

Abstract
We recently identified bis(amide) CCG-1423 (1) as a novel inhibitor of RhoA/C-mediated gene transcription that is capable of inhibiting invasion of PC-3 prostate cancer cells in a Matrigel model of metastasis. An initial structure-activity relationship study focusing on bioisosteric replacement of the amides and conformational restriction identified two compounds, 4g and 8, with improved selectivity for inhibition of RhoA/C-mediated gene transcription and attenuated cytotoxcity relative to 1. Both compounds were also capable of inhibiting cell invasion with equal efficacy to 1 but with less attendant cytotoxicity.
Cancer metastasis is a tremendous medical problem responsible for thousands of deaths every year.1 Metastases arise when dysregulation of one or more cellular processes allow malignant cells to escape the confines of the tissue of origin and establish themselves in alternate sites. Signaling through RhoA/C is important for invasion and metastasis of many cancers.2–4 In addition to the well-known role of RhoA/C in cytoskeletal function, there is a less well understood downstream action on gene transcription.5,6 The pathway involved in this has recently been elucidated and several components are related to cancer pathogenesis. The mitogenic G protein coupled receptor ligands bombesin, thrombin, and lysophosphatidic acid (LPA) and their receptors are well-known mitogens and stimulate tumor invasion. The novel Gα12 family of heterotrimeric G proteins (G12 and G13) activates RhoA and RhoC through guanine exchange factors such as leukemia-associated RhoGEF (LARG). Most relevant to the present work on Rho-transcriptional mechanisms are the megakaryoblastic leukemia transcriptional co-activator proteins (MKL1 & 2) which cooperate with the transcription factor, serum response factor (SRF), to increase expression of a number of genes potentially related to cancer progression and metastasis.5,6 Exciting recent knockout and siRNA data have shown a key in vivo role for RhoC in breast cancer metastasis7 and for MKL1 and SRF in melanoma and breast cancer metastases8. These studies provide important support for the idea that Rho signaling and specifically Rho-regulated gene transcription may be exciting targets for cancer therapy.
We recently identified a compound CCG-1423 (1) that blocks SRE-Luciferase gene transcription in response to activation of RhoA and RhoC signaling pathways.9 Consistent with its role as a Rho/SRF pathway inhibitor, 1 potently (<1 µM) inhibited LPA-induced DNA synthesis in PC-3 prostate cancer cells. It also inhibited the growth of RhoC-overexpressing melanoma lines (A375M2 and SK-Mel-147) at nanomolar concentrations, but was less active on related cell lines (A375 and SK-Mel-28) that express lower levels of RhoC. 1 inhibited Rho-dependent invasion by PC-3 prostate cancer cells, whereas it did not affect the Gαi-dependent invasion by the SKOV-3 ovarian cancer cell line. Thus, based on its profile, 1 is a promising lead compound for the development of novel pharmacologic tools to disrupt transcriptional responses of the Rho pathway in cancer.
Despite its favorable effects on cancer cell function, 1 did exhibit some modest acute cellular toxicity toward PC-3 cells at 24 hours as evidenced by some non-specific inhibition of gene expression (TK-Renilla) and a parallel decrease in a WST-1 cell viability readout. Consequently, we undertook initial molecular modifications of 1 with the goal of improving its potency and/or selectivity and attenuating its cytotoxicity. Three structural features of the lead were identified as potential areas of concern. First, the N-O bond in the tether between the two carboxamides is susceptible to reductive cleavage by thiols, thereby giving 1 the potential to non-selectively modify cysteine-containing proteins or to be cleaved by glutathione. Second, the two carboxamides could be expected to limit potency by impeding cell penetration.10,11 Finally, the relatively flexible nature of the tether between the two aromatic rings is likely not optimal for achieving both potency and selectivity.12 Our initial strategy to modify 1 thus included: removal of the N-O bond, bioisosteric replacement of the amides13, and conformational restriction14 of the tether between the aromatic rings. A limited survey of aromatic substitution was also undertaken to clarify the role of the lipophilic substituents.
The synthetic routes to new analogs of 1 are presented in Scheme 1 – Scheme 6. A preparation of bis(amides) 4 that was general for a variety of amino acids 2 (acyclic or cyclic, Table 1 and Table 4) is summarized in Scheme 1. Acylation with bis(trifluoromethyl)benzoyl chloride, either under Schotten-Baumann conditions or under anhydrous conditions, afforded the mono(amides) 3 in good yields. Final amidation with 4-chloroaniline was then effected with N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride (EDC) and 1-hydroxybenzotriazole (HOBt) to afford bis(amides) 4. N-methylanilide 4e, benzylamide 4f, and indoline amide 4p were made under similar conditions using N-methyl-4-chloroaniline, 4-chlorobenzylamine or 5-chloroindoline, respectively, in the final amidation step. Applying the same chemistry, bis(amides) 5a–g of aminoxyacetic acid (Table 2) with various aromatic substitution patterns were also prepared. Monoamine 8 was prepared by alkylation of 4-chloroaniline with 3-bromopropylamine 615, followed by acylation with bis(trifluoromethyl)benzoyl chloride (Scheme 2). The regioisomeric monoamine 11 was synthesized by alkylation of bis(trifluoromethyl)benzylamine with bromide 10, which was prepared by acylation of 4-chloroaniline with 3-bromopropionyl chloride (Scheme 3). Bis(amine) 12 (Table 3) could be obtained by exhaustive reduction of bis(amide) 4a (Table 1) with borane-THF complex at reflux.
Scheme 1.

Reagents and conditions: (a) 3,5-bis(CF3)PhCOCl, aq NaOH, RT, overnight; or 3,5-bis(CF3)PhCOCl, triethylamine (TEA), CH2Cl2 ; (b) 4-ClPhNH2, EDC, HOBt, DIPEA, THF, RT, overnight.
Scheme 6.

Reagents and conditions: (a) 4-ClPhNH2, HOBt, EDC, DIPEA, THF, RT, overnight, 87%; (b) 3,5-bis(CF3)COCl, TEA, CH2Cl2, RT, overnight, 97%; (c) i. TFA, −10 °C, NaNO2; ii. NaN3, −10 °C to RT, 2 h, 63%; (d) 25, CuSO4•5H2O, L-Ascorbic acid, H2O/tBuOH, 1:1, RT, overnight, 19%; (e) i. 6M HCl, 0 °C, NaNO2; ii. NaN3, 0 °C, 15 min., 56%; (f) 27, CuSO4•5H2O, L-Ascorbic acid, H2O/tBuOH, 1:1, RT, 48 h, 10%.
Table 1.
Effects of tether length and composition on transcription and cytotoxicity in transfected PC-3 cellsa
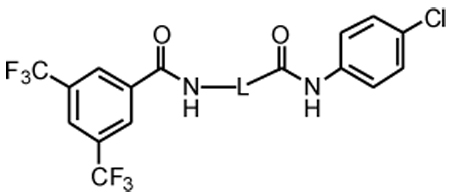 | |||||
|---|---|---|---|---|---|
| Cmpd | L | IC50 SRE.Lb (µ M) |
% inh SRE.Lb (10, 100 µM) |
% inh pRL-TKc (10, 100 µM) |
% inh WST-1d (10, 100 µM) |
| 1 | -OCH(CH3)- | 1.5 | 74, ND | 48, ND | 44, ND |
| 5a | -OCH2- | 4.7 | 71, 100 | 53, 89 | 42, 91 |
| 4a | -CH2CH2- | 38 | 38, 64 | 0, 22 | 0, 10 |
| 4b | -CH2- | 33 | 45, 85 | 15, 25 | 0, 30 |
| 4c | -CH2CH2CH2- | 21 | 37, 79 | 5, 42 | 0, 12 |
| 4d | -CH2CH2CH2CH2- | >100 | |||
For assay descriptions, see Ref 20. All values are mean of ≥ 3 experiments, each run in triplicate.
Inhibition of Rho-pathway selective serum response element-luciferase reporter.
Inhibition of control pRL-thymidine kinase Renilla luciferase reporter.
Inhibition of mitochondrial metabolism of WST-1.
Table 4.
Effects of conformational restriction on transcription and cytotoxicity in transfected PC-3 cellsa
| Cmpd | Structure | IC50 SRE.L (µM) |
% inh SRE.L (10, 100 µM) |
% inh pRL-TK (10, 100 µM) |
% inh WST-1 (10, 100 µM) |
|---|---|---|---|---|---|
| 4a | 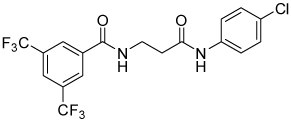 |
38 | 38, 64 | 0, 22 | 0, 10 |
| 4g | 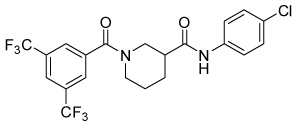 |
9.8 | 37, 78 | 19, 34 | 0, 14 |
| 4h | 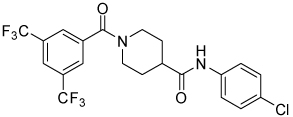 |
16 | 17, 87 | 0, 17 | 0, 39 |
| 4i | 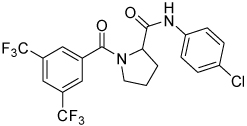 |
69 | 23, 83 | 12, 27 | 0, 22 |
| 4j | 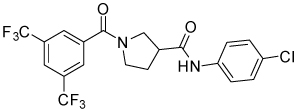 |
16.3 | 30, 100 | 8, 78 | 0, 67 |
| 4k | 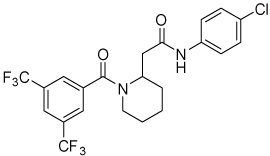 |
9.5 | 33, 86 | 0, 62 | 0, 14 |
| 4l | 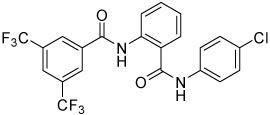 |
>100 | |||
| 4m | 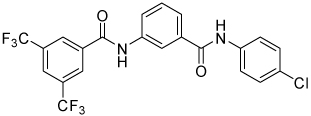 |
1.7 | 79, ND | 0, ND | 38, ND |
| 4n | 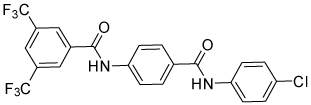 |
>100 | |||
| 4o | 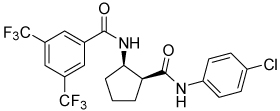 |
>100 | |||
| 4p | 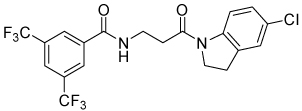 |
13 | 15, 40 | 0, 1 | 0, 0 |
| 4q | 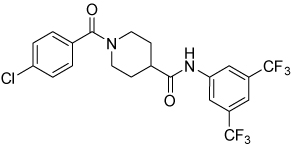 |
10 | 51, 100 | 30, 91 | 10, 88 |
| 19 | 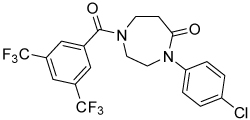 |
>100 | |||
| 20 | 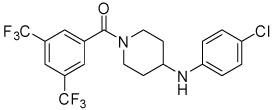 |
10.8 | 19, 54 | 0, 20 | 0, 0 |
Assays defined in Table 1. ND: not determined.
Table 2.
Effects of aromatic substitution on transcription and cytotoxicity in transfected PC-3 cellsa
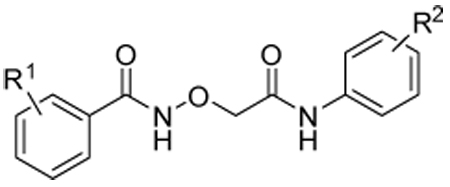 | ||||||
|---|---|---|---|---|---|---|
| Cmpd | R1 | R2 | IC50 SRE.L (µM) |
% inh SRE.L (10, 100 µM) |
% inh pRL-TK (10, 100 µM) |
% inh WST-1 (10, 100 µM) |
| 5a | 3,5-bis(CF3) | 4-Cl | 4.7 | 71, 100 | 53, 89 | 42, 91 |
| 5b | 3,5-bis(CF3) | 3-Cl | 5.9 | 65, 100 | 51, 89 | 49, 97 |
| 5c | 3,5-bis(CF3) | 4-H | 36 | 13, 65 | 33, 59 | 0, 37 |
| 5d | 3-CF3 | 4-Cl | 27 | 25, 86 | 5, 19 | 0, 58 |
| 5e | 4-CF3 | 4-Cl | 29 | 26, 91 | 6, 0 | 0, 56 |
| 5f | 4-H | 4-Cl | >100 | |||
| 5g | 4-Cl | 3,5-bis(CF3) | 8.6 | 58, 100 | 19, 87 | 11, 96 |
Assays defined in Table 1.
Scheme 2.

Reagents and conditions: (a) 4-ClPhNH2, PhMe, reflux, 45 min, 28%; (b) 3,5-bis(CF3)PhCOOH, HOBt, EDC, DIPEA, THF, RT, overnight, 65%.
Scheme 3.

Reagents and conditions: (a) 4-ClPhNH2, MeCN, reflux, 3 h, 94%; (b) 3,5-bis(CF3)PhCH2NH2, MeCN, reflux, 7 h, 99%.
Table 3.
Effects of amide modifications on transcription and cytotoxicity in transfected PC-3 cellsa
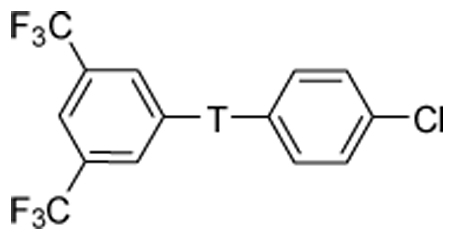 | |||||
|---|---|---|---|---|---|
| Cmpd | T | IC50SRE.L (µM) |
% inh SRE.L (10, 100 µM) |
% inh pRL-TK (10, 100 µM) |
% inh WST-1 (10, 100 µM) |
| 4a | -CONHCH2CH2CONH- | 38 | 38, 64 | 0, 22 | 0, 10 |
| 4e | -CONHCH2CH2CON(Me)- | >100 | |||
| 4f | -CONHCH2CH2CONHCH2 | >100 | |||
| 8 | -CONHCH2CH2CH2NH- | 5.1 | 70, 80 | 37, 35 | 0, 11 |
| 11 | -CH2NHCH2CH2CONH- | 8.1 | 64, 100 | 12, 91 | 0, 92 |
| 12 | -CH2NHCH2CH2CH2NH- | 9.1 | 65, 100 | 6, 90 | 0, 89 |
Assays defined in Table 1.
Thiazole 13 and ether 14 (Table 5) could be made by simple acylation of the respective commercially available amines with bis(trifluoromethyl)benzoyl chloride. Conformationally restricted lactam 19 required a multi-step synthesis (Scheme 4). Deprotection of commercially available N-(Boc)-4-piperidinone 15, followed by acylation, afforded benzamide 16. Oxime formation followed by Beckman rearrangement generated the ring-expanded intermediate diazepanone 18. Final N-arylation of the lactam with 4-iodo-chlorobenzene under Buchwald conditions16 then completed the synthesis. Amide 16 could also be reductively aminated with 4-chloroaniline and sodium cyanoborohydride to provide conformationally restricted amine 20 (Table 4).
Table 5.
Effects of amide replacement on transcription and cytotoxicity in transfected PC-3 cellsa
| Cmpd | Structure | IC50 SRE.L (µM)a |
% inh SRE.L (10, 100 µM)b |
% inh pRL-TK (10, 100 µM)b |
% inh WST-1 (10, 100 µM)b |
|---|---|---|---|---|---|
| 4a | 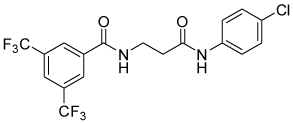 |
38 | 38, 64 | 0, 22 | 0, 10 |
| 13 | 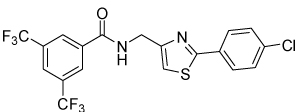 |
4.1 | 50, 45 | 39, 38 | 0, 0 |
| 14 | 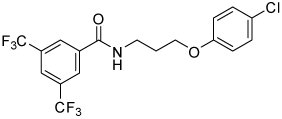 |
8.9 | 55, 86 | 28, 65 | 0, 38 |
| 23 | 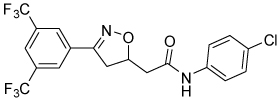 |
4.2 | 70, 84 | 51, 60 | 0, 38 |
| 29 | 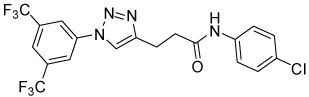 |
>100 | |||
| 31 | 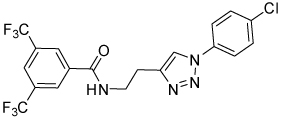 |
>100 |
Assays defined in Table 1.
Scheme 4.

Reagents and conditions: (a) TFA, CH2Cl2, 0°C to RT, 3h, quant.; (b) 3,5-bis(CF3)PhCOCl, aq NaOH, RT, overnight, 74%; (c) NH2OH•HCl, pyridine, 3Å sieves, RT, overnight, 72%; (d) Na2CO3, p-TsCl, acetone, RT, 3 h, 66%; (e) 4-I-PhCl, MeNHCH2CH2NHMe, CuI, Cs2CO3, dioxane, 100 °C, 24 h, 48%.
Isoxazoline 23 was prepared by a cycloaddition of the nitrile-oxide derived from oxime 2217 with N-(4-chlorophenyl)but-3-enamide18 (Scheme 5). Triazoles 29 and 31 were prepared from anilines 28 and 30, respectively, by azidation and copper-catalyzed Click cyclization with alkyne amides 25 and 27 (Scheme 6).19
Scheme 5.

Reagents and conditions: (a) HONH2•HCl, MeOH, reflux, 5 h, 48%; (b) N-(4-chlorophenyl)but-3-enamide, NaOCl, CH2Cl2, 0°C to RT, 24 h, 99%.
Effects on Rho-mediated gene expression, non-specific gene expression, and cytotoxicity of all new compounds were determined in transiently transfected PC-3 cells (Table 1–Table 5).20 The data in the tables is intended to illustrate both potency and efficacy. This was done due to the observation that some compounds had equivalent potencies (IC50s) against SRE.L, yet differed in their maximal responses (efficacies). There are a number of possible explanations for this, including differences in passive permeability into the cells, efflux out of the cells, or even the complement of Rho-pathway targets impacted. To optimize the likelihood for eventual in vivo activity, we elected to consider both potency and efficacy vs SRE.L in our structure-activity relationship (SAR) analysis. Effects against pRL-TK and WST-1 at high and low concentrations are included as an approximate indicator of selectivity. IC50s often could not be calculated from the generally weaker dose response data against these selectivity targets, and therefore are not included.
Table 1 summarizes the impact of changes on the tether between the two carboxamides of compound 1. Removing the methyl group (5a) had little effect on activity or selectivity. Replacement of the oxygen with carbon (4a) indeed removed acute cytotoxicity, even at high dose, and improved selectivity vs Renilla, but attenuated potency against SRE.L by over an order of magnitude. Decreasing or increasing the length of the carbon chain by one carbon (4b and 4c) did not greatly impact the potency or efficacy relative to 4a, but lengthening to four carbons (4d) resulted in a total loss of activity.
A brief survey of aromatic substitution is summarized in Table 2. Despite the diminished toxicity of 4a, we elected to use the more potent unsubstituted aminoxyacetic acid template of 5a to magnify any changes in activity. Moving the chloro group to the 3-position (5b) had no effect, but removing it altogether (5c) negatively affected both potency and efficacy. Removing one of the trifluoromethyl groups (5d and 5e) was similarly detrimental to removing the chloro group, and removing both trifluoromethyl groups (5f) led to a complete loss of activity. Reversing the position of the two aromatic rings (5g) gave a compound with activity similar to 5a. These limited data suggest that some degree of lipophilicity on the aromatic rings is crucial for activity, possibly to facilitate cell permeability. A more in-depth examination of aromatic substituent SAR, including whether electron deficiency is necessary, will be the subject of future work.
Table 3 summarizes our initial foray into modification of the secondary amides. N-methylation of the 4-chloroaniline amide (4e), intended to remove a hydrogen bond donor and improve cell permeability, resulted in a dramatic loss in activity. Replacement of one of the anilides with a benzylamide (4f) also was detrimental. Reducing the amides to the corresponding amines was considerably more successful. Both monoamines 8 and 11, as well as diamine 12, exhibited improved potency and efficacy against Rho-dependent gene expression. Of particular interest was monoamine 8, which had potency approaching that of the aminoxyacetic acid template 5a, retained the moderate selectivity of 4a vs non-specific gene expression, and showed very little cytotoxicity compared to the other monoamine and the diamine, even at high concentrations. The other two amines exhibited unacceptable levels of cytotoxicity.
Conformational restriction of the flexible bis(amide) tether was explored as a way to potentially improve both potency and selectivity (Table 4). In several cases, improved potency vs the corresponding acyclic analog was observed (e.g. 4g vs 4a, 4p vs 4e), but the goal of improving selectivity vs non-specific gene expression was not clearly achieved with any of the restricted analogs. Among a series of closely related five- and six-membered ring analogs (4g – 4j), it is interesting to note that a “meta”-like disposition of the two aromatic rings (4g, 4j) was favored over “para” or “ortho” in closely related analogs (4h, 4i). This pattern is dramatically reflected in the series of analogs constrained by a central aromatic ring (4l – 4n), wherein only the meta analog 4m is active. The inactivity of the vicinal substituted analog 4o and an analog with a “vicinal-like” disposition of the amides (19) is also consistent with this pattern. Compound 20, a restricted analog of monoamine 8, was inferior to the acyclic analog with regard to both potency and efficacy, perhaps another example of the disadvantageous arrangement of the aromatic rings in a “para”-like orientation. Finally, reversal of the aromatic rings (4q vs 4h) led to a significant loss in selectivity at high concentration vs Renilla and cytotoxicity.
Table 5 presents data for analogs incorporating potential bioisosteric replacements for one of the amides. Three of these compounds (13, 14 and 23) exhibited improved potency vs the bis(amide) 4a, but selectivity vs Renilla and/or cytotoxicity suffered. Triazoles 29 and 31 were completely inactive, perhaps suggesting a lack of permeability into the cells.
In our original publication, we showed that 1 selectively inhibited spontaneous PC-3 prostate cancer cell invasion through a Matrigel matrix, but not the Gαi-dependent LPA-stimulated SKOV-3 ovarian cancer cell invasion, in vitro.9 Therefore, we tested several of our analogs for their ability to inhibit PC-3 prostate cancer cell invasion with better selectivity for invasion versus toxicity (as measured by metabolism of WST-1) in comparison to 1. Data for selected compounds is presented in Table 621. Overall, a fairly consistent correlation was observed between inhibition of invasion and inhibition of SRE.L in PC-3 cells. For example, conformationally restricted analogs 4g – 4i, which all possessed roughly equivalent efficacies against SRE.L at 10 and 100 µM, were similarly active in the Matrigel assay. Compound 4g, however, was somewhat less toxic at the higher concentration, exhibiting an efficacy:toxicity profile at 100 µM that is superior to that of the original lead 1 at 10 µM. Also, a tight correlation between the selectivity for SRE.L:cytotoxicity and invasion:cytotoxicity can be seen in the series of amines 8, 11 and 12. Monoamine 8 had the most favorable ratio of efficacy to cytotoxicity in the transfected cell SRE-Luciferase studies, and this is reflected in the data in Table 6. This compound in fact inhibits invasion at 10 µM to an extent approaching that of the lead 1, with no observable toxicity. At 100 µM, nearly complete inhibition of invasion was achieved with a lesser degree of toxicity than that induced by 1 at 10 µM. Monoamine 11 and diamine 12, on the other hand, were much less selective for efficacy vs toxicity in the transfected cells (Table 3), and that is reflected in the data in Table 6 at the higher concentration.
Table 6.
Effects of new analogs on cell invasion and cytotoxicitya
| Cmpd | % inh invasionb (10 µM) |
% inh WST-1c (10 µM) |
% inh invasionb (100 µM) |
% inh WST-1c (100 µM) |
|---|---|---|---|---|
| 1 | 71 | 54 | ||
| 4a | 13 | 0 | 62 | 28 |
| 4b | 14 | 0 | 75 | 38 |
| 4c | 6 | 0 | 31 | 1 |
| 4d | 15 | 0 | 25 | 1 |
| 4g | 20 | 0 | 72 | 23 |
| 4h | 14 | 0 | 79 | 36 |
| 4i | 12 | 0 | 78 | 44 |
| 4m | 63 | 31 | 86 | 70 |
| 4n | 2 | 0 | 0 | 1 |
| 4p | 0 | 0 | 0 | 0 |
| 5a | 50 | 14 | 88 | 78 |
| 8 | 54 | 0 | 84 | 27 |
| 11 | 47 | 0 | 96 | 95 |
| 12 | 30 | 0 | 86 | 97 |
| 13 | 18 | 0 | 8 | 0 |
| 14 | 3 | 0 | 26 | 0 |
| 20 | 18 | 0 | 56 | 0 |
For assay descriptions, see Ref 21.
Inhibition of invasion by cultured PC-3 cells into a Matrigel matrix.
Inhibition of mitochondrial metabolism of WST-1.
Correlation graphs between the average inhibition of the PC-3 Matrigel invasion assay versus the average inhibition of the Gα12QL-stimulated SRE.L-luciferase expression in PC-3 cells at 10 µM and 100 µM are presented in Figure 1. A positive correlation can be seen at both concentrations, although there are a few outliers that strongly inhibit SRE.L without inhibiting invasion. Interestingly, there seems to be a threshold for inhibition of SRE.L (about 50%) that must be achieved before effects on invasion are observed.
Figure 1.

Correlation of Matrigel invasion and Gα12QL-stimulated SRE.L-luciferase expression inhibition in PC-3 cells. The data represent the average of experiments performed 3 separate times for an n = 3 in duplicate and triplicate, respectively.
In summary, an initial SAR survey of 1, an inhibitor of Rho-mediated gene expression, was undertaken with the goal of improving selectivity and/or potency, while attenuating cytotoxicity. In addition to removing the obviously labile N-O bond, two strategies were applied: 1) replacement of the secondary carboxamides to improve cell permeability; and 2) conformational restriction to improve potency and/or selectivity. These approaches afforded compounds with improved biological profiles relative to the lead 1, albeit at a cost of 5–10-fold lower potencies. Of particular interest are nipecotic amide 4g and monoamine 8, each of which are capable of inhibiting Gα12QL-stimulated SRE.L-luciferase expression with efficacy equal to that of 1, but with significantly less attendant cytotoxicity. Furthermore, we have for the first time demonstrated within this series of compounds a clear correlation between inhibition of Rho-mediated gene expression and cell invasion in a Matrigel matrix model of metastasis.
Future work will be aimed at expanding the diversity of the aromatic rings using one or both of the improved templates of 4g and 8, as well as constructing affinity reagents for the identification of the molecular target(s) of this novel class of inhibitors of the Rho-signaling pathway.
Acknowledgements
Supported by NIH grants R01GM39561 (RRN) and F31CA113268-04 (CRE) and support from the UM Comprehensive Cancer Center and the Prostate Cancer SPORE (P50CA069568).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Sawyer TK. Expert Opinion on Investigational Drugs. 2004;13:1–19. doi: 10.1517/13543784.13.1.1. [DOI] [PubMed] [Google Scholar]
- 2.Sahai E, Marshall C. J. Nat Cell Biol. 2003;5:711–719. doi: 10.1038/ncb1019. [DOI] [PubMed] [Google Scholar]
- 3.van Golen KL, Wu ZF, Qiao XT, Bao LW, Merajver SD. Cancer Res. 2000;60:5832–5838. [PubMed] [Google Scholar]
- 4.Liu N, Zhang G, Bi F, Pan Y, Xue Y, Shi Y, Yao L, Zhao L, Zheng Y, Fan D. J Mol Med. 2007;85:1149–1156. doi: 10.1007/s00109-007-0217-y. [DOI] [PubMed] [Google Scholar]
- 5.Cen B, Selvaraj A, Burgess RC, Hitzler JK, Ma Z, Morris SW, Prywes R. Mol Cell Biol. 2003;23:6597–6608. doi: 10.1128/MCB.23.18.6597-6608.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miralles F, Posern G, Zaromytidou AI, Treisman R. Cell. 2003;113:329–342. doi: 10.1016/s0092-8674(03)00278-2. [DOI] [PubMed] [Google Scholar]
- 7.Hakem A, Sanchez-Sweatman O, You-Ten A, Duncan G, Wakeham A, Khokha R, Mak TW. Genes Dev. 2005;19:1974–1979. doi: 10.1101/gad.1310805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Medjkane S, Perez-Sanchez C, Gaggioli C, Sahai E, Treisman R. Nat Cell Biol. 2009;11:257–268. doi: 10.1038/ncb1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Evelyn CR, Wade SM, Wang Q, Wu M, Iniguez-Lluhi JA, Merajver SD, Neubig RR. Molecular Cancer Therapeutics. 2007;6:2249–2260. doi: 10.1158/1535-7163.MCT-06-0782. [DOI] [PubMed] [Google Scholar]
- 10.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Advanced Drug Delivery Reviews. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 11.Burton PS, Conradi RA, Ho NF, Hilgers AR, Borchardt RT. J Pharm Sci. 1996;85:1336–1340. doi: 10.1021/js960067d. [DOI] [PubMed] [Google Scholar]
- 12.Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. J Med Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 13.Lima LM, Barreiro E. J. Curr Med Chem. 2005;12:23–49. doi: 10.2174/0929867053363540. [DOI] [PubMed] [Google Scholar]
- 14.Mann A, LeChatlier HL. Practice of Medicinal Chemistry. London: Elsevier; 2003. pp. 233–250. [Google Scholar]
- 15.Orelli LR, Garcia MB, Niemevz F, Perillo IA. Synthetic Communications. 1999;29:1819–1833. [Google Scholar]
- 16.Klapars A, Huang XH, Buchwald SL. Journal of the American Chemical Society. 2002;124:7421–7428. doi: 10.1021/ja0260465. [DOI] [PubMed] [Google Scholar]
- 17.Liu A, Liu A. Journal of Agricultural and Food Chemistry. 2008;56:6562. doi: 10.1021/jf800651z. [DOI] [PubMed] [Google Scholar]
- 18.Tellitu I, Urrejola A, Serna S, Moreno I, Herrero MT, Dominguez E, SanMartin R, Correa A. European Journal of Organic Chemistry. 2007:437–444. doi: 10.1021/jo062320s. [DOI] [PubMed] [Google Scholar]
- 19.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angewandte Chemie-International Edition. 2002;41:2596. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 20.PC-3 cells were seeded into 96-well plates at a cell density of 4 × 104 cells per well 24 hours prior to transient transfection with a Gα12Q231L activator expression plasmid along with both the SRE.L luciferase and pRL-TK Renilla luciferase reporter plasmids. The DNA plasmids were transfected using the lipid-based LipofectAMINE 2000 (Invitrogen) transfection reagent at a concentration of 1 µL per µg of DNA in antibiotic-free, Opti-MEM I medium. The total amount of DNA was kept constant by inclusion of the appropriate amount of the pcDNA3.1-zeo plasmid. Six hours after transfection, the transfection mixture was removed and cells were serum-starved overnight in DMEM medium containing 0.5% FBS and 1% penicillin-streptomycin. Firefly and Renilla luciferase activities were determined 19 hours later using the dual-luciferase assay kit (Promega) according to the manufacturer's instructions. Just before cell lysis, the viability of the cells was measured using a WST-1 cell proliferation reagent. Data are expressed as IC50 or percentage of inhibition (DMSO alone = 0%) in Table 1 – Table 5. Individual experiments were run in triplicate, and the values in the tables are the mean of at least three separate experiments. Because of the large degree of variability inherent in transient transfection assays, standard error measurements have not been included.
- 21.Compounds were tested as follows: PC-3 cells (2 × 105) were transferred to 24-well Matrigel inserts in low-serum DMEM medium (0.5% FBS) with DMSO or chemical compounds in the upper chamber. Low-serum DMEM medium (0.5% FBS) was added to the lower well and the invasion chambers were incubated at 37 °C in 5% CO2 for 24 hours. Inserts were fixed in methanol for 10 min and then stained for 60 min with 0.5% crystal violet in 20% methanol. After wiping the top surface of the filter with cotton swabs to remove non-invaded cells, the inserts were allowed to dry overnight. Inserts were incubated in 20% acetic acid on a plate shaker for 15 min to extract the crystal violet stain. The number of invaded cells was quantitated by measuring the absorbance of the extracted crystal violet stain at a wavelength of 595 nm with the Victor2 plate reader.