

Abstract
Human ITPase, encoded by the ITPA gene, and its orthologs (RdgB in Escherichia coli and HAM1 in Saccharomyces cerevisiae) exclude noncanonical nucleoside triphosphates (NTPs) from NTP pools. Deoxyinosine triphosphate (dITP) and 2′-deoxy-N-6-hydroxylaminopurine triphosphate are both hydrolyzed by ITPase to yield the corresponding deoxynucleoside monophosphate and pyrophosphate. In addition, metabolites of thiopurine drugs such as azathioprine have been shown to be substrates for ITPase. The ITPA 94C>A [P32T] variant is one of two polymorphisms associated with decreased ITPase activity. Furthermore, the ITPA 94C>A [P32T] variant is associated with an increased risk of adverse drug reactions for patients treated with azathioprine. The nature of the observed phenotypes for ITPA 94C>A [P32T] variant individuals is currently unclear. Our biochemical assays indicate the P32T ITPase has 55% activity with dITP compared to wild-type ITPase. Complementation experiments at 37°C show that N-6-hydroxylaminopurine sensitivity of E. coli rdgB mutants is reduced with a plasmid bearing the ITPA 94C>A [P32T] gene approximately 50% less than with a plasmid bearing the wild-type ITPA gene. The reduction in sensitivity is less at 42°C. Experiments with synthetic lethal E. coli recA(ts) rdgB mutants show that the ITPA 94C>A [P32T] gene also complements the recA(ts) rdgB growth deficiency at 42°C approximately 40% lower than wild-type ITPA gene. Western blot analysis indicates the expression level of P32T ITPase is reduced in these cells relative to wild-type. Our data support the idea that P32T ITPase is a functional protein, albeit with a reduced rate of noncanonical NTP pyrophosphohydrolase activity and reduced protein stability.
Keywords: ITPA, ITPase, RdgB, noncanonical purines, dITP, N-6-hydroxylaminopurine
Introduction
Human ITPase (encoded by the ITPA gene) is thought to exclude noncanonical (deoxy)nucleoside triphosphates ((d)NTPs) from DNA and RNA precursor pools [1–4]. Phosphorylation of inosine monophosphate (IMP), a precursor to adenosine monophosphate (AMP) and guanosine monophosphate (GMP), can produce deoxyinosine triphosphate (dITP) [5, 6]. Oxidative deamination of (deoxy)guanosine triphosphate ((d)GTP) forms (deoxy)xanthosine triphosphate ((d)XTP), another noncanonical (d)NTP that is a substrate for ITPase. In addition, 2′-deoxy-N-6-hydroxylaminopurine triphosphate (dHAPTP) is a substrate for ITPase [1]. Incorporation of these noncanonical dNTPs into DNA can result in mutation and genetic instability [5, 7]. dITP/dXTP/dHAPTP pyrophosphohydrolases, such as ITPase prevent this incorporation and are conserved throughout the three domains of life [1–3, 8, 9].
The RdgB protein in Escherichia coli is an ortholog of ITPase [1]. It has been shown that an E. coli recA200(ts) rdgB double mutant strain is inviable at 42°C [10]. When RdgB is not available, RecA is required due to the formation of double strand breaks resulting from endonuclease V initiated repair [7]. Adenylosuccinate synthase, which is coded for by the purA gene, initiates the conversion of IMP to AMP [6]. The temperature sensitivity of the recA200(ts) rdgB mutants can be overcome with overexpression of the purA gene, indicating that the role of RdgB may be to adjust the levels of nucleotide pools [11].
N-6-hydroxylaminopurine (HAP) is a noncanonical purine that is not known to arise spontaneously in E. coli, but serves as an excellent diagnostic tool to assess the functionality of the noncanonical purine repair system in E. coli [7]. E. coli moa strains are deficient in molybdopterin biosynthesis. Exposure of moa mutants to HAP results in a hypersensitive phenotype and an elevated level of mutagenesis relative to wild-type E. coli [12]. A moa rdgB mutant strain shows an even greater increase in HAP sensitivity and mutagenesis suggesting that a molybdoenzyme(s) and RdgB protein are required for the exclusion of HAP from DNA [7]. The HAP detoxifying molybdoenzyme activity has recently been attributed to the ycbX and yiiM gene products [13]. Incorporation of HAP into DNA stimulates endonuclease V to nick the DNA (unpublished results, M. Wan and R.P. Cunningham). If this nick is crossed by a replicative polymerase, a lethal double strand break will occur. Indeed, inactivation of the endonuclease V gene, nfi, renders moa rdgB strains viable at an elevated concentration of HAP, albeit with increased levels of mutagenesis [7].
A common ITPA mutation in human populations is the ITPA 94C>A [P32T] missense mutation which changes a proline residue at position 32 to threonine [14, 15]. Biochemical studies with erythrocytes from individuals homozygous for the ITPA 94C>A [P32T] mutation determined that these cells display 0% ITPase activity, while heterozygous individuals have about 25% ITPase activity [16]. These levels are consistent with and indicate ITPase activity levels depend on the integrity of both protomers of the ITPase dimer. The ITPA 94C>A [P32T] allele is present in all ethnic groups, being highest (11–19%) in Asian and lowest (1–2%) in Central and South American populations [17, 18]. ITPA deficiency is not linked to pathology in afflicted individuals, but perturbed (d)ITP levels may be harmful under circumstances of cellular stress.
ITPA deficiency may be responsible for adverse drug reactions in patients treated with azathioprine or 6-mercaptopurine [19–21]. Metabolites of these immunosuppressive thiopurine drugs are also substrates of ITPase [22]. These drugs have been used in the treatment of acute lymphocytic leukemias in adults [23], childhood acute myeloid leukemias [24], childhood non-Hodgkin’s lymphoma [25], Crohn’s disease [26], ulcerative colitis [27, 28], systemic lupus erythematosus [29], and solid organ transplantations [30]. A study of inflammatory bowel disease patients treated with azathioprine revealed that side effects such as rash, flu-like symptoms, and pancreatitis were correlated with the P32T mutation [19]. Other studies have linked side effects with azathioprine such as myelosuppression and hepatotoxicity to the ITPA 94C>A [P32T] mutation [31].
Currently two hypotheses exist that help to explain the decreased activity associated with the ITPA 94C>A [P32T] mutation. Stenmark et al. suggest that the mutation causes a shift of a loop in the protein which renders the protein catalytically inactive by disrupting substrate binding and/or catalysis [3]. Conversely Arenas et al. propose that the ITPA 94C>A [P32T] mutation causes missplicing at the mRNA level. They propose that missplicing is due to destruction of an exonic splicing silencing element in exon 2, and activation of two cryptic 5′ splice sites which results in the missplicing of exon 2 and 3 [14]. Our research aims to investigate the validity of these two models for explaining the observed deficiency in individuals with the ITPA 94C>A [P32T] mutation by measuring pyrophosphohydrolase activity in vitro and in vivo.
Materials and methods
E. coli strain construction and growth conditions
The growth medium was LB (10g bacto-tryptone, 5g yeast extract, 10g NaCl per liter dH20 at pH 7.2). The following concentrations of antibiotics were used: kanamycin 34μg/ml; chloramphenicol 34μg/ml; tetracycline 20μg/ml; ampicillin 50μg/ml [32]. Cell growth was monitored using a Shimadzu UV-1700 PharmaSpec spectrophotometer at 600nm. E. coli strains were constructed using P1-mediated transductions [33]. All parental strains were obtained from the Coli Genetic Stock Center unless noted. The moa rdgB strain designated EWU2, was made by first transducing the moa-250::Tn10d(Tc) allele [7] into KL16 cells and selecting for tetracycline resistance. This strain was then transduced with the rdgB3::kan allele [10] followed by selection for kanamycin resistance. Construction of EWU2 was verified by determining HAP sensitivity using a HAP gradient plate assay [7]. The recA200(ts) rdgB strain, designated EWU5, was constructed at 30°C by first cotransducing the recA200(ts) allele linked to slrD300::Tn10 into KL16 and selecting for transductants that were tetracycline resistant and UV sensitive at 42°C [34]. A second transduction followed with the rdgB3::kan allele [10] and selection for kanamycin resistance.
Plasmid construction
Construction of plasmids pET28a-ITPA, pBJH-ITPA and pBJH-EV (empty vector) was described previously [1]. Construction of pET28a-P32T ITPA was performed using the mega-primer method for site directed mutagenesis which employs a two-step PCR protocol using pET28a-ITPA as starting material [35]. Incorporation of a single C>A base change at position 94 was confirmed by DNA sequencing. Plasmid pBJH-P32T ITPA was constructed by NdeI – EcoRI double digestion of pET28a-P32T ITPA and pBJH-EV followed by ligation of the P32T ITPA fragment into pBJH. Construction of the pBJH plasmids in this manner allows for constitutive protein expression to be driven by the native E. coli promoter for endonuclease IV.
Overexpression and purification of His6-tagged P32T ITPase
Overexpression and purification of P32T ITPase was performed as described previously for wild-type ITPase [1].
Analysis of deoxyribonucleoside triphosphate pyrophosphohydrolase activity of P32T ITPase
Enzyme kinetics assays and data analysis for P32T ITPase was performed as described previously for wild-type ITPase [1].
HAP cytotoxicity complementation assay
HAP cytotoxicity was determined for E. coli moa rdgB double mutants (EWU2) transformed with pBJH-EV, pBJH-ITPA or pBJH-P32T ITPA as described previously [1].
Synthetic lethality complementation assay
Fresh 30°C overnight cultures of E. coli recA200(ts) rdgB double mutants (EWU5) transformed with pBJH-EV, pBJH-ITPA or pBJH-P32T ITPA were diluted into 25 mL of fresh LB-Ampicillin broth (250x dilution ratio) pre-warmed at 42°C. Cultures were incubated in a 42°C shaking water bath. Cell growth was monitored over a ten hour time course by determining optical density at 600 nm.
Western Blotting
EWU5 cells were transformed with pBJH-EV, pBJH-ITPA, and pBJH-P32T ITPA expression plasmids and grown at 30° and 42°C in fresh LB-Ampicillin broth to a density of approximately 2 × 108 cells/ml. The cells were collected by centrifugation and the bacterial pellets were washed with Tris-buffered saline (pH 7.6) (TBS), resuspended in TBS containing 0.1% Tween-20 (TBS-T) at a ratio of 2 ml/gram of cell paste and lysed by sonication using a Vibra-Cell sonicator (Sonics &Materials, Inc., Newtown, CT) set at 40% amplitude. The cell lysates were centrifuged for 20 minutes at 67,509 g. A Bradford Assay was performed on the supernatants to determine protein concentrations. BSA was used as a standard.
SDS-polyacrylamide gel electrophoresis was performed on a 12% separating gel using a discontinuous buffer system (Bio-Rad). Samples consisting of 5 μg of protein from each cleared lysate were diluted in gel buffer containing 4% SDS and 20% glycerin in 0.05 M Tris (pH 6.8) and were reduced with 2% β-mercaptoethanol prior to electrophoresis. For immunoblotting, electrophoretic transfer of proteins to nitrocellulose was performed in a Trans-Blot SD Semi-Dry apparatus (Bio-Rad) according to manufacturer’s instructions. Following transfer, the nitrocellulose blot was blocked overnight with 5% powdered milk in TBS-T at 4° C. The nitrocellulose blot was then incubated with primary antibody, Rabbit PolyAb Anti-ITPA (from Proteintech Group, Inc.), at a dilution of 1:800 for one hour at room temperature followed by three 15 minute washes in 5% powdered milk in TBS-T. The blot was incubated with HRP-conjugated secondary antibody (1:10,000 goat anti-rabbit), for one hour at room temperature followed by three 15 minute washes in 5% powdered milk in TBS-T. Immunoblots were developed using chemiluminescence (Super Signal Femto Maximum Sensitivity Kit from Pierce Protein Research Products) according to the manufacturer’s instructions. The blot was imaged using a Fluoro HD chemiluminescent imager from Alpha Innotech Corp. AlphaEase FC and FluorChem 9900 programs were used for protein detection and quantification.
Results
Kinetic analysis of P32T ITPase
Table 1 shows the results of our in vitro kinetic analysis with P32T ITPase using the presumed physiological substrate dITP. Enzyme preparations and experimental manipulations for wild-type and P32T ITPase were performed by the same experimenter under identical conditions. P32T ITPase obeyed Michaelis-Menten kinetics over the same substrate concentration previously specified for wild-type ITPase [1]. ITPase has previously been documented to be subject to substrate inhibition [1, 4], therefore data points not subject to substrate inhibition were chosen to determine kinetic constants using a standard nonlinear least squares estimation technique. From Table 1 it can be seen that the specificity constant (kA) for the P32T enzyme is approximately 55% of the value for wild-type enzyme. The difference in kA appears to be derived from the rate of catalysis (kcat) rather that the Michaelis constant (Km). In our case Km is reflective of substrate binding [36]. Therefore our data suggests that the mutant enzyme binds substrate appropriately, however, kcat is reduced approximately 50% for the mutant enzyme.
TABLE 1.
Kinetic constants.
| Genotype | Km | kcat | kA |
|---|---|---|---|
| μM | 1/s | 1/mM-s | |
| P32T | 27.9 ± 5.8 | 38.0 ± 1.8 | 1361 ± 289 |
| Wildtype[1] | 32.5 ± 2.8 | 79.6 ± 2.0 | 2448 ± 218 |
Each result is the mean ± S.D. from three independent experiments.
HAP cytotoxicity complementation assay
Cytotoxicity tests were performed to quantify the activity of the P32T mutant ITPase in vivo against exogenous noncanonical purine nucleoside triphosphates. For this assay plasmid expressing wild-type ITPase or the mutant P32T ITPase driven by a constitutively expressed E. coli promoter or empty vector were transformed into an E. coli moa rdgB double mutant. Figure 1A shows our cytotoxicity results after 48 hours of growth at 37°C on rich media containing various concentrations of HAP. We observed that moa rdgB cells transformed with empty vector are extremely sensitive to HAP, whereas transformation of these cells with plasmids bearing wild-type ITPA or ITPA 94C>A [P32T] complements the HAP sensitive phenotype to varying degrees. The level of survival for the cells transformed with wild-type plasmid is equivalent to the level reported for E. coli moa cells at these HAP concentrations [1]. This suggests that expression of wild-type ITPase from our plasmid fully complements the chromosomal rdgB mutation and indicates that ITPase activity from the plasmid is at a level equivalent to rdgB activity from the E. coli chromosome. Conversely, the observed level of complementation for the strain expressing P32T ITPase is between 100 and 25% of wild-type complementation. At 5 and 10 μg/ml HAP no statistically significant difference in survival is observed between strains expressing wild-type and P32T ITPase; however, at 25 μg/ml HAP we observed a roughly 50% decrease in survival for cells expressing P32T ITPase compared to cells expressing wild-type ITPase. At 50 μg/ml HAP the level of survival for cells expressing P32T ITPase is roughly 25% of cells expressing wild-type ITPase. At this concentration the level of survival for cells expressing P32T ITPase is equivalent to the level seen for cells transformed with empty vector at 10 μg/ml HAP. Therefore, it appears that the P32T ITPase has functional pyrophosphohydrolase activity in vivo against dHAPTP at a rate somewhat reduced compared to wild-type. To address the possibility that the P32T ITPase may have reduced stability compared to the wild-type enzyme, the HAP cytotoxicity assay was performed with incubation at 42°C. Figure 1B shows that for 5 and 10 μg/ml HAP the mean level of survival for cells expressing P32T ITPase is about 50% lower than the level for cells expressing wild-type ITPase. At 25 and 50 μg/ml HAP the mean level of survival for the cells expressing P32T ITPase is about 30% of cells expressing wild-type ITPase. Despite the level of error in the 42°C measurements it appears that a slight reduction in P32T ITPase activity is observed at elevated temperatures. Therefore, our data suggests that the P32T mutant may have reduced protein stability at elevated temperatures.
Figure 1.

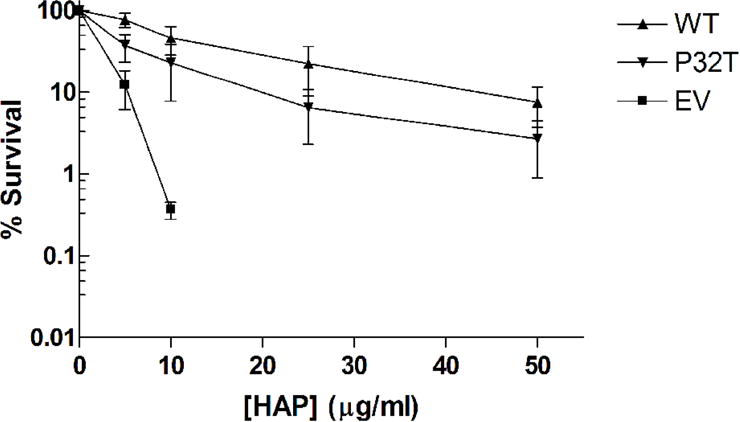
HAP cytotoxicity complementation assay. Fresh overnight cultures of EWU2 transformed with plasmids bearing constitutively expressed wild-type ITPase (WT), P32T ITPase (P32T) or empty vector (EV) were serially diluted and then plated on LB-ampicillin plates containing various concentrations of HAP. Surviving colonies were scored after 48 hours incubation at (A) 37°C or (B) 42°C. Each result is the mean ± S.E. of three to five independent experiments.
Synthetic lethality complementation assay
Complementation of synthetic lethality in recA rdgB cells was performed to quantify P32T ITPase activity in vivo against the presumed physiological substrate dITP. E. coli recA200(ts) rdgB double mutants are inviable at the non-permissive temperature of 42°C [10]. In recA200(ts) mutants the RecA protein is inactivated at 42°C rendering cells deficient in homologous recombination [37], an activity required for repair of the lethal double strand breaks that form in rdgB mutants [7]. For this assay we transformed E. coli recA200(ts) rdgB double mutants with the same plasmids used in the HAP cytotoxicity complementation assay (above). Figure 2 shows that the recA200(ts) rdgB double mutant transformed with empty vector does not grow at the non-permissive temperature over a ten hour time-course. In contrast, transformation of these cells with plasmids bearing either the wild-type ITPA or the ITPA 94C>A [P32T] gene reverses the synthetic lethal phenotype to varying degrees. Over the first half of the time-course, cells transformed with plasmid bearing wild-type ITPA or ITPA 94C>A [P32T] have growth curves that are indistinguishable from one another. For the latter half of the time-course the cells expressing P32T ITPase show a reduced rate of growth compared to the cells expressing wild-type ITPase. Nonetheless cells expressing P32T ITPase are viable and continue to grow over the duration of the time course. At the ten hour time point the level of cell growth for the cells expressing P32T ITPase is 60% of the level observed for the cells expressing wild-type ITPase, relative to the cells containing the empty vector plasmid. Figure 3 shows that these cells are viable after 48 hours incubation at the non-permissive temperature, however these cells appear to grow more slowly than those transformed with plasmid bearing wild-type ITPA. Note for the 42°C plate, that an approximately equal number of colonies are observed for cells transformed with plasmid bearing wild-type ITPA or ITPA 94C>A [P32T].
Figure 2.
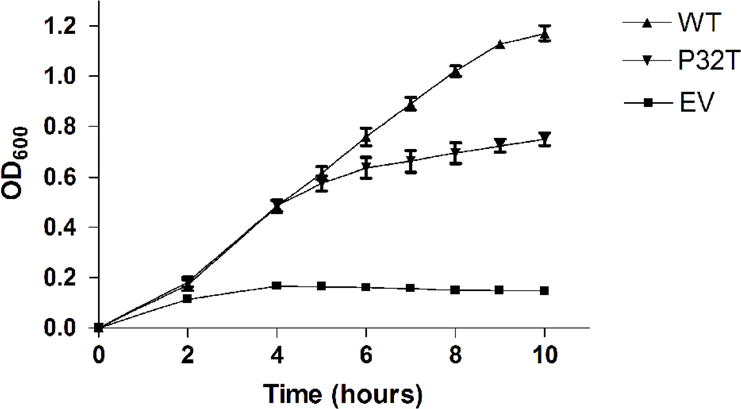
Synthetic lethality complementation assay. Fresh overnight cultures of EWU5 transformed with plasmids bearing constitutively expressed wild-type ITPase (WT), P32T ITPase (P32T) or empty vector (EV) were diluted in pre-warmed LB-ampicillin media and incubated in a shaking water bath at 42°C. Optical density measurements were taken over a ten hour time-course. Each result is the mean ± S.E. from three independent experiments.
Figure 3.

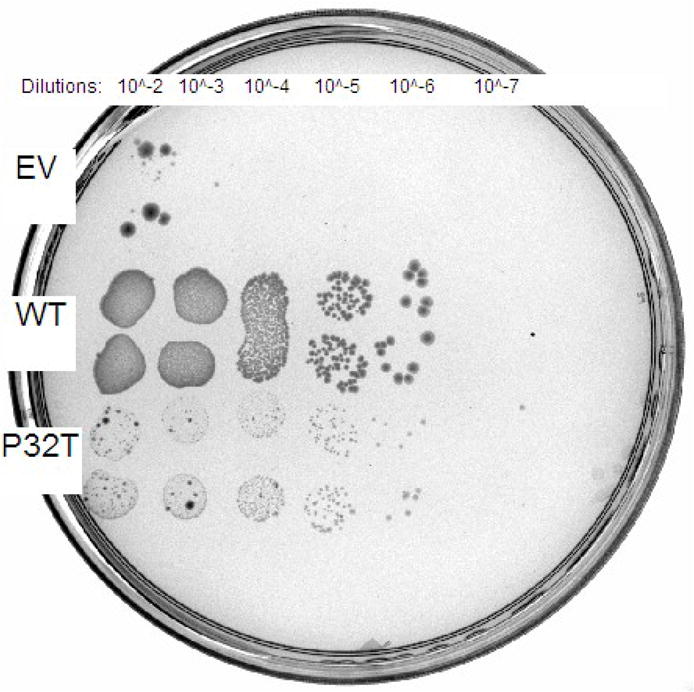
Chronic synthetic lethality complementation assay. Fresh overnight cultures of EWU5 transformed with plasmids bearing constitutively expressed wild-type ITPase (WT), P32T ITPase (P32T) or empty vector (EV) were serially diluted in LB media and 5 μl of each dilution was spotted in duplicate onto LB-ampicillin plates pre-warmed at 30 or 42°C. Plates were incubated at appropriate temperature for 48 hours and photographed under white light. (A) 30°C plate; (B) 42°C plate.
Investigation of the expression levels for wild-type and P32T ITPase in these cells indicate that decreased levels of P32T ITPase contribute to the slow growth phenotype for the cells transformed with plasmid bearing ITPA 94C>A [P32T]. The representative Western blot in Figure 4 shows that at 30°C the level of expression for wild-type ITPase is somewhat greater than the level seen for P32T ITPase. However, at 42°C the level of expression is noticeably reduced for the P32T ITPase, while the level for the wild-type ITPase is equivalent to the level seen at 30°C. Data from a semi quantitative analysis of duplicate Western blots is presented in Table 2. At 30°C the level of P32T ITPase is approximately 25% lower than the wild-type level, while the level of P32T ITPase is roughly 50% lower than the wild-type level at 42°C. Additionally, it appears that growth at 42°C results in an expression level of P32T ITPase that is 35% lower than the expression at 30°C, while the wild-type level remains unchanged. Altogether, our results suggest that the ITPA 94C>A [P32T] mutation results in a reduction in ITPase activity and ITPase stability. Presumably both defects contribute to an obvious slow growth phenotype for recA mutants due to an increase in the incorporation of deoxyinosine monophosphate into genomic DNA and resulting double strand breaks [1].
Figure 4.
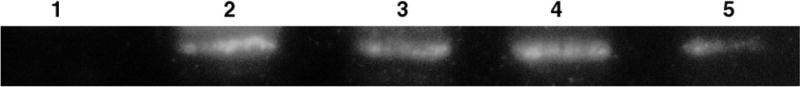
Western blot analysis. EWU5 cells were transformed with plasmids bearing constitutively expressed wild-type ITPase (WT), P32T ITPase (P32T) or empty vector (EV) and grown at 30° and 42°C. Cells were harvested and total protein concentration was determined. Western blotting was performed as described in Materials and Methods with 5 μg of total protein from each cleared cell lysate using Rabbit PolyAb Anti-ITPA and HRP-conjugated secondary antibody. Lane 1, EV at 30°C; lane 2, WT at 30°C; lane 3, P32T at 30°C; lane 4, WT at 42°C; lane 5, P32T at 42°C.
TABLE 2.
Semiquantitative Western blot analysis.
| Genotype | 30°C | 42°C |
|---|---|---|
| P32T | 73% ± 8% | 47% ± 8% |
| Wildtype | 100% | 112% ± 29% |
Each result is the mean ± error from two independent blots, relative to wild-type at 30°C.
Discussion
The experimental results presented herein suggest that the P32T ITPase is a functional protein, which has a reduced rate of catalysis and reduced stability compared to wild-type ITPase. Our in vitro and in vivo measurements suggest that the P32T ITPase has about 50% activity of the wild-type ITPase. From our kinetic assays we have measured a 45% reduction in catalytic efficiency with dITP as a substrate for P32T ITPase compared to wild-type ITPase. Likewise, with our synthetic lethal complementation assay we observed a rescue with the ITPA 94C>A [P32T] bearing plasmid that was approximately 60% of the wild-type ITPA level. Similarly, the 37°C HAP cytotoxicity complementation assays demonstrated that ITPA 94C>A [P32T] bearing plasmid reversed that HAP sensitive phenotype to a level that was about 50% that of wild-type ITPA bearing plasmid. For the synthetic lethal complementation assay dITP is the presumed lethal metabolite that ITPase is preventing from accumulating in DNA precursor pools. Therefore, from the data presented above we can infer that P32T ITPase has equivalent pyrophosphohydrolase activity with both dITP and dHAPTP in vivo. Indeed, wild-type ITPase has been shown to have equivalent pyrophosphohydrolase activity in vitro with dITP and dHAPTP as substrates [1].
Investigation of protein levels for wild-type and P32T ITPase suggest that the mutant form has reduced stability compared to wild-type. Interestingly, our Western blotting data indicates that the P32T ITPase is present at a lower level than wild-type ITPase. Protein expression from both wild-type ITPA and ITPA 94C>A [P32T] in the cells used for Western blot analysis is driven by the constitutively expressed native E. coli promoter for endonuclease IV. Therefore, we interpret a reduction in protein levels at 30°C to indicate that the P32T ITPase is an inherently less stable protein than the wild-type ITPase. The fact that lower levels of P32T ITPase are observed at 30°C than the level observed for wild-type suggests that the substitution of proline with threonine at this position leads to a protein conformation that is more susceptible to degradation that the wild-type structure. In addition, the stability of P32T ITPase appears to be further reduced at 42°C whereas the wild type protein levels remain constant over this temperature range. This is consistent with our results for the HAP cytotoxicity assay where, relative to wild-type expressing cells, we observed a greater reduction in survival for the P32T ITPase expressing cells at 42°C than we did at 37°C. Altogether we find that reduced catalysis and reduced protein stability contribute to a roughly 50% reduction in ITPase activity in vivo for P32T ITPase.
Our data has inconsistencies with the hypothesis postulated by Stenmark et al. who suggested that the P32T mutation would perturb ITPase structure in a way that would render the protein catalytically inactive [3]. Proline 32 is located in a loop on the outer surface of the protein, distal from the dimer interface and substrate binding pocket. Pro-32 is separated from an extended loop containing proposed active site residues Asp-41 and Glu-44 by a short five residue beta-strand [3]. We propose a model that is consistent with our kinetic and Western blot data in which a change from the conformationally restrictive proline to threonine may cause an alteration of the Asp-41/Glu-44 containing loop resulting in a reduced rate of catalysis (kcat) and reduced protein stability. However, this extended loop does extend into the dimer interface, therefore, our data does not rule out the possibility that “cross-talk” between subunits of this homodimeric protein may be perturbed. In fact, cooperativity has been reported for the E. coli ortholog [9]. Nonetheless, because Km was unchanged for the mutant it seems less likely that any possible cooperativity would be affected. In addition, this substitution may cause the Asp-41/Glu-44 containing loop to become more susceptible to misfolding or degradation by proteolysis.
The P32T mutation is understood to result in an absence of ITPase activity as measured in red blood cells derived from a patient with the ITPA 94C>A [P32T] mutation [16]. Insight into a potential mechanism for this deficiency has come from the analysis of mRNA transcripts from peripheral blood leukocytes of patients bearing the ITPA 94C>A [P32T] allele [14, 15]. These data suggest that a missplicing event in the ITPA 94C>A [P32T] transcript can result in decreased levels of mature ITPA 94C>A [P32T] transcripts. Assuming that missplicing of the ITPA 94C>A [P32T] transcript is not specific to peripheral blood leukocytes, our experimental data suggests the basis of ITPase deficiency in these patients is primarily the result of a defect in mRNA splicing, rather than the production of a grossly unproductive enzyme. We find that the P32T ITPase is a functional protein both in vivo and in vitro. Therefore, if normal levels of P32T ITPase protein were produced in an affected patient we could expect the level of ITPase activity in samples from that patient to be detectable. Because ITPase activity has been reported to be undetectable in individuals with the ITPA 94C>A [P32T] mutation it seems much more likely missplicing is occurring, and that the level of missplicing is varied among tissues, with red blood cells being severely affected. In addition several reports have indicated strong substrate inhibition for ITPase at elevated levels of (d)ITP [1, 38, 39]. Further analysis of our kinetic data obtained for wild-type ITPase under substrate inhibition conditions with dITP as a substrate [1] indicates that the inhibitor constant (Ki) is 277 μM, which is approximately nine times the value of the Km for this enzyme-substrate pair. Therefore, a reduction of ITPase activity in an individual with the ITPA 94C>A [P32T] mutation could potentially be compounded four-fold. In essence, a missplicing event would lead to decreased expression of an enzyme that is catalytically compromised and less stable, which in turn would result in an elevated concentration of ITP which would reduce ITPase activity via substrate inhibition. It is important to note the recent observation that Itpa homozygous knock-out mice die before weaning and have gross cardiac abnormalities [40]. In fact, the birth ratio indicates that over half of the homozygous null offspring died before birth [40]. Furthermore, ITPA has been found to be expressed ubiquitously in mice, supporting a need for functional protein in many tissues [41]. Therefore, barring some large differences among mammals, it seems unlikely that the ITPA 94C>A [P32T] mutation leads to a true null phenotype in humans. Instead we propose that the ITPA 94C>A [P32T] mutation results in a missplicing event that leads to the production of decreased levels of P32T ITPase which functions at a rate that is about half that of wild-type, is less stable than wild-type, and is subject to substrate inhibition1.
Acknowledgments
We are grateful for the Eastern Washington University J. Herman & Jean Swartz Undergraduate Biotechnology Scholarship awarded to G.H. This work was made possible by funding from a Faculty Grant for Research and Creative Work from Eastern Washington University awarded to N.E.B and NIH grants CA116318 and CRR1 C06RR0154464 awarded to R.P.C. We thank the Coli Genetic Stock Center for providing bacterial strains.
Footnotes
The authors wish to note that during the revision of our manuscript another manuscript has been published containing similar experimental results where the authors have reached conclusions which are consistent with the conclusions presented herein [42].
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Burgis NE, Cunningham RP. Substrate specificity of RdgB protein, a deoxyribonucleoside triphosphate pyrophosphohydrolase. J Biol Chem. 2007;282:3531–3538. doi: 10.1074/jbc.M608708200. [DOI] [PubMed] [Google Scholar]
- 2.Porta J, Kolar C, Kozmin SG, Pavlov YI, Borgstahl GE. Structure of the orthorhombic form of human inosine triphosphate pyrophosphatase. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2006;62:1076–1081. doi: 10.1107/S1744309106041790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stenmark P, Kursula P, Flodin S, Graslund S, Landry R, Nordlund P, Schuler H. Crystal structure of human inosine triphosphatase. Substrate binding and implication of the inosine triphosphatase deficiency mutation P32T. J Biol Chem. 2007;282:3182–3187. doi: 10.1074/jbc.M609838200. [DOI] [PubMed] [Google Scholar]
- 4.Lin S, McLennan AG, Ying K, Wang Z, Gu S, Jin H, Wu C, Liu W, Yuan Y, Tang R, Xie Y, Mao Y. Cloning, expression, and characterization of a human inosine triphosphate pyrophosphatase encoded by the ITPA gene. J Biol Chem. 2001;276:18695–18701. doi: 10.1074/jbc.M011084200. [DOI] [PubMed] [Google Scholar]
- 5.Galperin MY, Moroz OV, Wilson KS, Murzin AG. House cleaning, a part of good housekeeping. Mol Microbiol. 2006;59:5–19. doi: 10.1111/j.1365-2958.2005.04950.x. [DOI] [PubMed] [Google Scholar]
- 6.Zalkin H, Nygaard P. Biosynthesis of Purine Nucleotides. In: Neidhardt FC, editor. Escherichia coli and Salmonella Cellular and Molecular Biology. Vol. 1. ASM Press; Washington, DC: 1996. pp. 561–579. [Google Scholar]
- 7.Burgis NE, Brucker JJ, Cunningham RP. Repair system for noncanonical purines in Escherichia coli. J Bacteriol. 2003;185:3101–3110. doi: 10.1128/JB.185.10.3101-3110.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hwang KY, Chung JH, Kim SH, Han YS, Cho Y. Structure-based identification of a novel NTPase from Methanococcus jannaschii. Nat Struct Biol. 1999;6:691–696. doi: 10.1038/10745. [DOI] [PubMed] [Google Scholar]
- 9.Savchenko A, Proudfoot M, Skarina T, Singer A, Litvinova O, Sanishvili R, Brown G, Chirgadze N, Yakunin AF. Molecular basis of the antimutagenic activity of the house-cleaning inosine triphosphate pyrophosphatase RdgB from Escherichia coli. J Mol Biol. 2007;374:1091–1103. doi: 10.1016/j.jmb.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 10.Clyman J, Cunningham RP. Escherichia coli K-12 mutants in which viability is dependent on recA function. J Bacteriol. 1987;169:4203–4210. doi: 10.1128/jb.169.9.4203-4210.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clyman J, Cunningham RP. Suppression of the defects in rdgB mutants of Escherichia coli K-12 by the cloned purA gene. J Bacteriol. 1991;173:1360–1362. doi: 10.1128/jb.173.3.1360-1362.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kozmin SG, Pavlov YI, Dunn RL, Schaaper RM. Hypersensitivity of Escherichia coli Delta(uvrB-bio) mutants to 6-hydroxylaminopurine and other base analogs is due to a defect in molybdenum cofactor biosynthesis. J Bacteriol. 2000;182:3361–3367. doi: 10.1128/jb.182.12.3361-3367.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kozmin SG, Leroy P, Pavlov YI, Schaaper RM. YcbX and yiiM, two novel determinants for resistance of Escherichia coli to N-hydroxylated base analogues. Mol Microbiol. 2008;68:51–65. doi: 10.1111/j.1365-2958.2008.06128.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arenas M, Duley J, Sumi S, Sanderson J, Marinaki A. The ITPA c.94C>A and g.IVS2+21A>C sequence variants contribute to missplicing of the ITPA gene. Biochim Biophys Acta. 2007;1772:96–102. doi: 10.1016/j.bbadis.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 15.Cao H, Hegele RA. DNA polymorphisms in ITPA including basis of inosine triphosphatase deficiency. J Hum Genet. 2002;47:620–622. doi: 10.1007/s100380200095. [DOI] [PubMed] [Google Scholar]
- 16.Sumi S, Marinaki AM, Arenas M, Fairbanks L, Shobowale-Bakre M, Rees DC, Thein SL, Ansari A, Sanderson J, De Abreu RA, Simmonds HA, Duley JA. Genetic basis of inosine triphosphate pyrophosphohydrolase deficiency. Hum Genet. 2002;111:360–367. doi: 10.1007/s00439-002-0798-z. [DOI] [PubMed] [Google Scholar]
- 17.Maeda T, Sumi S, Ueta A, Ohkubo Y, Ito T, Marinaki AM, Kurono Y, Hasegawa S, Togari H. Genetic basis of inosine triphosphate pyrophosphohydrolase deficiency in the Japanese population. Mol Genet Metab. 2005;85:271–279. doi: 10.1016/j.ymgme.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 18.Marsh S, King CR, Ahluwalia R, McLeod HL. Distribution of ITPA P32T alleles in multiple world populations. J Hum Genet. 2004;49:579–581. doi: 10.1007/s10038-004-0183-y. [DOI] [PubMed] [Google Scholar]
- 19.Marinaki AM, Ansari A, Duley JA, Arenas M, Sumi S, Lewis CM, Shobowale-Bakre el M, Escuredo E, Fairbanks LD, Sanderson JD. Adverse drug reactions to azathioprine therapy are associated with polymorphism in the gene encoding inosine triphosphate pyrophosphatase (ITPase) Pharmacogenetics. 2004;14:181–187. doi: 10.1097/00008571-200403000-00006. [DOI] [PubMed] [Google Scholar]
- 20.Marinaki AM, Duley JA, Arenas M, Ansari A, Sumi S, Lewis CM, Shobowale-Bakre M, Fairbanks LD, Sanderson J. Mutation in the ITPA gene predicts intolerance to azathioprine. Nucleosides Nucleotides Nucleic Acids. 2004;23:1393–1397. doi: 10.1081/NCN-200027639. [DOI] [PubMed] [Google Scholar]
- 21.Okada Y, Nakamura K, Hiromura K, Nojima Y, Horiuchi R, Yamamoto K. Pro32Thr polymorphism of inosine triphosphate pyrophosphatase gene predicts efficacy of low-dose azathioprine for patients with systemic lupus erythematosus. Clin Pharmacol Ther. 2009;85:527–530. doi: 10.1038/clpt.2008.261. [DOI] [PubMed] [Google Scholar]
- 22.Bierau J, Lindhout M, Bakker JA. Pharmacogenetic significance of inosine triphosphatase. Pharmacogenomics. 2007;8:1221–1228. doi: 10.2217/14622416.8.9.1221. [DOI] [PubMed] [Google Scholar]
- 23.Gottlieb AJ, Weinberg V, Ellison RR, Henderson ES, Terebelo H, Rafla S, Cuttner J, Silver RT, Carey RW, Levy RN, et al. Efficacy of daunorubicin in the therapy of adult acute lymphocytic leukemia: a prospective randomized trial by cancer and leukemia group B. Blood. 1984;64:267–274. [PubMed] [Google Scholar]
- 24.Paton CM, Ekert H, Waters KD, Matthews RN, Toogood IR. Treatment of acute myeloid leukaemia in children. Aust N Z J Med. 1982;12:143–146. doi: 10.1111/j.1445-5994.1982.tb02446.x. [DOI] [PubMed] [Google Scholar]
- 25.Wollner N, Burchenal JH, Lieberman PH, Exelby P, D’Angio G, Murphy ML. Non-Hodgkin’s lymphoma in children. A comparative study of two modalities of therapy. Cancer. 1976;37:123–134. doi: 10.1002/1097-0142(197601)37:1<123::aid-cncr2820370119>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 26.Pearson DC, May GR, Fick GH, Sutherland LR. Azathioprine and 6-mercaptopurine in Crohn disease. A meta-analysis. Ann Intern Med. 1995;123:132–142. doi: 10.7326/0003-4819-123-2-199507150-00009. [DOI] [PubMed] [Google Scholar]
- 27.Bean RH. The treatment of chronic ulcerative colitis with 6-mercaptopurine. Med J Aust. 1962;49(2):592–593. [PubMed] [Google Scholar]
- 28.Mahadevan U, Tremaine WJ, Johnson T, Pike MG, Mays DC, Lipsky JJ, Sandborn WJ. Intravenous azathioprine in severe ulcerative colitis: a pilot study. Am J Gastroenterol. 2000;95:3463–3468. doi: 10.1111/j.1572-0241.2000.03362.x. [DOI] [PubMed] [Google Scholar]
- 29.Abu-Shakra M, Shoenfeld Y. Azathioprine therapy for patients with systemic lupus erythematosus. Lupus. 2001;10:152–153. doi: 10.1191/096120301676669495. [DOI] [PubMed] [Google Scholar]
- 30.Ponticelli C, Tarantino A, Vegeto A. Renal transplantation, past, present and future. J Nephrol. 1999;12(Suppl 2):S105–110. [PubMed] [Google Scholar]
- 31.Zelinkova Z, Derijks LJ, Stokkers PC, Vogels EW, van Kampen AH, Curvers WL, Cohn D, van Deventer SJ, Hommes DW. Inosine triphosphate pyrophosphatase and thiopurine s-methyltransferase genotypes relationship to azathioprine-induced myelosuppression. Clin Gastroenterol Hepatol. 2006;4:44–49. doi: 10.1016/j.cgh.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 32.Miller J. Experiments in Molecular Genetics. Cold Spring Harbor Laboratory; Cold Spring Harbor, N.Y: 1972. [Google Scholar]
- 33.Wechsler JA, Gross JD. Escherichia coli mutants temperature-sensitive for DNA synthesis. Mol Gen Genet. 1971;113:273–284. doi: 10.1007/BF00339547. [DOI] [PubMed] [Google Scholar]
- 34.Csonka LN, Clark AJ. Construction of an Hfr strain useful for transferring recA mutations between Escherichia coli strains. J Bacteriol. 1980;143:529–530. doi: 10.1128/jb.143.1.529-530.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barik S. Site-Directed Mutagenesis by Double Polymerase Chain Reaction. In: White BA, editor. Methods in Molecular Biology. Vol. 15. Humana Press, Inc; Totowa, NJ: 1993. PCR Protocols: Current Methods and Applications. [DOI] [PubMed] [Google Scholar]
- 36.Voet DJ, Voet JG. Biochemistry. 3. John Wiley & Sons, Inc; 2008. [Google Scholar]
- 37.Lloyd RG, Low B, Godson GN, Birge EA. Isolation and characterization of an Escherichia coli K-12 mutant with a temperature-sensitive recA- phenotype. J Bacteriol. 1974;120:407–415. doi: 10.1128/jb.120.1.407-415.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vanderheiden BS. Human erythrocyte “ITPase”: an ITP pyrophosphohydrolase. Biochim Biophys Acta. 1970;215:555–558. doi: 10.1016/0304-4165(70)90109-1. [DOI] [PubMed] [Google Scholar]
- 39.Holmes SL, Turner BM, Hirschhorn K. Human inosine triphosphatase: catalytic properties and population studies. Clin Chim Acta. 1979;97:143–153. doi: 10.1016/0009-8981(79)90410-8. [DOI] [PubMed] [Google Scholar]
- 40.Behmanesh M, Sakumi K, Abolhassani N, Toyokuni S, Oka S, Ohnishi YN, Tsuchimoto D, Nakabeppu Y. ITPase-deficient mice show growth retardation and die before weaning. Cell Death Differ. 2009 doi: 10.1038/cdd.2009.53. (Epub ahead of print). [DOI] [PubMed] [Google Scholar]
- 41.Behmanesh M, Sakumi K, Tsuchimoto D, Torisu K, Ohnishi-Honda Y, Rancourt DE, Nakabeppu Y. Characterization of the structure and expression of mouse Itpa gene and its related sequences in the mouse genome. DNA Res. 2005;12:39–51. doi: 10.1093/dnares/12.1.39. [DOI] [PubMed] [Google Scholar]
- 42.Stepchenkova EI, Tarakhovskaya ER, Spitler K, Frahm C, Menezes MR, Simone PD, Kolar C, Marky LA, Borgstahl GE, Pavlov YI. Functional study of the P32T ITPA variant associated with drug sensitivity in humans. J Mol Biol. 2009;392:602–613. doi: 10.1016/j.jmb.2009.07.051. [DOI] [PMC free article] [PubMed] [Google Scholar]