 1404 ± 1223 (199) | 1647 ± 1495 (44) | 0.90 | 0.91 |
Major/Minor alleles determined from the maximal set of unrelated (n=58)
MAF; Minor Allele Frequency determined from maximal set of unrelated (n=58)
The untransfomed mean trait value and standard deviation for each genotype (1/1, 1/2, 2/2; 1=major allele and 2=minor allele) and the number of individuals (n) with each genotype included in the analysis is listed. Diabetics were excluded for all measures of glucose homeostasis.
The P-value is listed comparing the means of each genotype. Numbers in bold indicate P ≤ 0.05. Numbers in italics trended toward association P ≤ 0.10.
The admixture-adjusted P-value is listed comparing the means of each genotype. Numbers in bold indicate P ≤ 0.05. Numbers in italics trended toward association P ≤ 0.10.
The extent to which variation at the DLG2 locus contributed to the evidence for linkage of DI on chromosome 11 is shown in Figure 3. Selection of a subset of highly associated, uncorrelated SNPs resulted in selection of seven SNPs to be used as covariates in the linkage analysis (rs725192, rs518143, 1367980, rs616414, rs10501545, rs1945309 and rs7119106; r2<0.41). Linkage of DI to chromosome 11 in the African American cohort containing only individuals with complete genotype data at the DLG2 SNPs of interest maximized at 73.0cM (95% CI: 69.0-77.0cM) with a LOD score of 4.37. The LOD score at the DLG2 locus, located at 90.8cM was 0.74. Inclusion of a subset of highly associated, uncorrelated SNPs as covariates in the analysis resulted in complete reduction of the LOD score at the DLG2 locus (LOD score of 0.04) and reduction of the maximal LOD score from 4.37 at 73.0cM to 3.71 at 72.0cM.
Figure 3.
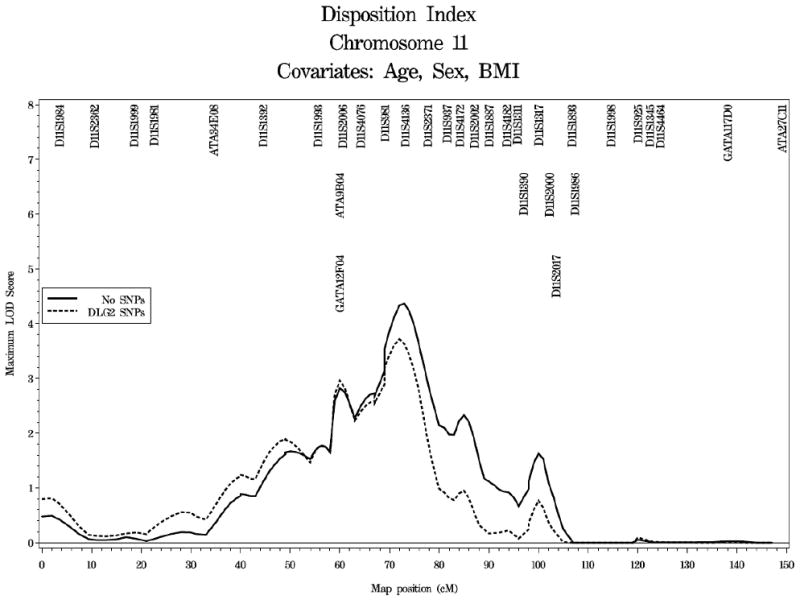
Linkage analysis of DI on chromosome 11 in African Americans from the IRAS-FS. The solid line represents individuals with complete genotype data at the DLG2 SNPs. The dashed line represents linkage analysis with inclusion of DLG2 SNPs as covariates.
Discussion
Using a positional cloning approach, evidence of DI linked to chromosome 11q has been observed. This result has been replicated in additional study samples and fine mapping resulted in a significant LOD score of 4.80 at 80cM near marker D11S937 in African Americans from the IRAS-FS [5]. The purpose of the current study was to evaluate discs large homolog 2 (DLG2) as a positional candidate gene for DI.
The longest of six DLG2 transcripts spans greater than 2.1Mb of chromosome 11q (Chr11: 82843701-85015962; NCBI Build 36.1 hg18; Figure 1A). The 193 SNPs typed tagged a large proportion of the known variation as assessed through HapMap Yoruba dataset (r2=0.71). The most significant evidence for association with DI was observed for SNP rs1945309 (admixture-adjusted P=8.72×10-5; Figure 1B) located in intron 13 of DLG2 (RefSeq Gene; NM_001364.2) with several additional, modestly correlated SNPs (r2<0.45) showing support for evidence of association within this region of DLG2. Using a significance threshold of P≤0.05 more SNPs reached statistical significance in association analysis with DI (n=21) than expected by chance alone (n=9.8). Additional genotyping and focused analysis within the intron 13 region of DLG2 identified several SNPs significantly associated with DI, albeit with a lesser magnitude than that observed at SNP rs1945309. Evaluation of association by independent sample set (Set 1: 320 subjects in 21 families and Set 2: 260 subjects in 21 families) in the IRAS-FS African-American population revealed association in both sets independently at the most significantly associated loci (rs10501545: admixture-adjusted PSet1=0.0094, PSet2=0.027 and PSet1+2=1.91×10-4 and rs1945309, admixture-adjusted PSet1=0.016, PSet2=0.0016 and PSet1+2=8.72×10-5; Appendices II and III). Variation at the most significant SNP rs1945309 followed a dominant mode of inheritance (admixture-adjusted P=2.55×10-5, Appendix IV) with one or two copies of the common allele (G) resulting in a 12% (213×10-5min-1) and 29% (512×10-5min-1) decrease in the disposition index, respectively. Variation at SNP rs10501545 followed an additive mode of inheritance (P=4.50×10-5) with the number of copies of the minor allele G resulting in a step-wise decrease in DI.
Evaluation of DLG2 SNPs for association with T2D resulted in modest evidence of association with 26 of 210 SNPs showing nominal evidence of association (P-Value<0.05; Appendix V). Notably, half of these SNPs clustered in the intron 2-6 region of the gene which was not strikingly associated with DI (Appendix I). Within the intron 13 region which was most strongly associated with DI, only one SNP was found to be associated with T2D (rs11233737, P=0.010). These associations should be viewed cautiously as the results could be attributed to the relatively modest sample size of diabetics (n=72) in this study.
In order to determine if variants of DLG2 contributed to the evidence of linkage observed for DI on chromosome 11, a subset of highly associated, uncorrelated SNPs were included in the linkage analysis as covariates. Linkage analysis limited to individuals with complete genotype data for the DLG2 SNPs of interest (n=7) increased the maximal LOD score to 4.37 at 73.0cM. Inclusion of DLG2 SNPs as covariates in the linkage analysis reduced the LOD score at the genetic position (90.76cM) of DLG2 from 0.74 to 0.04 with dominant and additive models showing a greater ability to explain the linkage as suggested by the model-specific single SNP analysis (Appendix IV). While this subset of SNPs reduced the maximal LOD score observed on chromosome 11 to 3.71 at 72.0cM, there still remains significant evidence of linkage which is not explained by SNPs in DLG2. This observation suggests that additional variants within this interval exist, which either independently or in concert with SNPs in DLG2, contribute to the linkage signal. A more thorough assessment of this linkage interval is warranted to identify these variants.
DLG2 encodes channel-associated protein of synapses-110 (chapsyn-110) which is a member of the ‘membrane-associated guanylate kinase’ (MAGUK) family of proteins. In vitro studies suggest that this protein may interact at postsynaptic sites to form a multimeric scaffold for the clustering of receptors, ion channels and associated signaling proteins [20], a critical process for processing and transmission of electrical signals in neurons. Increasing evidence has suggested a strong involvement of the central nervous system (CNS) in the maintenance of glucose homeostasis [21, 22] with specific examples demonstrated in the control of insulin sensitivity in peripheral tissues through the melanocortin pathway [23]. Along this line of reasoning, DLG2 could play a role in the regulation of glucose homeostasis by mediating feedback signals from the peripheral tissues (insulin sensitivity) and pancreatic β-cells (acute insulin response) through the neuroendocrine network.
In summary, using a positional cloning approach we identified a region of chromosome 11q linked to DI in African Americans from the IRAS-FS and with additional mapping efforts replicated and refined the linkage interval. While the DLG2 gene is distal to the LOD-1 support interval of the DI linkage peak, evaluation of DLG2 as a positional candidate gene revealed multiple SNPs associated with DI. These associations were concentrated within the intron 13 region and associated with DI in Set 1 and Set 2 with increasing magnitude of the effect observed in the combined population. Assessment of the contribution to linkage for SNPs observed to be highly associated with DI resulted in reduction of the linkage signal suggesting that variation at the DLG2 locus contributes but does not fully explain the DI linkage signal observed on 11q. While inclusion of DLG2 SNPs as covariates in the linkage analysis reduces the evidence for linkage, additional unidentified loci also contribute to the linkage signal. Studies are currently underway to evaluate additional genes within the DI linkage interval. Evaluation of DLG2 in additional studies will be necessary to confirm the association observed with DI and functional studies are needed to assess the possible role of DLG2 in the neuroendocrine component for the maintenance of glucose homeostasis.
Supplementary Material
Acknowledgments
We wish to thank participants of the IRAS Family Study. The IRAS Family Study was supported in part by NIH grants HL060894, HL060931, HL060944, HL061019, and HL061210. Genotyping services were provided by CIDR. CIDR is fully funded through a federal contract from the National Institutes of Health to The Johns Hopkins University, Contract Number N01-HG-65403.
Grant Support: The IRAS Family Study was supported in part by NIH grants HL060894, HL060931, HL060944, HL061019, and HL061210. Genotyping services were provided by the Center for Inherited Disease Research. CIDR is fully funded through a federal contract from the National Institutes of Health to The Johns Hopkins University, contract number N01-HG-65403.
Footnotes
Conflict of interest
The authors declare that they have no conflict of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kahn SE. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of Type 2 diabetes. Diabetologia. 2003;46(1):3–19. doi: 10.1007/s00125-002-1009-0. [DOI] [PubMed] [Google Scholar]
- 2.Bergman RN, Ader M, Huecking K, Van Citters G. Accurate assessment of beta-cell function: the hyperbolic correction. Diabetes. 2002;51 1:S212–20. doi: 10.2337/diabetes.51.2007.s212. [DOI] [PubMed] [Google Scholar]
- 3.Rich SS, Bergman RN. The genetic basis of glucose homeostasis. Curr Diabetes Rev. 2005;1(3):221–6. doi: 10.2174/157339905774574293. [DOI] [PubMed] [Google Scholar]
- 4.Rich SS, Bowden DW, Haffner SM, Norris JM, Saad MF, Mitchell BD, et al. Identification of quantitative trait loci for glucose homeostasis: the Insulin Resistance Atherosclerosis Study (IRAS) Family Study. Diabetes. 2004;53(7):1866–75. doi: 10.2337/diabetes.53.7.1866. [DOI] [PubMed] [Google Scholar]
- 5.Palmer ND, Langefeld CD, Campbell JK, Williams AH, Saad M, Norris JM, et al. Genetic mapping of disposition index and acute insulin response loci on chromosome 11q. The Insulin Resistance Atherosclerosis Study (IRAS) Family Study. Diabetes. 2006;55(4):911–8. doi: 10.2337/diabetes.55.04.06.db05-0813. [DOI] [PubMed] [Google Scholar]
- 6.Henkin L, Bergman RN, Bowden DW, Ellsworth DL, Haffner SM, Langefeld CD, et al. Genetic epidemiology of insulin resistance and visceral adiposity. The IRAS Family Study design and methods. Ann Epidemiol. 2003;13(4):211–7. doi: 10.1016/s1047-2797(02)00412-x. [DOI] [PubMed] [Google Scholar]
- 7.Steil GM, Volund A, Kahn SE, Bergman RN. Reduced sample number for calculation of insulin sensitivity and glucose effectiveness from the minimal model. Suitability for use in population studies. Diabetes. 1993;42(2):250–6. doi: 10.2337/diab.42.2.250. [DOI] [PubMed] [Google Scholar]
- 8.Bergman RN, Ider YZ, Bowden CR, Cobelli C. Quantitative estimation of insulin sensitivity. Am J Physiol. 1979;236(6):E667–77. doi: 10.1152/ajpendo.1979.236.6.E667. [DOI] [PubMed] [Google Scholar]
- 9.Bergman RN, Finegood DT, Ader M. Assessment of insulin sensitivity in vivo. Endocr Rev. 1985;6(1):45–86. doi: 10.1210/edrv-6-1-45. [DOI] [PubMed] [Google Scholar]
- 10.Pacini G, Bergman RN. MINMOD: a computer program to calculate insulin sensitivity and pancreatic responsivity from the frequently sampled intravenous glucose tolerance test. Comput Methods Programs Biomed. 1986;23(2):113–22. doi: 10.1016/0169-2607(86)90106-9. [DOI] [PubMed] [Google Scholar]
- 11.Carlson CS, Eberle MA, Rieder MJ, Yi Q, Kruglyak L, Nickerson DA. Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am J Hum Genet. 2004;74(1):106–20. doi: 10.1086/381000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.A haplotype map of the human genome. Nature. 2005;437(7063):1299–320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buetow KH, Edmonson M, MacDonald R, Clifford R, Yip P, Kelley J, et al. High-throughput development and characterization of a genomewide collection of gene-based single nucleotide polymorphism markers by chip-based matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Proc Natl Acad Sci U S A. 2001;98(2):581–4. doi: 10.1073/pnas.021506298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O'Connell JR, Weeks DE. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet. 1998;63(1):259–66. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21(2):263–5. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 16.Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, et al. The structure of haplotype blocks in the human genome. Science. 2002;296(5576):2225–9. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 17.Almasy L, Blangero J. Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet. 1998;62(5):1198–211. doi: 10.1086/301844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kong A, Gudbjartsson DF, Sainz J, Jonsdottir GM, Gudjonsson SA, Richardsson B, et al. A high-resolution recombination map of the human genome. Nat Genet. 2002;31(3):241–7. doi: 10.1038/ng917. [DOI] [PubMed] [Google Scholar]
- 19.de Bakker PI, Yelensky R, Pe'er I, Gabriel SB, Daly MJ, Altshuler D. Efficiency and power in genetic association studies. Nat Genet. 2005;37(11):1217–23. doi: 10.1038/ng1669. [DOI] [PubMed] [Google Scholar]
- 20.Kim E, Cho KO, Rothschild A, Sheng M. Heteromultimerization and NMDA receptor-clustering activity of Chapsyn-110, a member of the PSD-95 family of proteins. Neuron. 1996;17(1):103–13. doi: 10.1016/s0896-6273(00)80284-6. [DOI] [PubMed] [Google Scholar]
- 21.Schwartz MW. Progress in the search for neuronal mechanisms coupling type 2 diabetes to obesity. J Clin Invest. 2001;108(7):963–4. doi: 10.1172/JCI14127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwartz MW, Porte D., Jr Diabetes, obesity, and the brain. Science. 2005;307(5708):375–9. doi: 10.1126/science.1104344. [DOI] [PubMed] [Google Scholar]
- 23.Obici S, Feng Z, Tan J, Liu L, Karkanias G, Rossetti L. Central melanocortin receptors regulate insulin action. J Clin Invest. 2001;108(7):1079–85. doi: 10.1172/JCI12954. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.