

Abstract
High-throughput screening of 201,368 compounds revealed that 1-(3-(5-ethyl-5H-[1,2,4]triazino[5,6-b]indol-3-ylthio)propyl)-1H-benzo[d]imidazol-2(3H)-one (SID 7975595) inhibited RmlC a TB cell wall biosynthetic enzyme. SID 7975595 acts as a competitive inhibitor of the enzyme’s substrate and inhibits RmlC as a fast-on rate, fully reversible inhibitor. An analog of SID 7975595 had a Ki of 62 nM. Computer modeling showed that the binding of the tethered two-ringed system into the active site occurred at the thymidine binding region for one ring system and the sugar region for the other ring system.
Keywords: RmlC inhibitor, tuberculosis, TB cell wall, TB drug discovery
1. Introduction
Tuberculosis (TB) is a contagious disease and one of the leading causes of death world-wide. According to the World Health Organization (WHO), more than two billion people, one third the world’s total population, are infected with the TB bacterium, including 10–15 million in the United States alone. The emergence of drug-resistant strains has been a huge problem in recent years 1, and now strains resistant to both first and second line antibiotics have clearly been identified 2, 3. Although the incidence of tuberculosis has increased over the past decade, no tuberculosis-specific drugs have been discovered over the last 40 years.
Two of the first line TB drugs, isoniazid and ethambutol, attack the biosynthesis of the unique TB cell wall. Resistance against these drugs 1 makes the development of new drugs directed against the TB cell wall critical. A highly desirable cell wall target is the linker disaccharide that is instrumental in tethering the peptidoglycan layer to the mycolic acid inner leaflet of the outer membrane 4 as illustrated in Fig. 1. Specifically, the formation of the rhamnosyl residue (Fig. 1) was targeted using a high-throughput screen for compounds that inhibit the conversion of TDP-6-deoxy-D-xylo-hexopyranosid-4-ulose (TDP-KDX) to TDP-L-rhamnose (TDP-Rha), catalyzed by two enzymes, TDP-6-deoxy-D-xylo-4-hexulose 3,5-epimerase (RmlC) and TDP-deoxy-L-lyxo-4-hexulose reductase (RmlD) (Figs 1 & 2). As expected these enzymes have been shown to be essential 5, 6, and crystal structures for RmlC from M. tuberculosis with and without the substrate analog TDP-Rha 7 and RmlD from Salmonella enterica 8 have been published. An assay to search for inhibitors of them has been described 9 and was revamped for the studies herein.
Figure 1. M. tuberculosis cell wall structure and its biosynthesis.

The linker region being targeted in this work is shown in the lower center region of the figure and the rhamnosyl biosynthetic pathway is shown in the lower right hand region of the figure. The two enzymes, RmlC and RmlD, which were screened for inhibitors herein, are shown in the boxed region of the rhamnosyl biosynthetic pathway.
Figure 2. The assay for RmlC and/or RmlD inhibitors.

The enzymatic reactions screened and the detection of NADPH by fluorescence is shown.
As part of the National Institutes of Health Molecular Libraries Screening Centers Network (MLSCN), Penn Center for Molecular Discovery (PCMD) performs high-throughput screens against various targets and deposits data into PubChem. Herein we report the high-throughput screen (HTS) of 201,368 compounds from the NIH Molecular Libraries Small Molecule Repository (BioFocus DPI) against Mycobacterium tuberculosis RmlC and RmlD in the same assay. The results of this screen are available on PubChem as BioAssay IDs (AIDs) 1532, 1533, 1695 and 1696.
The most potent hit from the screen was further studied to determine which enzyme, RmlC or RmlD, was the target of inhibition, and its activity was confirmed by re-synthesis of the compound. Kinetic studies to identify its mode of inhibition were undertaken and structural analogs of the hit compound were also tested to initiate structure-activity relationships (SAR). Finally, preliminary studies of the action of this class of compounds against whole M. tuberculosis bacteria were performed.
2. Results
2.1 Characterization of M. Tuberculosis RmlB, RmlC, and RmlD Enzymes
Rhamnosyl biosynthetic enzymes, RmlB, RmlC and RmlD, were cloned and expressed in E. coli and purified to homogeneity. The purity of the enzymes was seen by single bands on polyacrylamide gel electrophoresis with SDS. The RmlC and RmlD enzymes exhibit single bands of molecular weights of approximately 25,000 and 40,000, respectively (data not shown).
2.2 Optimization of Assay
The assay measures the activity of the two enzymes RmlC and RmlD that act sequentially in the formation of TDP-rhamnose (TDP-Rha). The RmlC enzyme converts TDP-6-deoxy-D-xylo-hexopyranosid-4-ulose (TDP-KDX) to TDP-6-deoxy-L-lyxo-hexopyranosid-4-ulose (TDP-KDL) and RmlD converts TDP-KDL to TDP-Rha with the use of NADPH (Fig. 2). TDP-KDX was synthesized enzymatically by converting TDP-glucose to TDP-KDX using RmlB, allowing the reaction to go to completion (the reaction is irreversible), and TDP-KDX was used directly without purification. The final assay consisted of RmlC, RmlD, and NADPH, and was initiated by the addition of TDP-KDX. The activity of the enzymes was detected by the time-dependent decrease in fluorescence observed upon the oxidation of NADPH to NADP. The Km values of TDP-KDX and NADPH were determined to be 211and 12 μM, respectively (Fig. 3A and 3B). The assay was optimized by varying the concentrations of RmlC, RmlD, NADPH, and TDP-KDX to obtain a linear decrease in fluorescence over 90 min (data not shown) and to keep the substrate concentrations as near to their Km values as possible (200 μM TDP-KDX and 25 μM NADPH were selected for the screen). The amounts of RmlC and RmlD were balanced such that if the amount of either enzyme was decreased the rate of the overall reaction decreased (i.e. neither enzyme was in excess). This ensured that the assay was sensitive to inhibition of either enzyme. The IC50 of the known inhibitor, TDP, was determined to be 340 μM (Fig. 3C) The QC plate gave a Z′-value of 0.78 with TDP (Fig. 3D).
Figure 3. Assay optimization.

Km determinations of (A) TDP-KDX and (B) NADPH. Substrate versus rate plots are shown; Km values were determined from these data as described in materials and methods. (C) Determination of the IC50 of the known inhibitor TDP used for quality control. (D) HTS quality control plate; Z′ was calculated to be 0.78. The data with fluorescence values of ~200,000 were controls with full activity (with no TDP); the data points with values of ~0 were blanks with no activity (contained no TDP-KDX); and the data points at ~75,000 were positive controls for inhibition (containing TDP-KDX, NADPH, RmlC and TDP at a concentration of 500 μM). (E) Z′-values of HTS plates.
2.3 High-Throughput Screening
HTS of the 201,368 compounds of the MLSCN library is summarized in Table 1. The screen was performed with mixtures of four compounds per well in 384-well plates. When a given well showed greater than 30% inhibition the 4 individual compounds were retested in individual wells. The average Z′-factor for the screen was 0.67 ± 0.05, indicating good plate uniformity throughout the screen (Fig. 3E) 10. A total of 2328 wells showed greater than 30% inhibition and from these wells 5,266 individual compounds were obtained and tested as a single compound. Of these, 470 showed inhibition of greater than 30% and from these 388 were tested for dose response.
Table 1.
High-throughput screen of 201,368 compounds of the MLSCN library
| Screening Step | MLSCN library compounds |
|---|---|
| Mixture screen (4 compounds per well) | |
| Unique compounds tested | 201,3681 |
| Wells that showed >30% inhibition | 2,3282 |
| Initial hit rate3 | 1.2% |
| Retest (single compound per well) | |
| Compounds retested4 | 5266 |
| Number of compounds that showed >30% inhibition | 470 |
| Number of compounds that showed <30% inhibition | 4796 |
| Retest hit rate5 | 8.93% |
| Dose Response | |
| Compounds tested | 388 |
| Active compounds | 372 |
| Hits reclassified as inactive due to fluorescence increase in absence of enzyme | 358 |
| True hits | 14 |
| True hit rate6 | 0.007% |
64,020 compounds were tested twice and thus 265,388 compounds (including duplicates) were tested in total. See experimental procedures for details.
It is most likely that only one of the 4 compounds in each well was active.
Expressed as (2328/201,368)*100.
Active wells had 4 compounds (with minor exceptions) but not all compounds were available.
(470/5266)*100.
(14/201,368)*100
2.4 IC50 Determination of Hits
Compounds that gave greater than 30% inhibition in single well assays were tested in dose-response. Of the 388 compounds tested, 372 gave IC50 values of less than 55 μM and were selected as active compounds (Table 1). When tested in buffer alone in the IC50 format (i.e. without enzyme or substrates), most of the active compounds gave the same dose-response curves independent of the RmlC and RmlD enzymes, indicating that they are fluorescent at the excitation/emission wavelengths of 340/460 nm and that for an unknown reason this fluorescence increased with time leading to a false positive result. (Often these false positive compounds gave percent inhibition much greater than the theoretical maximum value.) All compounds with IC50 values less than 20 μM were tested in buffer alone and 14 non-fluorescent compounds were selected as true hits. The IC50 values of the 14 compounds ranged from 0.398 to 18.13 μM. 1-(3-(5-ethyl-5H-[1,2,4]triazino[5,6-b]indol-3-ylthio)propyl)-1H-benzo[d]imidazol-2(3H)-one, SID 7975595, with an IC50 of 0.398 μM (Fig. 4A) was the best hit and was further studied.
Figure 4. Characterization of SID 7975595.

(A) IC50 of SID 7975595 from screen. (B) Determination of which enzyme, RmlC or RmlD, was inhibited by the enzyme. In one case, (■) RmlD was held constant at 5.1 nM and RmlC was varied as indicated on the graph; in the other case (□) RmlC was held constant at 70 nM and RmlD was varied as indicated on the graph. The graph shows that the IC50 values were dependent on the amount of RmlC and independent of the amount of RmlD. (C) Calculation of Ki for SID 7975595. The Km(obs) was plotted against the concentration of SID 7975595 and the Ki calculated from the slope, which equals Km/Ki, where Km is given by the y-axis intercept. (D) Cytotoxicity of SID 7975595 against HAE cells. A titration of SID 7975595 (●) and doxorubicin (Δ) was performed at the concentrations indicated by the X axis. At each concentration of SID 7975595 and doxorubicin, a control was run with no additions (○) and with an amount of DMSO equal to that present in the compounds samples (---).
2.5 Determination of which enzyme, RmlC or RmlD), is being inhibited
The enzyme being inhibited by compound SID 7975595 was determined by varying the RmlC enzyme concentration while keeping the RmlD concentration constant and vice versa (Fig. 4B). The data show that the IC50 remains constant when the RmlD concentration is increased up to 80-fold, but the IC50 changes as the RmlC concentration is changed, demonstrating that the enzyme being inhibited by compound SID 7975595 is RmlC. These results were confirmed using a GC/MS based assay 11 in which the amount of product of RmlC, TDP-6-deoxy-L-lyxo-hexopyranosid-4-ulose is determined directly after 4-keto reduction and further derivatization. The percent inhibition of RmlC was calculated as 52, 72, and 83% at 0.25, 0.5, and 5 μM SID 7975595, respectively. Although this method is inherently less accurate (extremely low amounts of product must be quantitated because of the 2% conversion equilibrium value of product versus substrate 11) it clearly confirmed in a direct assay the action of SID 7975595 against RmlC.
2.6 Kinetic Characterization of Inhibition
The Km(obs) of TDP-KDX was determined in the presence and absence of compound SID 7975595 to determine if it was acting as a competitive or non-competitive inhibitor (Table 2). The data show it is a classical competitive inhibitor, where at increasing concentrations of the compound (0.1, 0.15, and 0.2 μM) the Km(obs) increases from 200 μM in the absence of compound to almost 700 μM in the presence of 0.2 μM compound, while the Vmax value changes only slightly compared with the value measured in the absence of compound (the slight increase may be an artifact of the inability to use a high enough concentration of TDP-KDX in the presence of inhibitor). A plot of Km(obs) versus [I], was used to determine the Ki of SID 7975595, which is equal to Km/slope (Fig. 4C). The Ki value for SID 7975595 was found to be 0.1 μM.
Table 2.
TDP-KDX Km(obs) and Vmax for the RmlC reaction at various concentrations of SID 7975595
| SID 7975595 (μM) | Km(obs) (SD) | Vmax (SD) |
|---|---|---|
| 0 | 211 (43) | 0.20 (.01) |
| 0.1 | 521 (133) | 0.24 (.03) |
| 0.15 | 561 (147) | 0.25 (.03) |
| 0.2 | 694 (194) | 0.27 (.03) |
To check for the rate of onset of inhibition of RmlC by SID 7975595, its IC50 was determined after various times of pre-incubation with RmlC before assaying for enzyme activity (Fig. 5A and 5B). If the onset of inhibition due to SID 7975595 is slow, the IC50 would be expected to decrease as the pre-incubation time is increased. No such decrease in IC50 was observed; the reason for the slight increase with pre-incubation is not clear. To determine whether or not the binding was reversible, SID 7975595 at 10 times its IC50 was pre-incubated with RmlC at 100 times its final assay concentration, before diluting the compound-RmlC mixture 100-fold and monitoring the enzyme activity over time. During the pre-incubation the concentration of inhibitor is much higher than the enzyme (20 μM versus 0.0012 μM); thus if inhibitor binding is irreversible essentially all of the enzyme will be inactivated and remain inhibited upon dilution. However, the results shown in Fig. 5C and 5D indicate that the compound is a fully reversible inhibitor; the slopes (taken from 80 to 200 min) observed in the assay after pre-incubation of compound and enzyme for 0.5 hr or 0 hr are very similar to that of the control with no SID 7975595 in the assay. For an additional control, SID 7975595 was included in the dilution buffer at 3.98 μM, and inhibition of the reaction occurred as expected (Fig. 5C and 5D). (The reason for the initial period of 40–50 minutes before the decrease in fluorescence begins is not understood; however the phenomenon occurs in this dilution experiment whether or not inhibitor is present and a similar increase of fluorescence early in the reaction also occurs in the absence of RmlC substrate, TDP-KDX.)
Figure 5. Characterization of Inhibition.

(A) The effect of pre-incubation of SID 7975595 on activity versus concentration of SID 7975595. Inhibitor was pre-incubated for 0, 1, 2, or 3 hr with RmlC before starting the assay and measuring the enzyme activity. (B) The IC50 values calculated from the data of (A). (C) Determination of the reversibility of SID 7975595 inhibition of RmlC. The compound at 10 times its IC50 was pre-incubated for 0 hr (□) and 0.5 hr (Δ) with enzyme at 100 times its usual assay concentration before diluting it 100-fold and measuring the activity over time. Control assays with no compound (●) and with SID 7975595 at 10 times its IC50 after dilution (○) are also shown. The delay before the decrease in fluorescence is commented on in the text. (D) The absolute values of the slopes calculated between 90 and 200 minutes from the experiments is shown in (C) indicate the reversibility of SID 7975595 inhibition of RmlC. The column “10x” refers to the control where the dilution was into 10 times the IC50 of SID 7975595 and inhibition occurred as expected.
2.7 Preliminary Structure-Activity Relationships
Analogs of SID 7975595 (77070, 77071, 77072, 77073, and 77074 in Table 3) were purchased from ChemBridge. In addition, to confirm their activity, SID 7975595 and 77074 were resynthesized (see materials and methods). The activity of the resynthesized compounds was indistinguishable from the ChemBridge compounds. Additional analogs, 78531, 78532, and 78533 (Table 3) were synthesized, and the IC50 values of all these triazinoindol-benzimidazolones are reported in Table 3. The IC50 values of 77070 shows that when the R1 ethyl group attached to the indol nitrogen is replaced by a benzyl group (77070) the IC50 increase to 3 μM or approximately 15 fold. However, when R1 is H (77073) the IC50 also increases about the same amount. The IC50 is also affected by subtle changes to the R1 group; most dramatically the saturation of an allyl group (77074) to an n-propyl group (77071) caused a 10 fold increase in the IC50. Altogether these data suggested that the R1 portion of the compound must be involved in the binding of the compound in the active site (see modeling studies below). Consistently, the allyl substituent (77074) is the best inhibitor, and use of the Cheng-Prusoff equation 12, Ki=IC50/(1+[S]/Km), to convert the IC50 to Ki (an approach using less experimental data than that of Fig. 4C) yields a Ki of 62 nM. Oxidation of the thioether to sulfone did not substantially alter the enzyme inhibition (Table 3), however, the resulting compounds did show some chemical instability when stored for longer periods of time, disfavoring their further follow up. Interestingly, the introduction of methyl group in the R2 position in 78531 (Table 3) substantially reduced the ability of the triazinoindol-benzimidazolone to inhibit RmlC. Finally, variation of the spacer region between the two rings from 4 atoms (77074) to 2 atoms (SID 7972845) dramatically reduced the activity of the inhibitor. With this SAR in hand, we modeled the compounds in the active site of the known structure of RmlC 7.
Table 3.
IC50 values for inhibition of RmlC by substituted triazinoindol-benzimidazolones, and the MIC values of these compounds against M. tuberculosis in culture.
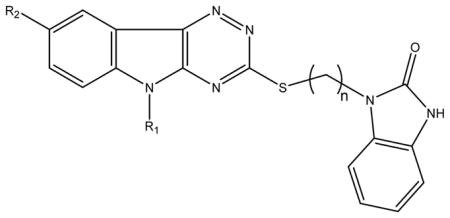 | ||||||
|---|---|---|---|---|---|---|
| Compound ID | R1 | R2 | n | Sulfone1 | IC50 (μM) | MIC (μg/ml)2 |
| 77074 | CH 2=CH-CH2 | H | 3 | No | 0.12 | 13 |
| SID 7975595 | CH3-CH2 | H | 3 | No | 0.2 | 19 |
| 77072 | CH3- | H | 3 | No | 0.5 | 9 |
| 77071 | CH3-CH2-CH2 | H | 3 | No | 1.25 | 9 |
| 77070 | 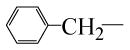 |
H | 3 | No | 2.9 | >50 |
| 77073 | H | H | 3 | No | 3.3 | ND |
| SID 7972845 | CH2=CH-CH2 | H | 1 | No | 10.3 | ND |
| 78531 | CH2=CH-CH2 | CH3 | 3 | No | 20 | 25 |
| 78532 | CH3-CH2 | H | 3 | Yes | 0.83 | ND |
| 78533 | CH2=CH-CH2 | H | 3 | Yes | 0.43 | ND |
Refers to oxidation of the thioether to a sulfone.
The MIC values were from at least 2 separate tests.
IC50 values markedly increased with time after compounds were dissolved in DMSO suggesting lack of stability.
2.8 Modeling of triazinoindol-benzimidazolones into the active site of RmlC
Prior to the docking of triazino-indols, the substrate analog, TDP-Rha, was re-docked into RmlC using both Glide and Autodock Vina. The docking poses generated by both programs were very similar to the crystal structure, a root mean square deviation (RMSD) of 0.9 Å (Glide SP), 1.7 Å (Glide XP), and 2.0 Å (Autodock Vina), respectively. As the RmlC-TDP-Rha complex was not part of either program’s training set 13, the relatively small RMSD values suggest that both programs performed well in the re-docking of TDP-Rha (see Fig. 6A).
Figure 6. Model of SID 7975595 docked in the active site.

(A) Binding pose of TDP-Rha as revealed by crystal structure 7 (thick sticks) and by Autodock Vina (thin sticks) (B) Docking pose of SID7975595 produced by Autodock Vina, (C) docking pose of SID7975595 produced by Glide XP. (D) Overlay of 78531 on 77074 docking pose. The extra methyl group (red surface) can cause steric clash with His119 and Phe121 (green surface) in the active site. In all panels, the protein backbone is shown in cartoon representation, with key residues in the active site shown in stick representation. Tyr132 which is commented on in the text is shown in (B) next to the triazinoindol ring of SID 7975595 but is not labeled due to lack of space.
The docking of SID 7975595 and its structural analogs yielded two different poses using Glide and Autodock Vina (Fig. 6B and 6C). In both poses, one ring system interacts with the thymidine binding regions of the active site and the other ring system with the sugar binding ring of the site (Figs. 6A, 6B, and 6C); the difference of the two poses being which ring system is in which position. In the Glide pose, the tricylic ring of SID 7975595 forms a stacking interaction with Tyr138 (thymidine binding region), whereas in the Autodock Vina pose, this ring is deeply inserted into the active site near Lys72 (sugar binding region). Although we cannot completely rule out either of the two poses without a crystal structure of RmlC-SID 7975595 complex, the Autodock Vina pose appears to be more consistent with the experimental data. As mentioned above, an extra methyl group on 78531 dramatically reduced its inhibitory activity and increased its IC50 from 0.12 μM of the parent compound 77074 to 20 μM. In the Autodock Vina pose, the tricyclic ring is inserted into the active site, where an extra methyl group at the R2 position will cause steric clash with His119 and Phe121 (Fig. 6D). Therefore, the Autodock Vina pose (Fig. 6B) better explains the reduced activity of 78531, and will be our focus in the following discussions.
As shown in Fig. 6B, SID 7975595 forms a large hydrophobic contact with the protein, preventing water molecules from accessing residues Phe26 and Tyr132, as well as residues at the bottom of the active site, i.e., Val74, Phe121, and Val130. These hydrophobic interactions appear to be the main contribution to the affinity of the compound, although hydrogen bond interaction is also observed between a nitrogen atom on the tricyclic ring and residue Arg59 which are 2.3A apart. The R1 substitutions on the tricyclic ring may help the compounds to secure a tight hydrophobic seal with the protein. As shown in Fig. 6B and 7A, the ethyl and allyl groups form abundant hydrophobic contact with the protein near Tyr132, whereas a methyl or hydrogen R1 group is unable to achieve such an effect. Interestingly, the large benzyl group of 77070 can be accommodated by a pocket near Glu143 and His62 (Fig. 7B). However, this conformation puts the benzyl ring near Lys72 and completely displaces water molecules hydrating this charged residue. This unfavorable desolvation effect may explain the reduced activity of the compound. Both Glide and Autodock Vina produced similar poses for SID 7972845, which is two carbons shorter than the highly active 77074 in the linker region connecting the two ring systems. The docking poses suggest that the shortened linker forces the compound to reorient in order to maintain hydrophobic contacts with the protein at both the bicyclic and tricyclic rings (Fig. 7C).
Figure 7. Docking poses of SID 7975595 structural analogs.

(A) Overlay of the docking poses of 77073 (thick sticks) and 77074 (thin sticks). The allyl group of the latter is highlighted in green surface. (B) Overlap of the docking poses of 77070 (thick sticks) and 77074 (thin sticks). The benzyl ring of 77070 is accommodated in a pocket formed by His62 and Glu143, where it is in close contact with Lys72 (red surface). (C) Overlay of the docking poses of SID 7972845 (thick sticks) and 77074 (thin sticks). In all three panels, the protein is shown in gray surface.
2.9. MIC of Compound in M. tuberculosis Cells
Minimum inhibitory concentrations of the hit compound (SID 7975595) and some of its analogs against M. tuberculosis are shown in Table 3. Clearly the compounds are active, and there is a rough correlation between the activity against whole M. tuberculosis cells and the activity against RmlC. However, future studies, such as the determination of the MIC values in strains overexpressing RmlC and the search for the build up of intermediates such as TDP-Glc in cells treated with the RmlC inhibitors are required before the action of these compounds at the whole bacterial cell level can be concluded to be due to inhibition of RmlC.
2.9 Cytotoxicity of SID 7975595 in HAE Cells
Toxicity of compound SID 7975595 in mammalian cells was determined in human aortic endothelial cells (Fig. 4D). The data show that SID 7975595 has an IC50 of approximately 75 μM in HAE cells while the positive control, doxorubicin, has an IC50 of ~10 μM. The percent of DMSO introduced into the assay during the addition of SID 7975595 or doxorubicin has no effect on the cells, as shown in Fig. 4D.
3. Discussion
A screen of 201,368 compounds from the NIH Molecular Libraries Small Molecule Repository for molecules that have the potential to be developed into new drugs against tuberculosis has identified a new series of compounds that inhibit the activity of one of the essential cell wall biosynthetic enzymes of M. tuberculosis. The most potent hit from the screen, 1-(3-(5-ethyl-5H-[1,2,4]triazino[5,6-b]indol-3-ylthio)propyl)-1H-benzo[d]imidazol-2(3H)-one (SID 7975595), has a “two-headed” structure (triazinoindol as the larger head group and benzimidazolone as the smaller head group) so based on this parent structure we have called this series of compounds triazinoindol-benzimidazolones.
SID 7975595 acts as a classical competitive inhibitor of the RmlC enzyme by increasing the Km for TDP-KDX without affecting the Vmax and has a Ki of 100 nM. Comparison of the Km of TDP-KDX and the Ki of the inhibitor suggests that the inhibitor bindings with an approximately 2000-fold higher affinity. SID 7972845 (Table 3) is identical to the most potent triazinoindol-benzimidazolone, 77074, except that the linker distance is two atoms in total between the two head groups in SID 7972845, versus four atoms in total in 70774. The IC50 of SID 7972845 compared to77074 increases 86-fold to 10.3 μM, suggesting this linker distance between the two head groups has a significant effect on the occupancy of the active site by the inhibitor. This result is perhaps expected as the shorter linker drastically constrains the position of the two aromatic groups as shown in the docking experiments (Fig. 7C).
Interestingly, the docking result shows that one of the aromatic head groups occupies the thymidine binding region and the other group the sugar binding region in the active site. Indeed, the difference between the two docking poses from Glide and Autodock Vina revolves around which aromatic head group occupies which position and in reality this could be compound dependent. The docking results of the triazinoindol-benzimidazolones also suggest that hydrophobic interaction plays an important role in their binding to RmlC. The different substitutions at the R1 position may affect the activity of the compounds by controlling the amount of hydrophobic contact with the protein. A small R1 group (H atom) reduces the area of such contact, whereas a large R1 group (benzyl ring) can cause an unfavorable interaction with charged and polar residues in the active site.
The ~10-fold increase in IC50 when the R1 allyl group is reduced from an allyl to an n-propyl group is at least partially explained by the loss of rotational entropy of the latter compound. Docking programs often assign a 0.65 kcal/mol penalty to the entropic loss of every rotatable bond, which corresponds to approximately a 3-fold difference in IC50. However, the binding does exhibit a 10-fold difference and other factors may be involved as well.
Unfortunately, in both Glide and Autodock Vina results, the docking scores have a poor correlation with the measured IC50 values (data not shown). Therefore, we could only establish a preliminary structure activity-relationship from the docking poses of SID 7975595 and its analogs. The poor correlation between docking scores and the IC50, albeit disappointing, may be explained by a standard error of 2–3 kcal/mol often associated with docking scores 13, 14. In addition, the binding free energy differences of the SID 7975595 analogs, as estimated from their IC50 values, are only 0.1 to 2.0 kcal/mol. Therefore, more accurate free energy calculations will be performed to obtain a better correlation with IC50 and to guide compound design in our future work.
Analysis of the docking poses shows that the charged Lys72 is poorly hydrated upon binding of SID 7975595 and its analogs. As a consequence, it may be desirable to introduce a polar group on the tricyclic ring to provide a hydrogen bond partner for Lys72. Such a hydrogen bond enclosed in a hydrophobic environment has been found particularly favorable in many cases 14. Modeling and synthesis of these compounds are currently under way in our labs.
The triazinoindol-benzimidazolone SID 7975595 has been shown to be relatively non-toxic (Fig. 4D). The triazinoindol-benzimidazolones are also active against M. tuberculosis in culture (Table 3) although the activity is modest. Future studies will involve producing compounds with higher activities at the whole cell level by combining modeling and medicinal chemical efforts. With such compounds in hand efforts will focus on confirming that the target in whole bacteria is RmlC using genetic and precursor isolation approaches.
4. Experimental procedures
4.1 Materials
MOPS, Triton X-100 and TDP-Glucose were purchased from Sigma. MgCl2 and glycerol were from Fisher. NADPH was purchased from Roche. Black 384-well low volume non-binding polystyrene plates were from Corning (Corning 3676). The 384-well v-bottom polypropylene plates (Greiner 781280), 1536-well v-bottom polypropylene plates (Greiner 782270), and 384-well white sterile tissue culture treated plates (Greiner 781080) were all from Greiner.
4.2 Protein Expression and Purification
The clone expressing Salmonella enterica RmlB used to make the TDP-KDX 15 was a gift from Jim Naismith, and RmlB was expressed and purified as described 15. (The RmlB from M. tuberculosis requires the addition of NAD for activity whereas NAD is tightly bound to the S. enterica RmlB as purified and no additional NAD is required. As we preferred not to introduce NAD into the system and were not testing for inhibition of RmlB, the S. enterica enzyme was used for substrate preparation). RmlC and RmlD from M. tuberculosis were cloned and expressed in Escherichia coli and purified as described 9.
4.3 Synthesis of TDP-KDX
The substrate, TDP-6-deoxy-D-xylo-hexopyranosid-4-ulose (TDP-KDX), for the RmlC enzyme was synthesized enzymatically by converting d-TDP-glucose (2.5 mg) to TDP-KDX using 2.4 μg of purified RmlB from Salmonella typhimurium 15 in 50 mM MOPS buffer, pH 7.4, at 30 °C for 1 hr. Aliquots of TDP-KDX were stored at −80 °C.
4.4 Assay Optimization
The activity of the M. tuberculosis cell wall enzymes, RmlC and RmlD, towards TDP-KDX was measured by the decrease in fluorescence observed upon the oxidation of NADPH to NADP, at excitation and emission wavelengths of 340 and 460 nm, respectively. The Km for TDP-KDX (substrate) was determined by varying the TDP-KDX concentration from 0–1000 μM (10 point, 1.5-fold serial dilutions), while maintaining the NADPH concentration at 15 μM in a total volume of 10 μl. The NADPH (cofactor) Km was determined by varying NADPH concentration from 0–100 μM (12 point, 1.5-fold serial dilutions), while maintaining TDP-KDX concentration at 200 μM in a total volume of 10 μl. The data were fit using the computer program XLfit using the “General pharmacology model 250” (Michaelis-Menten model).
Based on the Km values of TDP-KDX and NADPH, assays were set up varying the TDP-KDX (100 and 200 μM) and NADPH (15 and 25 μM) concentrations as well as the concentrations of RmlC (8.75 × 10−5 to 5.25 × 10−4 μg/μl) and RmlD (2.175 × 10−4 to 1.31 × 10−3 μg/μl) in 50 mM MOPS, pH 7.4, containing 1 mM MgCl2, 10% glycerol and 0.01% Triton X-100, at 25 °C (data not shown). The assay was monitored by the decrease in fluorescence over 3 hr on a plate reader (Perkin Elmer Envision 2102 Multilabel reader). Based on concentrations that gave a linear assay time course, final assay conditions for the HTS in 384-well plates (Corning 3676) were selected: 200 μM TDP-KDX, 25 μM NADPH, 2.63 × 10−4 μg/μl RmlC and 6.55 × 10−4 μg/μl RmlD in a volume of 10 μl, and monitoring of the decrease in 340/460 fluorescence over 90 minutes at 25° C. Under these conditions the amounts of RmlC and RmlD were balanced as determined by a significant decrease in rate when the amount of either enzyme was diminished.
TDP, a known inhibitor of the RmlC/RmlD enzymes, was used in a quality control (QC) plate to monitor the sensitivity of the enzymes towards inhibition throughout the course of the HTS. The IC50 value of TDP was determined by dose response (16 point, 2-fold serial dilution, highest concentration of 30 mM) against 200 μM TDP-KDX, 15 μM NADPH, 1.8 × 10−4 μg/μl RmlC, and 4.4 × 10−4 μg/μl RmlD in a 384-well assay plate (Corning 3676). The change in fluorescence over 3 hr was monitored, and the IC50 was calculated by a non-linear fit of the rate of change in fluorescence against TDP concentration. Based on the observed IC50 of 340 μM, a solution of TDP in DMSO was added to a compound storage plate (Greiner 781280), set up to deliver a final assay concentration of 500 μM TDP, by pintool transfer of 0.12 μl (384-pin, V&P Scientific), expected to give slightly greater than 50% inhibition. The TDP storage plate contained 20 μl of 40.3 mM TDP in DMSO in columns 3–22, and 20 μl DMSO in columns 2 and 24 for controls (100% activity) and 20 μl DMSO in columns 1 and 23 for blanks (0% activity, no TDP-KDX). A TDP-containing QC plate was included in each HTS run and assayed using the same protocol as the test compound plates (see below).
4.5 High-Throughput Screening (HTS)
HTS of 201,368 compounds from the Molecular Libraries Screening Center Network (MLSCN) library was conducted in 384-well assay plates. The compounds were supplied as a smaller library (106,290 compounds) and as a larger library (214,178 compounds) that fully contained all of the 106,290 compounds of the first library but with no straightforward way to only screen the compounds not present in the smaller library. The entire smaller library was screened and 159,098 compounds from the larger library were screened of which 95,078 were not present in the smaller library leading to the screening of 201,368 unique compounds. The initial library was supplied by BioFocus DPI in 384-well storage plates and the expanded library supplied by BioFocus DPI in 1536-well compound storage plates. The 384-well library (supplied as 10 mM solutions in DMSO in v-bottom plates) was screened as mixtures of four compounds per well, by transfer of compounds from four 384-well compound storage plates into a single 384-well assay plate. Thus, 384-well dilution compound plates (Greiner 781280) were made at 0.5 mM in 20 μl DMSO (one compound per well) from the 10 mM plates supplied by BioFocus DPI. Mixtures were generated during HTS, using a pintool (384-pin, V & P Scientific) to transfer 0.12 μl of compound from the same well number (e.g. A3) from four different 384-well 0.5 mM dilution compound plates into a single well of a 384-well assay plate containing 4 μl of H2O (again the same well number, e.g., A3). The final compound concentration was 5.5 μM per compound (22.0 μM total per well). A Multidrop reagent dispenser (Thermo) was used to dispense 5 μl of 83.5 mM MOPS buffer containing 2x RmlC, RmlD and NADPH, and a Multidrop Micro (Thermo) was used to dispense 1 μl of 83.5 mM MOPS buffer containing 10x TDP-KDX. Blank columns (1 and 23) contained 1 μl of 83.5 mM MOPS buffer instead of TDP-KDX. The final RmlC, RmlD, and NADPH concentrations in the 10.5 μl assay were 2.63 × 10−4 μg/μl, 6.53 × 10−4 μg/μl, and 25 μM, respectively, and the final concentration of TDP-KDX was 200 μM. The final buffer concentration was 50 mM MOPS buffer, pH 7.4 with 1 mM MgCl2, 10% glycerol, and 0.01% Triton X-100. The assay was monitored by the change in NADPH fluorescence at 25 °C, measured at time 0 and after 90 minutes, at excitation and emission wavelengths of 340 and 460 nm, respectively. Data were analyzed in IDBS ActivityBase, and the percent inhibition of each compound was calculated from the change in fluorescence over 90 min (ΔSignal), calculated from t=0 and t=90 min reads, and the mean of the change in plate controls and blanks over 90 min, using the following equation:
The 159,098 compounds in the 1536-well library were supplied by BioFocus DPI as 2.5 mM solutions in DMSO, stored in 1536-well v-bottom compound plates. These compounds were also screened as mixtures of four compounds per well in 384-well assay plates. Mixtures were generated by pintool (384-pin) transfer of 0.091 μl of compound from each of the four quadrants of a 1536-well compound plate into a single well of a 384-well compound dilution plate (Greiner 781280) containing 25 μl of H2O. For example, compounds in wells A5, A6, B5, and B6 of a 1536-well compound plate were mixed in well A3 of a 384-well compound dilution plate. Four μl from each well of the compound dilution plate were transferred into a 384-well assay plate (Corning 3676) using a 384-tip pipetting head (PerkinElmer Evolution P3 Pipetting Platform). The well location of the compounds was unchanged during the transfer into the assay plate; for example, the mixture of four compounds in well A3 of a compound dilution plate was transferred to well A3 of the corresponding assay plate. The 384-well mixture dilution compound plate contained 9.1 μM of each compound and a total of 36.8 μM in 25 μl H2O, and the final assay plate contained 3.64 μM of each compound and a total mixture concentration of 14.6 μM in 10 μL in each well. The additions of RmlC, RmlD, and NADPH (5 μl) and TDP-KDX (1 μl), and the HTS assay were the same as in the HTS of the 384-well compound plates. The data were analyzed in IDBS ActivityBase, as described above.
The compounds that gave greater than 30 % inhibition in the HTS of the 384- and 1536-well compound plates were selected as hits and retested as single compounds. Compounds were ordered from BioFocus DPI, diluted into 384-well compound plates (0.5 mM), transferred by pintool into 4 μl of H2O in assay plates to give a final concentration of 5.5 μM in a final assay volume of 10 μl, and screened as described above. Each compound was tested in duplicate, the data were analyzed by IDBS ActivityBase, and compounds that gave >30% inhibition in both well locations were selected for further study in dose-response.
4.6 IC50 determination of HTS hits
Compounds for the dose-response testing were reordered from BioFocus DPI in 384-well plates, with 20 compounds at 10 mM in 10 μl DMSO in each plate in wells A3-A22. Dose-response compound plates were created by diluting each 10 mM compound solution to 2.5 mM with the addition of 30 μl of DMSO, followed by a 16-point 2-fold serial dilution using 30 μl disposable tips on the Evolution pipetting platform, to a final volume of 20 μl in each well with the highest concentration at 2.5 mM. Compounds from the dose-response compound plate were transferred twice by pintool (2 × 0.11 μl) into 4 μl of H2O in an assay plate, followed by addition of 5 μl of RmlC, RmlD, and NADPH and 1 μl of TDP-KDX as above for the HTS assay. Each dose-response plate was tested in triplicate using the protocol described above for HTS. The data were analyzed using ActivityBase. Each dose-response assay plate contained compounds in columns 3–22, controls (100% activity) in columns 2 and 24, and blanks (0% activity) in columns 1 and 23. Each compound column (3–22) contained 16 two-fold dilutions of a single compound, ranging in concentration from 55 μM to 1.7 nM. Percent activity was calculated for each concentration of each compound from the change in fluorescence over 90 min, as described above under HTS. IC50 values were calculated from a 4-parameter logistic fit of the change in percent activity with compound concentration using XLfit (IDBS).
Compounds that gave IC50 values of less than 20 μM were tested in buffer alone, in the dose-response assay format, to determine the fluorescence due to the compounds in the absence of enzymes, substrate, and NADPH. One μl of compound from each well of the dose-response compound plate was mixed with 18 μl of H2O in a V-bottom plate (Greiner 781280), and 4 μl of this dilution mixture was transferred into an assay plate (Corning 3676) containing 6 μl of 83.5 mM MOPS buffer, and the fluorescence at 340/460 nm was monitored over 90 min. The data were analyzed by Excel and dose-response curves were plotted
4.7 Acquisition and synthesis of compounds
The active compound SID 7975595 was obtained three ways: re-supply from the Molecular Libraries Screening Small Molecule Repository, from ChemBridge, and by chemical synthesis (see below). The highly active 77074 was obtained from ChemBridge and chemically synthesized (see below); the remaining compounds shown in Table 2 were prepared by chemical synthesis.
4.8 Chemical synthesis
All solvents and chemicals were purchased from Sigma-Aldrich and Fisher Scientific except that 5-ethyl-5H-[1,2,4]triazino[5,6-b]indole-3-thiol was purchased from ChemBridge Corporation, CA, USA; 5-allyl-5H-[1,2,4]triazino[5,6-b]indole-3-thiol was purchased from Ryan Scientific, Inc., SC, USA; and 5-allyl-8-methyl-5H-[1,2,4]triazino[5,6-b]indole-3-thiol was purchased from Sigma-Aldrich (rare chemical library). 1H NMR spectra were recorded at 300 MHz on a Bruker ARX NMR instrument or 500 MHz on a Varian Inova NMR instrument. Accurate mass mass spectrometry in positive mode was performed on an Agilent 6220 TOF mass spectrometer equipped with a MultiMode source selected to be in the dual atmospheric pressure chemical ionization/electrospray ionization mode. Analytical RP-HPLC was conducted on a Shimadzu HPLC system with a Phenomenex C18 column (100Å, 3 μm, 4.6 × 50 mm), flow rate 1.0 mL/min and a gradient of solvent A (water with 0.1% TFA) and solvent B (acetonitrile): 0–2.00 min 100% A; 2.00–8.00 min 0–100% B (linear gradient). UV detection at 218 and 254 nm was used.
4.8.1. Synthesis of triazinoindol-benzimidazolones SID7975595, 77074, and 78531
Alkylation of the corresponding triazino-indol-3-thiols was performed as reported previously 16, 17 using optimized reaction conditions (Scheme 1). 5-Allyl-5H-[1,2,4]triazino[5,6-b]indol-3-thiol (0.242 g, 1 mmol), potassium hydroxide (65 mg, 1 mmol, 86.4%), and water (5 mL) were stirred at room temperature for 2h, followed by the addition of 1-(3-chloropropyl)-1,3-dihydro-2H-benzimidazol-2-one (0.234 g, 1 mmol, 90%). The resulting solution was stirred at 90°C for 24h. The reaction mixture was evaporated, and the crude product was purified by Biotage flash column chromatography to yield product 77074 as a light yellow powder.
Scheme 1.

1-(3-(5-ethyl-5H-[1,2,4]triazino[5,6-b]indol-3-ylthio)propyl)-1H-benzo[d]imidazol-2(3H)-one, SID7975595, 0.38 g (1 mmol scale), 93.9% yield. 1H NMR, 300 MHz (CDCl3): δ9.37 (s, 1H, NH), 8.42 (d, J = 7.6 Hz, 1H), 7.64–7.73 (m, 1H), 7.39–7.49 (m, 2H), 7.00–7.18 (m, 4H), 4.34 (q, J = 7.2 Hz, 2H, NCH2CH3), 4.14 (t, J = 6.9 Hz, 2H, NCH2), 3.46 (t, J = 7.0 Hz, 2H, SCH2), 2.40 (quintet, J = 7.0 Hz, CH2), 1.45 (t, J = 7.2 Hz, 3H, CH3). Mass spectrum m/z (M+Na)+ 427.1314 (427.1312 predicted). HPLC purity: >96% (monitored at 218 and 254 nm), tR = 5.92 min.
1-(3-(5-allyl-5H-[1,2,4]triazino[5,6-b]indol-3-ylthio)propyl)-1H-benzo[d]imidazol-2(3H)-one, 77074, 0.23 g (1 mmol scale), 55.2% yield. 1H NMR, 300 MHz (CDCl3): δ 8.41–8.47 (m, 1H), 8.29 (s, 1H, NH), 7.64–7.71 (m, 1H), 7.43–7.49 (m, 2H), 7.03–7.10 (m, 4H), 5.89–6.03 (m, 1H, CH=(Allyl)), 5.27 (d, J = 10.4 Hz, 1H, =CHa(Allyl)), 5.18 (d, J = 17.7 Hz, 1H, =CHb(Allyl)), 4.91 (d, J = 5.3 Hz, 2H, CH2(Allyl)), 4.12 (t, J = 6.9 Hz, 2H, NCH2), 3.44 (t, J = 7.0 Hz, 2H, SCH2), 2.38 (quintet, J = 7.0 Hz, 2H, CH2). Mass spectrum m/z (M+Na)+ 439.1315 (439.1312 predicted). HPLC purity: 100% (monitored at 218 and 254 nm), tR = 6.02 min.
1-(3-(5-allyl-8-methyl-5H-[1,2,4]triazino[5,6-b]indol-3-ylthio)propyl)-1H-benzo[d]imidazol-2(3H)-one, 78531, 40 mg (0.195 mmol scale), 47.6% yield. 1H NMR, 500 MHz (CDCl3): δ 8.52 (s, 1H, NH), 8.20 (s, 1H), 7.46 (d, J = 8.3 Hz, 1H), 7.31 (d, J = 8.3 Hz, 1H), 7.02–7.08 (m, 4H), 5.89–5.97 (m, 1H, CH=(Allyl)), 5.24 (d, J = 10.5 Hz, 1H, =CHa(Allyl)), 5.13 (d, J = 17.1 Hz, 1H, =CHb(Allyl)), 4.85 (d, J = 5.4 Hz, 2H, CH2(Allyl)), 4.10 (t, J = 6.8 Hz, 2H, NCH2), 3.41 (t, J = 7.1 Hz, 2H, SCH2), 2.36 (quintet, J = 7.1 Hz, 2H, CH2). Mass spectrum m/z (M+Na)+ 453.1469 (453.1458 predicted). HPLC purity: 100% (monitored at 218 and 254 nm), tR = 6.23 min.
4.8.2. Synthesis of triazinoindol-benzimidazolone sulfonation derivatives 78532 and 78533
Sulfonation reaction was carried out according to a reported procedure 18. Catalytic amounts (ca. 1 mg each) of sodium tungstate dihydrate (Na2WO4·2H2O), methyltrioctylammonium hydrogen sulfate ([CH3(n-C8H17)3N]HSO4), and phenylphosphonic acid (C6H5PO3H2) were added to a 25 mL flask, followed by the addition of 150 mg of aqueous 30% H2O2. After the mixture was stirred vigorously at room temperature for 10 min, 1-(3-(5-allyl-5H-[1,2,4]triazino[5,6-b]indol-3-ylthio)propyl)-1H-benzo[d]imidazol-2(3H)-one (77074, 70 mg, 0.168 mmol) was added and stirred at room temperature for 24h. The resulting mixture was evaporated and purified by reverse phase Biotage flash C18 column chromatography to give product 78533 as a light yellow powder.
1-(3-(5-ethyl-5H-[1,2,4]triazino[5,6-b]indol-3-ylsulfonyl)propyl)-1H-benzo[d]imidazol-2(3H)-one, 78532, 15 mg (0.0989 mmol scale), 34.7% yield. 1H NMR, 500 MHz (CDCl3): δ 9.34 (s, 1H, NH), 8.57 (d, J = 7.8 Hz, 1H), 7.86 (t, J = 7.8 Hz, 1H), 7.62 (d, J = 8.3 Hz, 1H), 7.58 (t, J = 7.6 Hz, 1H), 6.99–7.10 (m, 4H), 4.53 (q, J = 7.3 Hz, 2H, NCH2CH3), 4.14 (t, J = 6.1 Hz, 2H, NCH2), 3.87 (t, J = 6.8 Hz, 2H, SO2CH2), 2.50 (quintet, J = 6.4 Hz, CH2), 1.52 (t, J = 7.3 Hz, 3H, CH3). Mass spectrum m/z (M+Na)+ 459.1213 (459.1210 predicted). HPLC purity: 100% (monitored at 218 and 254 nm), tR = 5.60 min.
1-(3-(5-allyl-5H-[1,2,4]triazino[5,6-b]indol-3-ylsulfonyl)propyl)-1H-benzo[d]imidazol-2(3H)-one, 78533, 40 mg (0.168 mmol scale), 53.1% yield. 1H NMR, 500 MHz (CDCl3): δ 9.75 (s, 1H, NH), 8.55 (d, J = 7.6 Hz, 1H), 7.82 (t, J = 7.6 Hz, 1H), 7.54–7.62 (m, 2H), 7.00–7.15 (m, 4H), 5.93–6.02 (m, 1H, CH=(Allyl)), 5.31 (d, J = 10.3 Hz, 1H, =CHa(Allyl)), 5.23 (d, J = 17.1 Hz, 1H, =CHb(Allyl)), 5.09 (d, J = 4.2 Hz, 2H, CH2(Allyl)), 4.15 (br s, 2H, NCH2), 3.87 (br s, 2H, SO2CH2), 2.50 (br s, 2H, CH2). Mass spectrum m/z (M+Na)+ 471.1212 (471.1215 predicted). HPLC purity: 100% (monitored at 218 and 254 nm), tR = 5.72 min.
4.9 IC50 determination of triazinoindol-benzimidazolones
A slightly different method than that described above was used for determining the IC50 values of triazinoindol-benzimidazolones that were obtained after the screen was complete. Thus the IC50 values of compounds obtained from ChemBridge and by chemical synthesis were determined in 96-well plates by monitoring the conversion of NADPH to NADP via absorbance at 340 nm. The total assay volume was 200 μl of 50 mM 3-(N-morpholino)propanesulfonic acid buffer (MOPS) with 10% glycerol, 0.01% Triton X-100, 1 mM Mg2Cl2, at pH 7.4. Test compounds were added in 4 μl of DMSO at a range of concentrations. The remaining components were added to the following concentrations: RmlC (70 nM), RmlD (5 nM), TDP-KDX (200 μM), and NADPH (25 μM). The reaction was started by the addition of RmlC and continuously monitored for 80 min, after which the initial reactions velocities were determined and the IC50 values calculated using the program GraFit (Erithacus Software limited).
4.10 Determination of which Enzyme (RmlC or RmlD) is the Target of Inhibition
The 96-well plate method used to determine IC50 values of triazinoindol-benzimidazolones was performed. Thus, to determine which enzyme is inhibited by SID 7975595, the concentration of one enzyme was varied while maintaining the other constant, and vice versa, and the IC50 of compound SID 7975595 was monitored for changes in IC50 that correlated with changes in enzyme concentration. The RmlC enzyme concentration was held constant at 70 nM while varying RmlD concentration from 5 to 412 nM, and the RmlD concentration was held constant at 5 nM while varying RmlC concentration from 70 to 350 nM, in the presence of 200 μM TDP-KDX and 25 μM NADPH.
4.11 Direct Confirmation of inhibition of RmlC using GC/MS analysis to quantify the product TDP-6-deoxy-L-lyxo-hexopyranosid-4-ulose
An assay to confirm the inhibition of RmlC by SID 7975595 (at the concentrations of 0.25, 0.5, and 5 μM) in the absence of RmlD in which the RmlC product is measured by GC/MS after reduction of the 4-ketogroup and derivatization was performed as previously described 11.
4.12 Determination of the mode of Inhibition by SID 7975595
To determine the mode of inhibition of SID 7975595, the Km of TDP-KDX was determined in the absence of inhibitor and in the presence of several concentrations of inhibitor. TDP-KDX concentration was varied from 0–100 μM in the presence of SID 7975595 concentrations of 0.1, 0.15, and 0.2 μM. The Km and Vmax were determined by non-linear regression using GraFit. The Km(obs) was plotted against the concentration of SID 7975595 and the Ki calculated from the slope, which equals Km/Ki, where Km is given by the y-axis intercept.
4.13 Determination of time dependence of the onset of inhibition of SID 7975595
Time-dependence of the onset of inhibition by compound SID 7975595 was studied by pre-incubating the compound with RmlC and RmlD enzymes (and NADPH) in 384 well plates for times ranging from 0 to 3 hr before adding TDP-KDX to initiate the enzymatic assay. One μl of compound SID 7975595 from each well of the dose-response compound plate (see above) was mixed with 18 μl per well of H2O in a 384-well plate (Greiner 781280) and 4 μl of the resulting diluted compound was transferred into a 384-well assay plate (Corning 3676) containing 5 μl of 2x RmlC, RmlD, and NADPH in 83.5 mM MOPS buffer and pre-incubated for 0–3 hr. After pre-incubation, TDP-KDX was added and the enzymes were assayed over 90 min, and IC50 data were analyzed as described above for IC50 determination of HTS hits.
4.14 Determination of the reversibility of SID 7975595 inhibition
The reversibility of inhibition by SID 7975595 was determined using the procedure of Copeland 19. Thus SID 7975595 at 10 times its IC50 was incubated for 0 and 0.5 hr at 25°C with RmlC at 100 times its final assay concentration in 2 μl buffer, in Eppendorf microcentrifuge tubes. After incubation, the compound-RmlC mixture was diluted 100-fold to a final volume of 200 μl, RmlD, TDP-KDX, and NADPH were added, and the activity of the enzyme was measured by transferring 10 μl of the assay mixture into a 384-well assay plate and monitoring the decrease in NADPH fluorescence over 200 min as described above. Control assays were set-up in the absence of compound.
4.15 Modeling the binding of triazinoindol-benzimidazolones in the active site of RmlC
Docking of SID 7975595 and its analogs was performed using both Glide 20 and Autodock Vina 13. As a control experiment, TDP-Rha was re-docked into RmlC using both programs. The RmlC dimer structure was taken from pdb entry 2ixc (5). Protonation states of protein residues were initially assigned based on pKa calculations using PROPKA 21, 22. Key residues at the active site were then inspected visually to make sure that their protonation states were consistent with experimental findings. In particular, Lys72 was given a +1 charge despite of a predicted pKa of ~7, and His62, His119 were kept neutral with protons on their ε-nitrogen. The protein was then prepared using the Schrodinger Protein Preparation Wizard 23–25 and AutodockTools 26 for Glide and Autodock Vina docking, respectively. Initial structures of SID 7975595 and its analogs listed in Table 1 were prepared using Ligprep 27 or DiscoveryStudio 28. Docking with Glide was performed using both the standard precision (SP) and the extra precision (XP) modes, with the ligand van der Waals radii scaled by 0.8. Docking with Autodock Vina was performed with an exhaustiveness factor of 8.
4.16 MIC Determination
The MIC of SID 7975595 and structural analogs against Mycobacterium tuberculosis (H37Rv) was determined by the microbroth dilution method as described by Sun et al 29.
4.17 Cytotoxicity Studies with Human Aortic Endothelial (HAE) Cells
Cytotoxicity of compound SID 7975595 in mammalian cells was tested in HEA cells by seeding 1000 cells/25 μl/well in white 384-well tissue culture treated plates and incubating at 37 °C for 24 hr before treating with compound and incubating for an additional 24 hr at 37 °C and measuring luminescence. To prepare SID 7975595, 3μl of 10 mM SID 7975595 in DMSO was mixed with 47 μl of EGM-2 endothelial cell media and 25 μl of this mixture was transferred into 25 μl of media for a 16-point, 2-fold serial dilution ranging in concentration from 600 μM to 18 nM. As controls, 6 μl of doxorubicin (positive control) and DMSO (solvent control) were mixed with 94 μl of media and 25 μl of each was transferred into 25 μl media for a 16 point, two-fold serial dilution, where the doxorubicin concentrations were the same as the SID 7975595 concentrations (600 μM to 18 nM) and the DMSO only concentration was the same as the DMSO concentration in both the SID 7975595 and doxorubicin tests. Five μl of each of the serial dilutions were added to cells in 25 μl media for final compound concentrations of 100 μM to 3 nM. Luminescence was measured on the Envision microplate reader after the addition of 30 μl of CellTiter-Glo and incubation for 10 min. Each plate was tested in triplicate.
Acknowledgments
We gratefully acknowledge the support of NIH grant P01 AI 057846 (MRM, REL); NIH grant U54-HG003915 (SLD); and grants from NSF, NIH, HHMI, CTBP and NBCR (JAM). We also thank Jim Naismith for helpful discussions and for providing the E. coli strain over expressing RmlB from Salmonella typhimurium. We thank Huiyan Jing for assistance with the cytotoxicity assay.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.O’Brien RJ, Nunn PP. Am J Respir Crit Care Med. 2001;163:1055–1058. doi: 10.1164/ajrccm.163.5.2007122. [DOI] [PubMed] [Google Scholar]
- 2.Gandhi NR, Moll A, Sturm AW, Pawinski R, Govender T, Lalloo U, Zeller K, Andrews J, Friedland G. Lancet. 2006;368:1575–1580. doi: 10.1016/S0140-6736(06)69573-1. [DOI] [PubMed] [Google Scholar]
- 3.Van Rie A, Enarson D. Lancet. 2006;368:1554–1556. doi: 10.1016/S0140-6736(06)69575-5. [DOI] [PubMed] [Google Scholar]
- 4.Barry CE, Crick DC, McNeil MR. Infect Disorders-Drug Targets. 2007;7:182–202. doi: 10.2174/187152607781001808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li W, Xin Y, McNeil MR, Ma Y. Biochem Biophys Res Commun. 2006;342:170–178. doi: 10.1016/j.bbrc.2006.01.130. [DOI] [PubMed] [Google Scholar]
- 6.Ma Y, Pan F, McNeil MR. J Bacteriol. 2002;184:3392–3395. doi: 10.1128/JB.184.12.3392-3395.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dong C, Major LL, Srikannathasan V, Errey JC, Giraud MF, Lam JS, Graninger M, Messner P, McNeil MR, Field RA, Whitfield C, Naismith JH. J Mol Biol. 2007;365:146–159. doi: 10.1016/j.jmb.2006.09.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blankenfeldt W, Kerr ID, Giraud MF, McMiken HJ, Leonard G, Whitfield C, Messner P, Graninger M, Naismith JH. Structure (Camb) 2002;10:773–786. doi: 10.1016/s0969-2126(02)00770-0. [DOI] [PubMed] [Google Scholar]
- 9.Ma Y, Stern RJ, Scherman MS, Vissa V, Yan W, Jones VC, Zhang F, Franzblau SG, Lewis WH, McNeil MR. Antimicrob Agents Chemother. 2001;45:1407–1416. doi: 10.1128/AAC.45.5.1407-1416.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang JH, Chung TDY, Oldenburg KR. Journal of Biomolecular Screening. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 11.Stern RJ, Lee TY, Lee TJ, Yan W, Scherman MS, Vissa VD, Kim SK, Wanner BL, McNeil MR. Microbiology. 1999;145:663–671. doi: 10.1099/13500872-145-3-663. [DOI] [PubMed] [Google Scholar]
- 12.Cheng Y, Prusoff WH. Biochemical Pharmacology. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 13.Trott O, Olson AJ. J Comput Chem. 2009 doi: 10.1002/jcc.21334. On line ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. Journal of Medicinal Chemistry. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 15.Allard STM, Giraud MF, Whitfield C, Messner P, Naismith JH. Acta Crystallogr D Biol Crystallogr. 2000;56(Pt 2):222–225. doi: 10.1107/s0907444999016200. [DOI] [PubMed] [Google Scholar]
- 16.Hamid HA, Mousaad A, Ramadan ES, Ashry ESHE. Heterocyclic Communications. 1999;5:473–480. [Google Scholar]
- 17.Kgokong JL, Smith PP, Matsabisa GM. Bioorganic & Medicinal Chemistry. 2005;13:2935–2942. doi: 10.1016/j.bmc.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 18.Sato K, Hyodo M, Aoki M, Zheng XQ, Noyori R. Tetrahedron. 2001;57:2469–2476. [Google Scholar]
- 19.Copeland RA. Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists. J. Wiley; Hoboken, N.J: 2005. [PubMed] [Google Scholar]
- 20.Glide, version 5.5. Schrodinger LLC; New York: 2009. [Google Scholar]
- 21.Li H, Robertson AD, Jensen JH. Proteins-Structure Function and Bioinformatics. 2005;61:704–721. doi: 10.1002/prot.20660. [DOI] [PubMed] [Google Scholar]
- 22.Bas DC, Rogers DM, Jensen JH. Proteins-Structure Function and Bioinformatics. 2008;73:765–783. doi: 10.1002/prot.22102. [DOI] [PubMed] [Google Scholar]
- 23.Prime version 2.1. Schrodinger, LLC; New York, NY: 2009. [Google Scholar]
- 24.Impact version 5.5. Schrodinger, LLC; New York, NY: 2009. [Google Scholar]
- 25.Epik version 2.0. Schrodinger, LLC; New York, NY: 2009. Schroding Suite 2009 Protein Preparation Wizard. [Google Scholar]
- 26.Sanner MF. Journal of Molecular Graphics & Modelling. 1999;17:57–61. [PubMed] [Google Scholar]
- 27.LigPrep, version 2.3. Schrodinger, LLC; New York, NY: 2009. [Google Scholar]
- 28.DiscoveryStudio, version 2.1. Accelrys Software Inc; 2009. [Google Scholar]
- 29.Sun D, Scherman MS, Jones V, Hurdle JG, Woolhiser LK, Knudson SE, Lenaerts AJ, Slayden RA, McNeil MR, Lee RE. Bioorganic & Medicinal Chemistry. 2009;17:3588–3594. doi: 10.1016/j.bmc.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]