

Abstract
Forecasting disease susceptibility requires detection of maladaptive signatures prior to onset of overt symptoms. A case-in-point are cardiac ATP-sensitive K+ (KATP) channelopathies, for which the substrate underlying disease vulnerability remains to be identified. Resolving molecular pathobiology, even for single genetic defects, mandates a systems platform to reliably diagnose disease predisposition. High-throughput proteomic analysis was here integrated with network biology to decode consequences of Kir6.2 KATP channel pore deletion. Differential two-dimensional gel electrophoresis reproducibly resolved > 800 protein species from hearts of asymptomatic wild-type and Kir6.2-knockout counterparts. KATP channel ablation remodeled the cardiac proteome, significantly altering 71 protein spots, from which 102 unique identities were assigned following hybrid linear ion trap quadrupole-Orbitrap tandem mass spectrometry. Ontological annotation stratified the KATP channel-dependent protein cohort into a predominant bioenergetic module (63 resolved identities), with additional focused sets representing signaling molecules (6), oxidoreductases (8), chaperones (6), and proteins involved in catabolism (6), cytostructure (8), and transcription and translation (5). Protein interaction mapping, in conjunction with expression level changes, localized a KATP channel-associated subproteome within a nonstochastic scale-free network. Global assessment of the KATP channel deficient environment verified the primary impact on metabolic pathways and revealed overrepresentation of markers associated with cardiovascular disease. Experimental imposition of graded stress precipitated exaggerated structural and functional myocardial defects in the Kir6.2-knockout, decreasing survivorship and validating the forecast of disease susceptibility. Proteomic cartography thus provides an integral view of molecular remodeling in the heart induced by KATP channel deletion, establishing a systems approach that predicts outcome at a presymptomatic stage.
Keywords: channelopathy, individualized medicine, KATP channel, KCNJ11, Kir6.2, network, predictive medicine, proteomics, systems biology
Introduction
The tenets of predictive medicine offer a paradigm shift from reactive traditional approaches to manage overt heart disease toward proactive interventions tailored to prevent or mitigate disease manifestation. Predictive medicine is enabled by mechanistic insights extracted from divergence of disease-prone cellular networks from their normal counterparts.1,2 Decoding inherent maladaptive signatures prior to symptom onset would guide rational forecasting of individual disease susceptibility.3,4 To date, however, subclinical molecular profiles predictive of heart disease remain only partially characterized, limiting realization of anticipatory solutions.
To this end, resolving cardioprotective processes and their underlying systems organization is a critical step in comprehending cardiac homeostasis in health and disease. A case-in-point is the metabolism-gated ATP-sensitive K+ (KATP) channel, a prototypic biosensor that enables high fidelity readout of metabolic distress signals.5–8 Unique among ion channels, this checkpoint performs a rheostat-like operation adjusting membrane potential-dependent functions to match energetic demands of the working heart.9–12 Abundant in the myocardial sarcolemma, KATP channel complexes form through assembly of the KCNJ11-encoded Kir6.2 pore with the regulatory ATP-binding cassette sulfonylurea receptor subunits.13–16 KATP channel dysfunction compromises vital homeostatic functions leading to life-threatening conditions.17–20 While KATP channelopathies are increasingly implicated in human cardiac pathologies,21–23 the molecular consequences of channel deficiency that predispose to disease vulnerability have yet to be dissected.
Systems platforms offer a means of reliably integrating pathobiologic complexity to predict disease predisposition.24–27 In this regard, proteomics methods provide a high-throughput, unbiased approach for large-scale identification of proteins responsible for (patho)physiological processes. Herein, we demonstrate extensive protein alterations in response to KATP channel ablation, which collectively comprise an integrated, scale-free network, with an overarching function of energy metabolism and overrepresentation of markers associated with cardiovascular disease. Subsequent experimental validation of KATP channel knockout cardiomyopathic predisposition underscores the utility of a systems-based approach to anticipate outcome from a presymptomatic profile.
Materials and Methods
KATP Channel Knockout and Cardiac Tissue Fractionation
KATP channel-deficient mice (Kir6.2-KO) were generated by targeted disruption of KCNJ11, encoding the pore-forming Kir6.2 channel subunit of myocardial KATP channels, and backcrossed for 5 generations to a C57BL/6 background.28 Animal protocols followed NIH guidelines, and received Mayo Clinic Institutional Animal Care and Use Committee approval. Cardiac function of adult age-matched wild-type (WT) and Kir6.2-KO male mice was compared by M-mode echocardiography, a standard measure of heart contractility.19,21 Animals were weighed, anesthetized with isoflurane, and hearts were removed and weighed with the left ventricle including septum excised and snap-frozen in liquid N2. Cytosolic tissue extracts were prepared by homogenization at 4 °C in extraction buffer, consisting of (in mM) HEPES 25 (pH 7.4), PMSF 0.25, DTT 50, 1.25 μM pepstatin A, Mini-Complete protease inhibitor cocktail (Roche Applied Science), and 1% phosphatase inhibitor cocktails 1 and 2 (Sigma). Homogenized samples were centrifuged (16 000g) at 4 °C for 10 min, and supernatant protein content quantified in triplicate by microassay procedure.29–31
Two-Dimensional Gel Electrophoresis and Image Analysis
Protein extracts (100 μg) were resolved by immobilized pH gradient (IPG) 2-DE following addition to isoelectric focusing (IEF) rehydration buffer (7 M urea, 2 M thiourea, 2% w/v CHAPS, 50 mM DTT, 1× Bio-Rad pH 3–10 ampholytes). IPG Ready Strips (pH 3–10 170 mm, Bio-Rad) were actively rehydrated, followed by IEF using a series of voltage ramping steps to a total of 60 kVh.32 Focused IPG strips were reduced in equilibration buffer (50 mM Tris-HCl, pH 8.8, 6 M urea, 30% v/v glycerol, 2% w/v SDS) supplemented with 10 mg/mL DTT, followed by alkylation in equilibration buffer containing 25 mg/mL iodoacetamide. Proteins were resolved orthogonally by 12.5% SDS-PAGE in a Bio-Rad Protean II XL system.33 Resolved gels were silver-stained and digitized for image analysis conducted with Bio-Rad PDQuest v.7.4.0.32–34 Gel images were normalized by densitometric quantitation of valid spots for fold-change determination. For proteins identified in more than one spot, the sum of values for all spots was used to determine a weighted average.32
Nanoelectrospray Linear Ion Trap Tandem Mass Spectrometry
Altered protein species were isolated, destained, and prepared for LC-MS/MS.32–34 Peptides were reconstituted in 0.15% formic acid, 0.05% TFA, and trap injected onto a 75 μm × 5 cm ProteoPep C18 PicoFrit nanoflow column (New Objective). Chromatography was performed using an Eksigent nanoHPLC system (MDS Sciex) coupled to an LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific). Continuous scanning for eluting peptide ions was carried out between 375–1600 m/z, automatically switching to MS/MS CID mode on ions exceeding an intensity of 8000. Raw MS/MS spectral data were converted to .dta files using Bioworks 3.2 (Thermo Fisher Scientific), and merged files correlated to theoretical tryptic fragments in Swiss-Prot (v.53.0 comprised of 269 293 sequences) using Mascot v.2.2.32 Searches were conducted on mammalian sequences (53 539 entries), with up to 2 missed cleavages, mass tolerances of ±0.01 Da for precursor ions (with 13C peak correction) and ±0.6 Da for product ions, and allowance for protein N-terminal acetylation, methionine oxidation or cysteine carbamidomethylation. Protein identities were confirmed by matching multiple peptide spectra at P < 0.05, with proteins accepted at P < 0.01.26 Proteins identified by a single peptide were thus subject to a higher stringency level (P < 0.01), and were also confirmed by manual spectrum inspection, with detected fragment ions from the MS/MS spectrum required to be above baseline noise, have demonstrable continuity in b- or y-ion series, and proline residues yielding intense y-ions.35 Positive matches to nonmouse species lacking Swiss-Prot mouse homologues were screened against the TrEMBL database to confirm a match to a mouse protein (indexed 2007–01–23, comprising 3 633 676 sequences). Protein assignments were further validated by congruence of observed versus predicted pI/Mr, using the ExPASy pI/Mr tool (us.expasy.org/tools/pi_tool.html), taking into consideration protein processing, known post-translational modifications, and predicted mitochondrial localization and signal peptide cleavage sites as determined using the established algorithm TargetP 1.1.36
Protein Interaction Network Analysis
Identified proteins, with their fold change ratios, were submitted as focus proteins for network analysis using Ingenuity Pathways Analysis (IPA, Ingenuity Systems) to predict a composite outcome through connection of functional subnetworks.32,33 The composite was mapped using the network visualization algorithm Cytoscape 2.5.1,37 with network topology assessed using Network Analyzer.38 Node degree (k), the number of node connections, and node degree distribution (P[k]), the probability that a node has k links, where P[k] = X[k]/n, when X[k] is the number of nodes with degree k and n equals total nodes were used to define network architecture.32 P[k] versus k discriminates between random and scale-free topographies,39 defined by normal and power law distributions, respectively. The Anderson-Darling normality test40 was used to rule out a normal distribution, so P[k] versus k was calculated as a power law relationship using a cumulative distribution function41 to determine γ in the power law distribution (P[k] ∼ k−γ), using eq 1:
| (1) |
where γ is the power law exponent, n is the number of network nodes, xi is the node degree, and xmin is the minimum node degree within the network, with statistical error σ41 for eq 1 defined by eq 2:
| (2) |
Interrogation of the composite network was carried out with the Cytoscape module BiNGO 2.0 (Bioinformatic Network Gene Ontology), to assess Gene Ontology (GO) biological process enrichment,42 providing an unbiased bioinformatic approach to interpret functional interdependencies within the composite network. IPA was also used to identify prioritized physiological or pathophysiological categories with connections to proteins comprising the resolved interaction network. Individual categories are considered for overrepresentation by IPA on the basis of statistical analysis of their proportional occurrence within the network relative to their overall occurrence in the entire proteome.
Imposition of Cardiac Stress
Mild, moderate or severe stress load was imposed, respectively, by aquatic endurance training,43 volume overload induced hypertension,19 and transverse aortic banding induced pressure overload21 in adult age and sex-matched WT and Kir6.2-KO mice. In endurance training, collective swimming was carried out for 90 min, twice daily, over 28 days.43 In volume overload, following unilateral nephrectomy, mineralocorticoid-hypertension was induced by 21-day release of a subcutaneous 50 mg deoxycorticosterone acetate pellet (Innovative Research of America) with 1% NaCl, 0.2% KCl drinking water supplementation.19 In pressure overload, thoracic aortae were uniformly constricted to the diameter of a 27-gauge needle.21 Left ventricular fractional shortening (FS) was calculated by M-mode echocardiography as a standard parameter of heart contractility, and left ventricle mass was measured to evaluate cardiomegaly, as previously described.19,21,43
Statistical Analysis
Unless otherwise indicated, comparison between groups was performed using a standard t-test of variables with 95% confidence intervals, and data expressed as mean ± SE. For BiNGO, a hypergeometric test with Benjamini and Hochberg false discovery rate correction was used to assess the protein network for significant overrepresentation of GO biological processes. Kaplan–Meier analysis with logrank testing was applied for survival analysis, with P < 0.05 predetermined.44
Results
Kir6.2 Ablation Transforms the Cardiac Proteome
Targeted disruption of KCNJ11 generates Kir6.2-deficient mice that lack functional KATP channels in ventricular myocytes.45 Stress-free KATP channel knockout (Kir6.2-KO) animals did not display apparent differences in cardiac function or gross heart morphology when compared to age- and sex-matched wild-type (WT) counterparts (Figure 1A). To assess the potential spectrum of molecular consequences precipitated by KATP channel deficiency, the left ventricle of WT (n = 4) and Kir6.2-KO (n = 4) were subjected to comparative proteomic analysis. Separation of cytoplasmic tissue extracts by broad pH range 2-DE resolved >800 unique protein species (Figure 1B), with high gel-to-gel reproducibility (Figure 1C). Densitometric quantification of relative spot intensities as exemplified for spot 58 (Figure 1D) revealed that 71 of 804 (8.8%) protein species were significantly altered by Kir6.2 deletion (P < 0.05), a third of which increased while two-thirds decreased (Figure 1E). Thus, ablation of KCNJ11 transforms the ventricular protein expression pattern unmasking a KATP channel-dependent cardiac subproteome.
Figure 1.
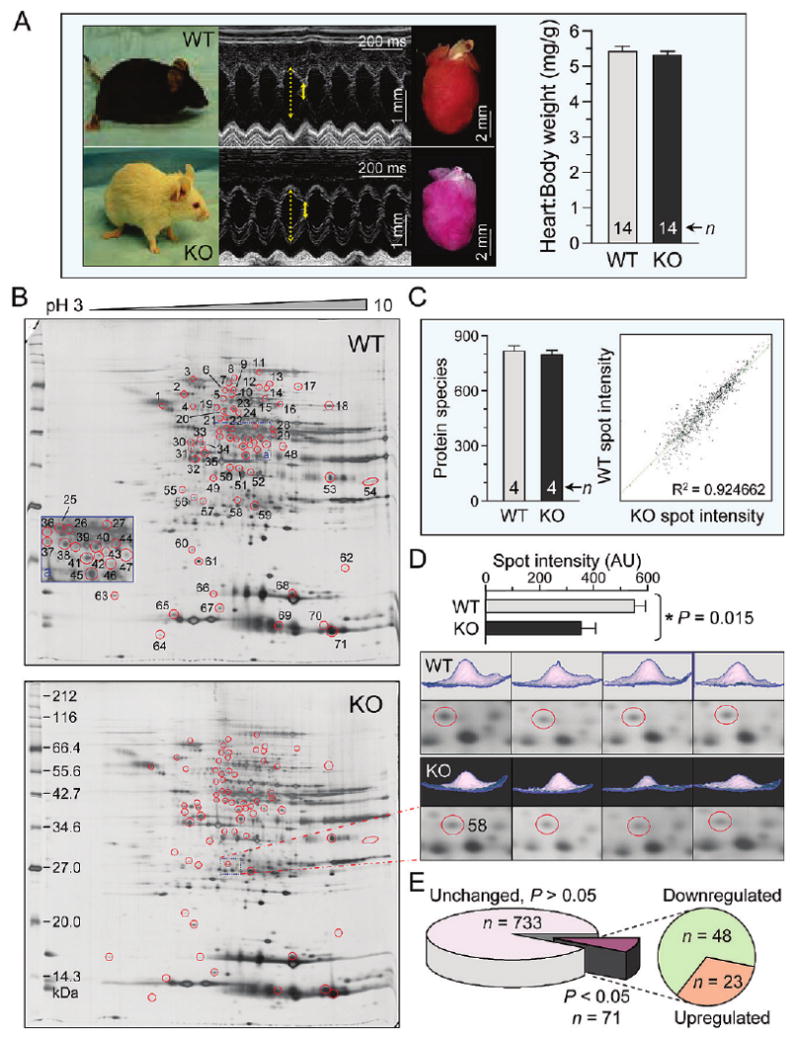
Kir6.2 deletion remodels the ventricular proteome. (A) Adult age and sex-matched wild-type (WT) and Kir6.2-knockout (KO) mice assessed under stress-free conditions by ventricular M-mode echocardiography exhibited no apparent differences in function (solid and dotted yellow lines indicate extent of systolic and diastolic wall motion, respectively), nor were there differences in gross heart morphology or heart to body weight ratio. (B) Silver-stained 2-D gels of left ventricular cytoplasmic protein extracts (100 μg per gel) from WT and KO mice resolved by pH 3–10 IEF, 12.5% SDS-PAGE. Differentially expressed spots are circled and numbered to cross-reference with corresponding protein assignments determined by LTQ-Orbitrap MS/MS analysis of excised tryptic digests. (C) Gel reproducibility was demonstrated by consistent numbers of detected protein species and strong correlation (R2 = 0.925) across treatments between average normalized intensities of matching protein spots from WT and KO gels. (D) Example of spot quantification (top), indicating 2-D gel position and 3-D rendering of the quantified spot from all resolved gels (bottom, n = 4 for WT and KO). (E) Densitometric analysis comparing WT to KO revealed that 71 of 804 spots differed (P < 0.05), including 23 up-regulated and 48 down-regulated protein species.
Cartography of Kir6.2-Knockout Proteome
To determine the identity of protein alterations induced by Kir6.2 deletion, a discriminatory analysis of cytoplasmic subproteomes was carried out by linear ion trap quadrupole (LTQ) Orbitrap MS/MS of in-gel tryptic digests. Using a stringent cutoff (P < 0.01) for protein identity, assignments were obtained for 62 of the 71 altered protein species, matching derived spectra to multiple unique peptides per protein or, for proteins identified by a single peptide match, by subsequent manual inspection of the MS/MS spectrum (Supplementary Table 1, Supporting Information). Collectively, a total of 102 intracellular protein assignments were obtained (Figure 2 and 3). Ontological annotation stratified the KATP channel-dependent protein cohort into a predominant bioenergetic module (63/102 proteins), including mitochondrial proteins, primarily dehydrogenases and transferases involved in the tricarboxylic acid cycle or fatty acid β-oxidation pathway, cytoplasmic proteins primarily functioning within the glycolytic pathway, as well as a variety of enzymes contributing to amino acid or nucleic acid metabolism (Figure 2). Multiple changes (39/102 proteins) were also detected in nonmetabolic functional classes, comprising signaling molecules, oxidoreductases and chaperones, as well as proteins involved in catabolism, cytostructure, and transcription/translation (Figure 3). Thus, resolution of the identity of specific proteins contributing to the cardiac KATP channel subproteome revealed a broad spectrum of compromised cellular processes.
Figure 2.

Metabolic remodeling is the primary effect of KATP channel deficiency. Proteins assigned by LTQ-Orbitrap MS/MS analysis from significantly altered spots were functionally categorized by Swiss-Prot ontological annotations. The primary association was with metabolic processes, as 63/102 protein assignments encompassed mitochondrial, cytoplasmic, and amino or nucleic acid metabolic functions. Protein names are listed with their Swiss-Prot gene name (for Node Symbol) to locate them in the protein interaction network, and with their corresponding spot numbers from their 2-D gel positions. Protein accession number, Mascot score, number of unique identified peptides, % sequence cov. (coverage), predicted Mr and pI for each protein (following expected post-translational processing, e.g., removal of a known or predicted mitochondrial signal peptide), and fold change (KO versus WT) are indicated. For proteins detected in more than one spot, maximum score and number of unique peptides are reported. Fold change was calculated as described in experimental procedures, and for proteins detected in both increasing and decreasing spots (**), both values are indicated. Complete MS/MS data for all proteins is outlined in Supplementary Table 1 (Supporting Information). (*TrEMBL entry; ** Contains both up- and down-regulated spots; † Node not detected for network analysis by Ingenuity Pathways; ‡ Mr/pI calculated after TargetP 1.1 prediction of mitochondrial localization and signal peptide cleavage site).
Figure 3.
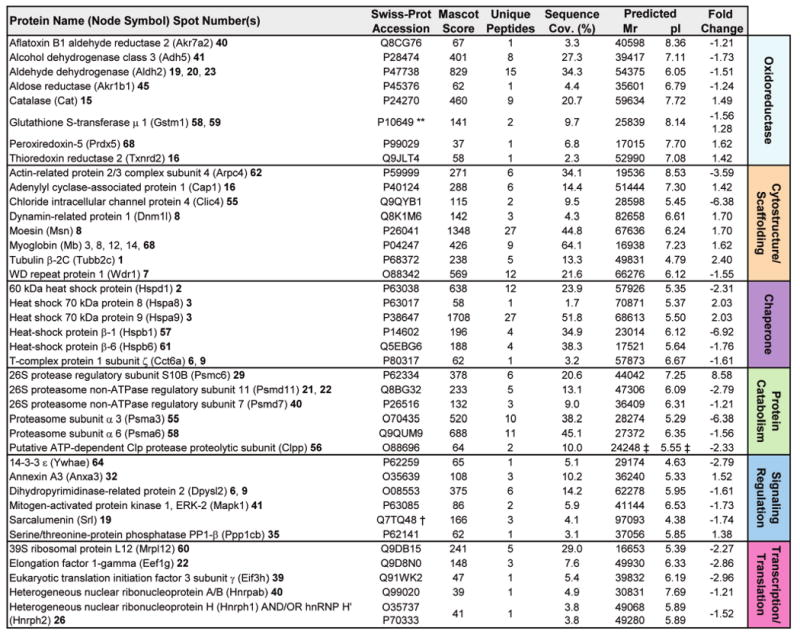
Secondary functions associated with KATP channel deficiency form an infrastructure supporting metabolism. Ontological categorization indicated that a secondary functional group consisting of the remaining identified proteins (39/102) comprises a number of functions supporting cellular metabolic activity. These proteins function as oxidoreductases, in cytostructure and scaffolding, as stress-related chaperones, or in protein catabolism, signaling regulation, or transcription and translation. Protein names are listed with their Swiss-Prot gene name (for Node Symbol) to locate them in the protein interaction network, and with their corresponding spot numbers from their 2-D gel positions. Protein accession number, Mascot score, number of unique identified peptides, % sequence cov. (coverage), predicted Mr and pI for each protein (following expected post-translational processing, for example, removal of a mitochondrial signal peptide), and fold change (KO versus WT) are indicated. For proteins detected in more than one spot, maximum score and number of unique peptides are reported. Fold change was calculated as described in experimental procedures, and for proteins detected in both increasing and decreasing spots (**), both values are indicated. Complete MS/MS data for all proteins is outlined in Supplementary Table 1 (Supporting Information). (**Contains both up- and down-regulated spots; † Node not detected for network analysis by Ingenuity Pathways; ‡ Mr/pI calculated after TargetP 1.1 prediction of mitochondrial localization and signal peptide cleavage site).
KATP Channel-Dependent Subproteome Network Topography and Ontological Annotation
Network analysis was carried out to obtain a collective understanding of processes associated with the remodeled proteome in the setting of KATP channel deficiency. Protein–protein interaction mapping clustered KATP channel-dependent proteins into an organized network comprised of 221 nodes linked by 928 edges. Ontological assessment of individual proteins validated the primary functional impact on metabolic pathways observed with the proteomic data, with all network elements accounted for by identical classifications (Figure 4A and B). The nonrandomness of network topology was confirmed by derivation of network degree distribution, the interrelationship between the network variables node degree (k) and node degree distribution [P(k)]. After ruling out a random distribution using the Anderson-Darling normality test (P = 1.52 × 10−9), a cumulative distribution function was applied to define a nonstochastic scale-free architecture, following a power law distribution where P(k) ∼ k−γ, with γ = 1.653 ± 0.044 (Figure 4C), which falls within parameters characteristic of biological networks.39
Figure 4.
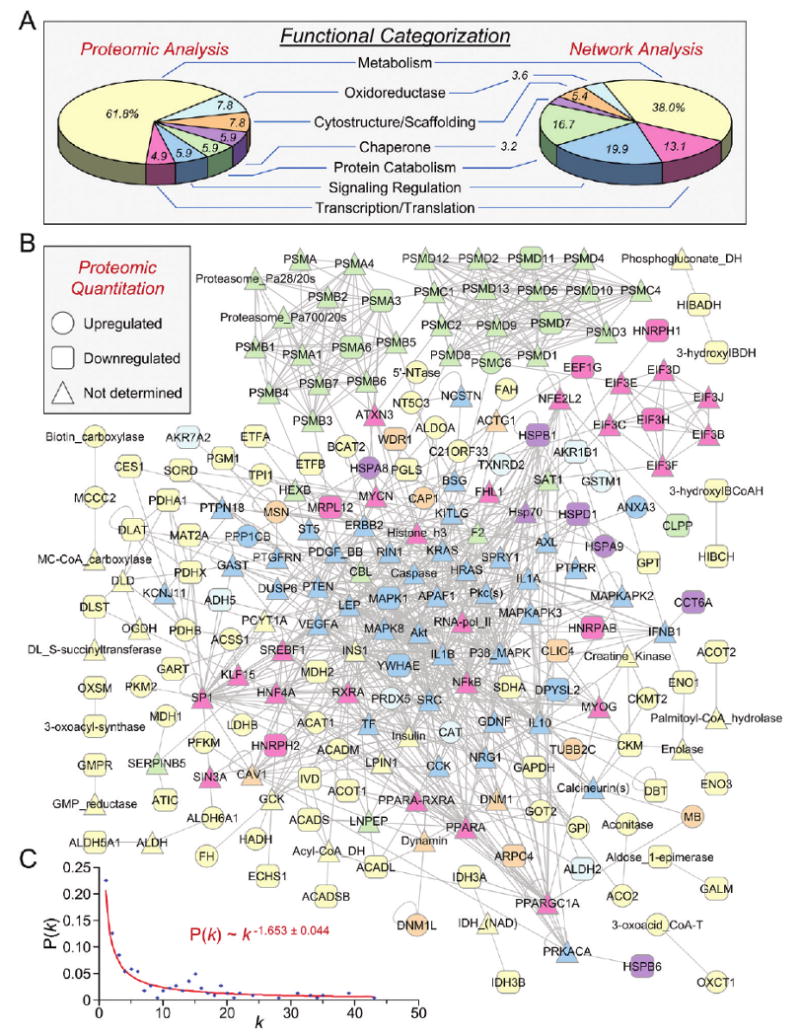
Network analysis of the Kir6.2-dependent proteome. (A) The distribution of Swiss-Prot annotated primary ontological functions of altered proteins (Proteomic Analysis) carry through to the expanded protein interaction network (Network Analysis), with metabolism predominant in both. (B) Differentially expressed proteins of the Kir6.2-dependent subproteome submitted to Ingenuity Pathways Analysis as focus nodes generated a 221 protein interaction network. Nodes are listed by Swiss-Prot gene designations, with the exception of nodes representing protein families. Node color corresponds to ontological function (from A), while node shape (legend) indicates directionality of expression level change for proteins characterized during proteomic analysis. (C) Network degree distribution, (P[k]) versus degree (k), followed a power law distribution indicating nonstochastic scale-free network architecture characteristic of biological networks.
Network assessment with BiNGO to identify network node GO biological process enrichment verified the overarching prevalence of bioenergetic components, as 87% of unique network protein nodes were linked to metabolism, as were all 55 biological processes overrepresented (P < 0.001) within the full ontological spectrum of 962 potential processes associated with the derived KATP channel-dependent network (Figure 5). Indeed, biological processes linked to glycolysis, fatty acid metabolism, the tricarboxylic acid cycle, and protein catabolism demonstrated the greatest extent of statistical overrepresentation. Dissection of the composite network into discrete functional subnetworks further corroborated the massive contribution to bioenergetics, as amino acid metabolism, carbohydrate metabolism, and small molecule biochemistry were designated by Ingenuity Pathways Analysis as top functions associated with the most prominent subnetwork, comprising 24 of the 102 focus proteins, while carbohydrate metabolism, amino acid metabolism, and lipid metabolism were top functions for the second most prominent subnetwork, containing 18 focus proteins. Thus, the network encompassing the resolved KATP channel-dependent subproteome organizes into a bona fide biological system with an overwhelming bioenergetic ontological linkage to metabolic and metabolism related processes.
Figure 5.
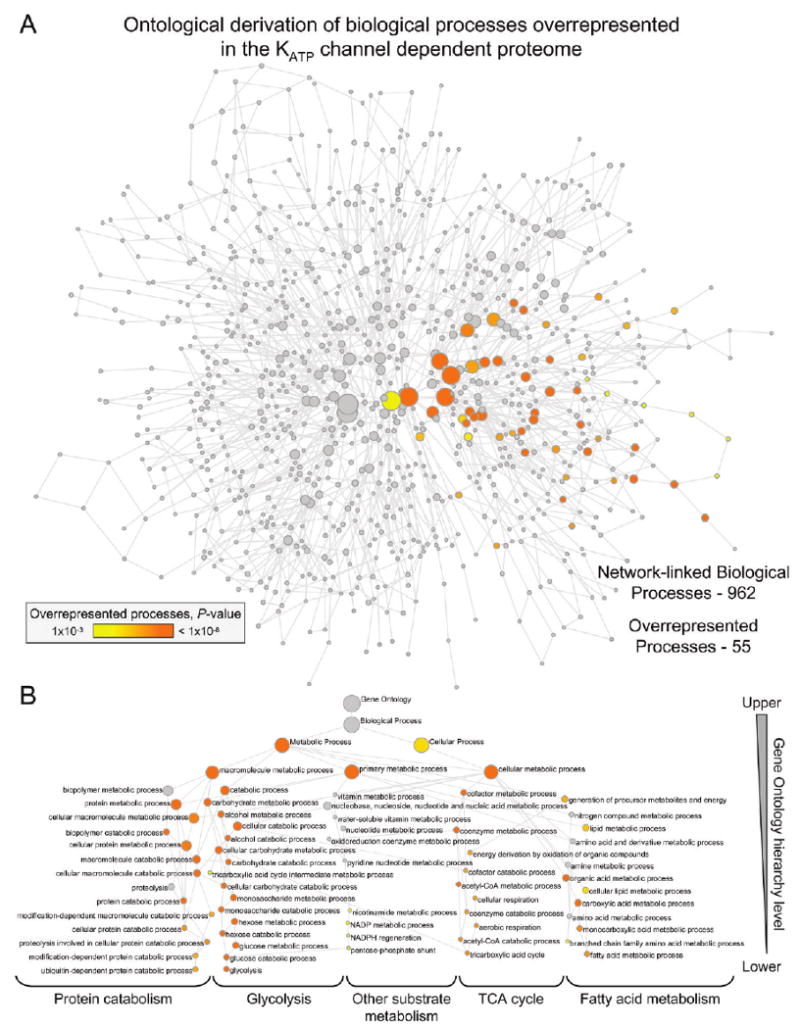
Biological processes linked to the KATP channel-dependent protein interaction network. (A) KATP channel-dependent protein interaction network was interrogated with the Cytoscape module BiNGO to identify network-linked Gene Ontology (GO) biological processes, including evidence of overrepresentation. The derived GO network consisted of 962 processes (nodes), of which 55 were significantly overrepresented (colored nodes, P < 0.001), with nodes connected by GO hierarchical relationships. Node size is proportional to the number of network proteins annotated to that biological process, while node shading represents a 5-log gradient of significant, false discovery rate-corrected P-values, with gray nodes not significantly overrepresented. (B) Hierarchical layout of overrepresented biological processes from panel A, with node size/color retained and identities indicated. Of note, all 55 colored nodes involve metabolic processes. Functional interdependencies within the GO hierarchy translated into significant overrepresentation by entire hierarchical branches, with the most significant nodes furthest down the hierarchy offering the strongest interpretive explanation. Processes linked to “Glycolysis”, the “TCA cycle”, “Fatty acid metabolism” and “Protein catabolism” demonstrated the greatest overrepresentation.
Forecast of Disease Predisposition Validated through Stress Imposition
The subproteome and associated network neighborhood database that underlie Kir6.2 deficiency provided a systems framework to screen for putative pathophysiological patterns arising from proteome remodeling. Collective bioinformatic interrogation of the KATP channel-dependent subproteome demonstrated an overrepresentation for the “cardiovascular disease” category predicting, in silico, heart disease susceptibility, a pattern reiterated and accentuated in the broader network interactome context (Figure 6A). To validate this prediction in vivo, WT and Kir6.2-KO cohorts were subjected to graded levels of imposed cardiac stress. Whereas no difference was evident prior to stress, with progressively greater stress challenge the absence of KATP channels was associated with aggravated increases in left ventricular mass and decreases in fractional shortening (Figure 6B and C). Ultimately, proteomic remodeling in response to Kir6.2 deletion precipitated a highly maladaptative cardiac stress response with decreasing survival arising from progressively increased stress (Figure 6D). In contrast, WT exhibited significant protection when subject to mild (0% mortality, n = 18), moderate (3%, n = 24) or severe stress (44%, n = 28) as compared to Kir6.2-KO counterparts - mild (21%, n = 18), moderate (45%, n = 24), severe (74%, n = 108, all P < 0.01 versus WT). Thus, while the KATP channel knockout does not display an overt cardiac disease phenotype in the absence of stress, proteomic remodeling in response to Kir6.2 deletion predisposes to aggravated outcome, establishing a molecular substrate for maladaptive response to stress that leads to increased morbidity and mortality in the setting of KATP channel deficit.
Figure 6.
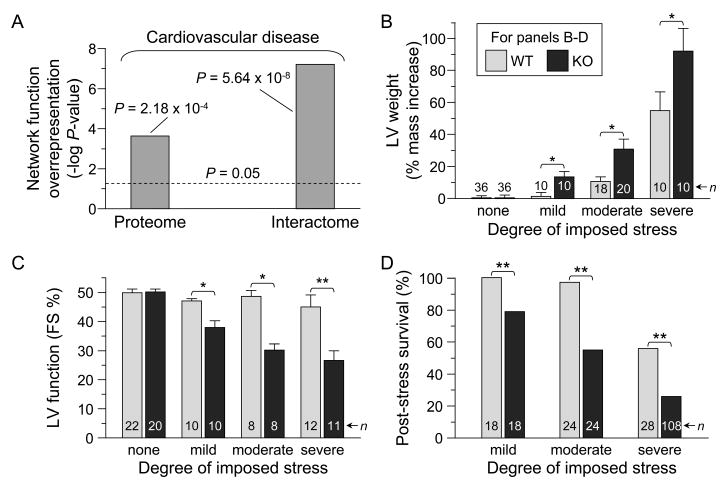
Predicted outcome of KATP channel deficit and functional validation. (A) “Cardiovascular disease” was significantly overrepresented as a consequence of KATP channel deficit based on Ingenuity Pathways Analysis of both the KATP channel-dependent altered proteins (Proteome) and their derived protein network (Interactome). (B–D) Functional confirmation of predicted outcome. While wild-type (WT) and Kir6.2-knockout (KO) counterparts exhibited no difference in structural or functional parameters in the absence of imposed stress, progressively greater challenge by mild (chronic repetitive aquatic exercise), moderate (deoxycorticosterone acetate/salt-induced hypertension) or severe (transverse aortic constriction) stress led to aggravated increases in left ventricular (LV) mass (B; *P < 0.05) and decreases in LV fractional shortening, FS (C; *P < 0.05, **P < 0.01), with KATP channel deletion ultimately leading to decreasing post-stress survival (D; **P < 0.01 versus WT).
Discussion
This study establishes a previously unrecognized physiological role for KATP channels in supporting a broad subproteome vital to the well-being of the ventricular mammalian myocardium. Systems analysis of the cardiac subproteome remodeled in response to ablation of the KATP channel pore extracted a subclinical signature predicting susceptibility to cardiovascular disease at an otherwise asymptomatic state. By decoding molecular profiles predictive of disease manifestation, the present study offers a paradigm toward realizing the prospect of anticipatory medicine prior to symptom onset.
Traditionally, KATP channels have been associated with protection of the myocardium under stress.8,17,18,46 Despite their discovery nearly three decades ago within the cardiac sarcolemma where they were found to be particularly abundant, understanding of KATP channel contributions has been limited to conditions of ischemic and more recently nonischemic challenge.5,19,21,47,48 Their role, however, in the stress-free heart has remained largely an enigma.49 The present demonstration that KATP channel deficiency, produced by ablation of the KCNJ11-encoding Kir6.2 pore, alters nearly 9% of all detected protein species within left ventricular extracts offers the first definitive evidence for a homeostatic requirement of KATP channels in ensuring stability of the normal cardiac proteome.
While cardiac KATP channels have been previously implicated in individual energy metabolism pathways,50–54 herein disruption of channel biogenesis combined with high-throughput proteomic cartography diagnosed extensive changes across a spectrum of metabolic processes, involving over 100 unique identities. Over 60% of the altered subproteome comprised enzymes implicated in mitochondrial metabolism via the tricarboxylic acid cycle and fatty acid β-oxidation, cytoplasmic metabolism primarily involving glycolysis, as well as contributors to both amino acid and nucleic acid metabolism. Among this broad spectrum of energetic molecules are proteins with established links to the cardiac KATP channel, including the glycolytic enzymes triosephosphate isomerase, pyruvate kinase, and glyceraldehyde-3-phosphate dehydrogenase,52,53 the fatty acid biosynthesis enzyme long chain acyl-CoA dehydrogenase,54 and contributors to cardiac energy shuttling, including creatine kinase isoforms and lactate dehydrogenase.51,55,56 Beyond metabolism-centric protein partners, discrete clusters of signaling proteins, oxidoreductases, chaperones, catabolic proteins, and proteins supporting cytostructure, transcription and translation were also affected by disruption of KATP channel function. This study thereby demonstrates, under normal cellular physiological conditions, the identity of multiple molecular components constituting a robust KATP channel-dependent cardiac subproteome.
Charting protein–protein relationships authenticated the primary impact on metabolic pathways, and encompassed a wide influence of the KATP channel upon a 221 node network neighborhood. The resolved interactome fulfilled criteria of nonstochastic topography typical of authentic biological networks, which confers the capability of readily adopting optimal functional states.39,57 Unbiased gene ontology-based network interrogation further extended the significance of the underlying KATP channel-dependent infrastructure, revealing bioenergetic ontological linkage of nearly 90% of network nodes to metabolism and metabolism-related processes. This iterative process of proteomic cartography and functional ontology thus details the KATP channel network as a biologically relevant, nonrandom, protein assembly.
Networks arising from established protein interactions provide a means for defining phenotypic outcome following perturbation.58 In this study, removal of KATP channels corrupted the baseline proteome which translated into a prioritized susceptibility to “cardiovascular disease” within not only the remodeled subproteome but also the resultant interactome. There are, however, diverse examples of cardiac dysfunction whereby associated proteomic remodeling is either absent prior to symptom manifestation59 or detectable but not extensive until disease onset,60 suggesting the critical nature of KATP channels in endowing stress tolerance.
The bioinformatic forecast was directly validated through prospective exposure of the KATP channel knockout to increasing stress load, which led to progressive worsening of cardiac function, structure and, ultimately, diminished survival relative to the equivalently stressed wild-type. The observed stress intolerance is consistent with a fundamental protective role for KATP channels enabled through coordinated interactions with metabolic partners integrating ion homeostasis with energetic demands of the cell.55,56,61 In fact, the pronounced metabolic remodeling in presymptomatic KATP channel knockouts, in concert with the inability to precondition the Kir6.2-KO against subsequent injury,62 is in line with findings establishing extensive proteomic changes in response to distinct pharmacological agents29 or hemodynamic stress26 known to interact with63–65 or involve19 the cardioprotective KATP channel. A distinct example of change deciphered in the KATP channel deficient milieu is the downregulation of aldehyde dehydrogenase-2, recently implicated in cardiac protection against damage.66,67 More broadly, metabolic remodeling ensuing from gene deletion of distinct cardioprotective molecules compromises the ability of the heart to match energy production with utilization, precipitating maladaptation to stress.68–70 As heart muscle requires great dynamic flexibility to maintain cardiac performance in the face of continuous energetic demands in both health and disease,71–73 acquired or, as shown here, congenital inability to adequately maintain metabolic integrity would preclude the adaptive flexibility required for cardioprotection.
The cardiac KATP channel has previously been established as a rheostat-like protein complex that matches cellular metabolic supply with demand in response to disease. Based on an unbiased systems biology approach, the present study establishes a critical, previously unrecognized, role for KATP channels in the stability of the normal cardiac proteome such that the heart can adequately respond to stress. Monogenic deletion of the KCNJ11-encoded Kir6.2 channel pore precipitated complex proteomic remodeling of a broad and interrelated KATP channel-dependent infrastructure within heart muscle. Beyond proteins with previously established links to the KATP channel, bioinformatic interrogation of the affected KATP channel subnetwork unmasked extensive metabolic restructuring associated with overrepresentation of “cardiovascular disease” markers. Validated experimentally by the differential response of the KATP channel knockout to graded stress, this approach to detect, identify and forecast disease-prone networks offers the opportunity to elicit a paradigm shift away from traditional post-onset disease management and toward subclinical detection, thus enabling earlier, presymptomatic intervention.
Supplementary Material
Acknowledgments
We thank Jonathan Nesbitt for surgical expertise and the Mayo Proteomics Research Center staff, particularly Benjamin Madden, Kenneth Johnson, Christopher Mason and Roman Zenka, for expert guidance in mass spectrometry and bioinformatic analysis. Support was provided by the National Institutes of Health, Marriott Heart Disease Research Program, and Mayo Clinic. D.K.A. is the recipient of a Marriott Individualized Medicine Career Development Award. J.Z. holds a Mayo Graduate School fellowship. S.Y. is the recipient of an American Society for Clinical Pharmacology and Therapeutics Young Investigator Award. A.T. holds the Marriott Family Professorship in Cardiovascular Research at Mayo Clinic.
Footnotes
Supporting Information Available: Supplementary Table 1, which contains acquired mass spectral data for all identified proteins. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hood L, Heath JR, Phelps ME, Lin B. Systems biology and new technologies enable predictive and preventative medicine. Science. 2004;306:640–3. doi: 10.1126/science.1104635. [DOI] [PubMed] [Google Scholar]
- 2.Weston AD, Hood L. Systems biology, proteomics, and the future of health care: toward predictive, preventative, and personalized medicine. J Proteome Res. 2004;3:179–96. doi: 10.1021/pr0499693. [DOI] [PubMed] [Google Scholar]
- 3.Bell J. Predicting disease using genomics. Nature. 2004;429:453–6. doi: 10.1038/nature02624. [DOI] [PubMed] [Google Scholar]
- 4.Waldman SA, Terzic A. Therapeutic targeting: a crucible for individualized medicine. Clin Pharmacol Ther. 2008;83:651–4. doi: 10.1038/clpt.2008.65. [DOI] [PubMed] [Google Scholar]
- 5.Alekseev AE, Hodgson DM, Karger AB, Park S, Zingman LV, Terzic A. ATP-sensitive K+ channel channel/enzyme multimer: Metabolic gating in the heart. J Mol Cell Cardiol. 2005;38:895–905. doi: 10.1016/j.yjmcc.2005.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ashcroft FM. ATP-sensitive potassium channelopathies: Focus on insulin secretion. J Clin Invest. 2005;115:2047–58. doi: 10.1172/JCI25495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miki T, Seino S. Roles of KATP channels as metabolic sensors in acute metabolic changes. J Mol Cell Cardiol. 2005;38:917–25. doi: 10.1016/j.yjmcc.2004.11.019. [DOI] [PubMed] [Google Scholar]
- 8.Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature. 2006;440:470–6. doi: 10.1038/nature04711. [DOI] [PubMed] [Google Scholar]
- 9.Zingman LV, Alekseev AE, Bienengraeber M, Hodgson DM, Karger AB, Dzeja PP, Terzic A. Signaling in channel/enzyme multimers: ATPase transitions in SUR module gate ATP-sensitive K+ conductance. Neuron. 2001;31:233–45. doi: 10.1016/s0896-6273(01)00356-7. [DOI] [PubMed] [Google Scholar]
- 10.Liu XK, Yamada S, Kane GC, Alekseev AE, Hodgson DM, O'Cochlain DF, Jahangir A, Miki T, Seino S, Terzic A. Genetic disruption of Kir6.2, the pore-forming subunit of ATP-sensitive K+ channel, predisposes to catecholamine induced ventricular dysrhythmia. Diabetes. 2004;53:S165–8. doi: 10.2337/diabetes.53.suppl_3.s165. [DOI] [PubMed] [Google Scholar]
- 11.Selivanov VA, Alekseev AE, Hodgson DM, Dzeja PP, Terzic A. Nucleotide-gated KATP channels integrated with creatine and adenylate kinases: amplification, tuning and sensing of energetic signals in the compartmentalized cellular environment. Mol Cell Biochem. 2004;256–7:243–56. doi: 10.1023/b:mcbi.0000009872.35940.7d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gumina RJ, O'Cochlain DF, Kurtz CE, Bast PH, Pucar D, Mishra P, Miki T, Seino S, Macura S, Terzic A. KATP channel knockout worsens myocardial calcium stress load in vivo and impairs recovery in stunned heart. Am J Physiol Heart Circ Physiol. 2007;292:H1706–13. doi: 10.1152/ajpheart.01305.2006. [DOI] [PubMed] [Google Scholar]
- 13.Inagaki N, Gonoi T, Clement JP, Wang CZ, Aguilar-Bryan L, Bryan J, Seino S. A family of sulfonylurea receptors determines the pharmacological properties of ATP-sensitive K+ channels. Neuron. 1996;16:1011–7. doi: 10.1016/s0896-6273(00)80124-5. [DOI] [PubMed] [Google Scholar]
- 14.Lorenz E, Terzic A. Physical association between recombinant cardiac ATP-sensitive K+ channel subunits Kir6.2 and SUR2A. J Mol Cell Cardiol. 1999;31:425–34. doi: 10.1006/jmcc.1998.0876. [DOI] [PubMed] [Google Scholar]
- 15.Zingman LV, Hodgson DM, Bienengraeber M, Karger AB, Kathmann EC, Alekseev AE, Terzic A. Tandem function of nucleotide binding domains confers competence to sulfonylurea receptor in gating ATP-sensitive K+ channels. J Biol Chem. 2002;277:14206–10. doi: 10.1074/jbc.M109452200. [DOI] [PubMed] [Google Scholar]
- 16.Park S, Lim BB, Perez-Terzic C, Mer G, Terzic A. Interaction of asymmetric ABCC9-encoded nucleotide binding domains determines KATP channel SUR2A catalytic activity. J Proteome Res. 2008;7:1721–8. doi: 10.1021/pr7007847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zingman LV, Hodgson DM, Bast PH, Kane GC, Perez-Terzic C, Gumina RJ, Pucar D, Bienengraeber M, Dzeja PP, Miki T, Seino S, Alekseev AE, Terzic A. Kir6.2 is required for adaptation to stress. Proc Natl Acad Sci USA. 2002;99:13278–83. doi: 10.1073/pnas.212315199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kane GC, Liu XK, Yamada S, Olson TM, Terzic A. Cardiac KATP channels in health and disease. J Mol Cell Cardiol. 2005;38:937–43. doi: 10.1016/j.yjmcc.2005.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kane GC, Behfar A, Dyer RB, O'Cochlain DF, Liu XK, Hodgson DM, Reyes S, Miki T, Seino S, Terzic A. KCNJ11 gene knockout of the Kir6.2 KATP channel causes maladaptive remodeling and heart failure in hypertension. Hum Mol Genet. 2006;15:2285–97. doi: 10.1093/hmg/ddl154. [DOI] [PubMed] [Google Scholar]
- 20.Kane GC, Lam CF, O'Cochlain DF, Hodgson DM, Reyes S, Liu XK, Miki T, Seino S, Katusic ZS, Terzic A. Gene knockout of the KCNJ8-encoded Kir6.1 KATP channel imparts fatal susceptibility to endotoxemia. FASEB J. 2006;20:2271–80. doi: 10.1096/fj.06-6349com. [DOI] [PubMed] [Google Scholar]
- 21.Yamada S, Kane GC, Behfar A, Liu XK, Dyer RB, Faustino RS, Miki T, Seino S, Terzic A. Protection conferred by myocardial ATP-sensitive K+ channels in pressure overload-induced congestive heart failure revealed in KCNJ11 Kir6.2-null mutant. J Physiol. 2006;577:1053–65. doi: 10.1113/jphysiol.2006.119511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bienengraeber M, Olson TM, Selivanov VA, Kathmann EC, O'Cochlain DF, Gao F, Karger AB, Ballew JD, Hodgson DM, Zingman LV, Pang YP, Alekseev AE, Terzic A. ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic KATP channel gating. Nat Genet. 2004;36:382–7. doi: 10.1038/ng1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olson TM, Alekseev AE, Moreau C, Liu XK, Zingman LV, Miki T, Seino S, Asirvatham SJ, Jahangir A, Terzic A. KATP channel mutation confers risk for vein of Marshall adrenergic atrial fibrillation. Nat Clin Pract Cardiovasc Med. 2007;4:110–6. doi: 10.1038/ncpcardio0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reyes S, Terzic A, Mahoney DW, Redfield MM, Rodeheffer RJ, Olson TM. KATP channel polymorphism is associated with left ventricular size in hypertensive individuals: a large-scale community-based study. Hum Genet. 2008;123:665–7. doi: 10.1007/s00439-008-0519-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Drake TA, Ping P. Thematic review series: systems biology approaches to metabolic and cardiovascular disorders. Proteomics approaches to the systems biology of cardiovascular diseases. J Lipid Res. 2007;48:1–8. doi: 10.1194/jlr.R600027-JLR200. [DOI] [PubMed] [Google Scholar]
- 26.Zlatkovic J, Arrell DK, Kane GC, Miki T, Seino S, Terzic A. Proteomic profiling of KATP channel-deficient hypertensive heart maps risk for maladaptive cardiomyopathic outcome. Proteomics. 2009;9:1314–25. doi: 10.1002/pmic.200800718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hwang D, Lee IY, Yoo H, Gehlenborg N, Cho JH, Petritis B, Baxter D, Pitstick R, Young R, Spicer D, Price ND, Hohmann JG, Dearmond SJ, Carlson GA, Hood LE. A systems approach to prion disease. Mol Syst Biol. 2009;5:252. doi: 10.1038/msb.2009.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miki T, Nagashima K, Tashiro F, Kotake K, Yoshitomi H, Tamamoto A, Gonoi T, Iwanaga T, Miyazaki J, Seino S. Defective insulin secretion and enhanced insulin action in KATP channel-deficient mice. Proc Natl Acad Sci USA. 1998;95:10402–6. doi: 10.1073/pnas.95.18.10402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arrell DK, Elliott ST, Kane LA, Guo Y, Ko YH, Pedersen PL, Robinson J, Murata M, Murphy AM, Marbán E, Van Eyk JE. Proteomic analysis of pharmacological preconditioning. Novel protein targets converge to mitochondrial metabolism pathways. Circ Res. 2006;99:706–14. doi: 10.1161/01.RES.0000243995.74395.f8. [DOI] [PubMed] [Google Scholar]
- 30.Arrell DK, Niederländer NJ, Perez-Terzic C, Behfar A, Terzic A. Embryonic stem cell cardiac differentiation: a proteomic perspective. Adv Mol Med. 2006;2:149–56. [Google Scholar]
- 31.Arrell DK, Neverova I, Fraser H, Marbán E, Van Eyk JE. Proteomic analysis of pharmacologically preconditioned cardiomyocytes reveals novel phosphorylation of myosin light chain 1. Circ Res. 2001;89:480–7. doi: 10.1161/hh1801.097240. [DOI] [PubMed] [Google Scholar]
- 32.Arrell DK, Niederländer N, Faustino RS, Behfar A, Terzic A. Cardioinductive network guiding stem cell differentiation revealed by proteomic cartography of TNFα-primed endodermal secretome. Stem Cells. 2008;26:387–400. doi: 10.1634/stemcells.2007-0599. [DOI] [PubMed] [Google Scholar]
- 33.Arrell DK, Niederländer NJ, Perez-Terzic C, Behfar A, Terzic A. Pharmacoproteomics: advancing the efficacy and safety of regenerative therapeutics. Clin Pharmacol Ther. 2007;82:316–9. doi: 10.1038/sj.clpt.6100310. [DOI] [PubMed] [Google Scholar]
- 34.Behfar A, Perez-Terzic C, Faustino RS, Arrell DK, Hodgson DM, Yamada S, Pucéat M, Niederländer N, Alekseev AE, Zingman LV, Terzic A. Cardiopoietic programming of embryonic stem cells for tumor-free heart repair. J Exp Med. 2007;204:405–20. doi: 10.1084/jem.20061916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Link AJ, Eng J, Schieltz DM, Carmack E, Mize GJ, Morris DR, Garvik BM, Yates JR., 3rd Direct analysis of protein complexes using mass spectrometry. Nat Biotechnol. 1999;17:676–82. doi: 10.1038/10890. [DOI] [PubMed] [Google Scholar]
- 36.Emanuelsson O, Nielsen H, Brunak S, von Heijne G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol. 2000;300:1005–16. doi: 10.1006/jmbi.2000.3903. [DOI] [PubMed] [Google Scholar]
- 37.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Assenov Y, Ramírez F, Schelhorn SE, Lengauer T, Albrecht M. Computing topological parameters of biological networks. Bioinformatics. 2007;24:282–4. doi: 10.1093/bioinformatics/btm554. [DOI] [PubMed] [Google Scholar]
- 39.Barabási AL, Oltvai ZN. Network biology: understanding the cell's functional organization. Nat Rev Genet. 2004;5:101–13. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 40.Anderson TW, Darling DA. Asymptotic theory of certain “goodness-of-fit” criteria based on stochastic processes. Ann Math Stat. 1952;23:193–212. [Google Scholar]
- 41.Newman MEJ. The structure and function of complex networks. SIAM Rev. 2003;45:167–256. [Google Scholar]
- 42.Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of Gene Ontology categories in biological networks. Bioinformatics. 2005;21:3448–9. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- 43.Kane GC, Behfar A, Yamada S, Perez-Terzic C, O'Cochlain DF, Reyes S, Dzeja PP, Miki T, Seino S, Terzic A. ATP-sensitive K+ channel knockout compromises the metabolic benefit of exercise training, resulting in cardiac deficits. Diabetes. 2004;53:S169–75. doi: 10.2337/diabetes.53.suppl_3.s169. [DOI] [PubMed] [Google Scholar]
- 44.Yamada S, Nelson TJ, Crespo-Diaz RJ, Perez-Terzic C, Liu XK, Miki T, Seino S, Behfar A, Terzic A. Embryonic stem cell therapy of heart failure in genetic cardiomyopathy. Stem Cells. 2008;26:2644–53. doi: 10.1634/stemcells.2008-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Suzuki M, Li RA, Miki T, Uemura H, Sakamoto N, Ohmoto-Sekine Y, Tamagawa M, Ogura T, Seino S, Marbán E, Nakaya H. Functional roles of cardiac and vascular ATP-sensitive potassium channels clarified by Kir6.2-knockout mice. Circ Res. 2001;88:570–7. doi: 10.1161/01.res.88.6.570. [DOI] [PubMed] [Google Scholar]
- 46.Terzic A, Jahangir A, Kurachi Y. Cardiac ATP-sensitive K+ channels: regulation by intracellular nucleotides and K+ channel-opening drugs. Am J Physiol. 1995;269:C525–45. doi: 10.1152/ajpcell.1995.269.3.C525. [DOI] [PubMed] [Google Scholar]
- 47.Noma A. ATP-regulated K+ channels in cardiac muscle. Nature. 1983;305:147–8. doi: 10.1038/305147a0. [DOI] [PubMed] [Google Scholar]
- 48.Zingman LV, Alekseev AE, Hodgson-Zingman DM, Terzic A. ATP-sensitive potassium channels: metabolic sensing and cardioprotection. J Appl Physiol. 2007;103:1888–93. doi: 10.1152/japplphysiol.00747.2007. [DOI] [PubMed] [Google Scholar]
- 49.Zingman LV, Hodgson DM, Alekseev AE, Terzic A. Stress without distress: homeostatic role for KATP channels. Mol Psychiatr. 2003;8:253–4. doi: 10.1038/sj.mp.4001323. [DOI] [PubMed] [Google Scholar]
- 50.Carrasco AJ, Dzeja PP, Alekseev AE, Pucar D, Zingman LV, Abraham MR, Hodgson DM, Bienengraeber M, Pucéat M, Janssen E, Wieringa B, Terzic A. Adenylate kinase phosphotransfer communicates cellular energetic signals to ATP-sensitive potassium channels. Proc Natl Acad Sci USA. 2001;98:7623–8. doi: 10.1073/pnas.121038198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Crawford RM, Ranki HJ, Botting CH, Budas GR, Jovanoví A. Creatine kinase is physically associated with the cardiac ATP-sensitive K+ channel in vivo. FASEB J. 2002;16:102–4. doi: 10.1096/fj.01-0466fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dhar-Chowdhury P, Harrell MD, Han SY, Jankowska D, Parachuru L, Morrissey A, Srivastava S, Liu W, Malester B, Yoshida H, Coetzee WA. The glycolytic enzymes, glyceraldehyde-3-phosphate dehydrogenase, triose-phosphate isomerase, and pyruvate kinase are components of the KATP channel macromolecular complex and regulate its function. J Biol Chem. 2005;280:38464–70. doi: 10.1074/jbc.M508744200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jovanović S, Du Q, Crawford RM, Budas GR, Stagljar I, Jovanović A. Glyceraldehyde 3-phosphate dehydrogenase serves as an accessory protein of the cardiac sarcolemmal KATP channel. EMBO Rep. 2005;6:848–52. doi: 10.1038/sj.embor.7400489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu GX, Hanley PJ, Ray J, Daut J. Long-chain acyl-coenzyme A esters and fatty acids directly link metabolism to KATP channels in the heart. Circ Res. 2001;88:918–24. doi: 10.1161/hh0901.089881. [DOI] [PubMed] [Google Scholar]
- 55.Abraham MR, Selivanov VA, Hodgson DM, Pucar D, Zingman LV, Wieringa B, Dzeja PP, Alekseev AE, Terzic A. Coupling of cell energetics with membrane metabolic sensing: Integrative signaling through creatine kinase phosphotransfer disrupted by M-CK gene knock-out. J Biol Chem. 2002;277:24427–34. doi: 10.1074/jbc.M201777200. [DOI] [PubMed] [Google Scholar]
- 56.Crawford RM, Budas GR, Jovanović S, Ranki HJ, Wilson TJ, Davies AM, Jovanović A. M-LDH serves as a sarcolemmal KATP channel subunit essential for cell protection against ischemia. EMBO J. 2002;21:3936–48. doi: 10.1093/emboj/cdf388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Albert R, Jeong H, Barabási AL. Error and attack tolerance of complex networks. Nature. 2000;406:378–82. doi: 10.1038/35019019. [DOI] [PubMed] [Google Scholar]
- 58.Loscalzo J, Kohane I, Barabási AL. Human disease classification in the postgenomic era: a complex systems approach to human pathobiology. Mol Syst Biol. 2007;3:124. doi: 10.1038/msb4100163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Meng C, Jin X, Xia L, Shen SM, Wang XL, Cai J, Chen GQ, Wang LS, Fang NY. Alterations of mitochondrial enzymes contribute to cardiac hypertrophy before hypertension development in spontaneously hypertensive rats. J Proteome Res. 2009;8:2463–75. doi: 10.1021/pr801059u. [DOI] [PubMed] [Google Scholar]
- 60.Sutak R, Xu X, Whitnall M, Kashem MA, Vyoral D, Richardson DR. Proteomic analysis of hearts from frataxin knockout mice: marked rearrangement of energy metabolism, a response to cellular stress and altered expression of proteins involved in cell structure, motility and metabolism. Proteomics. 2008;8:1731–41. doi: 10.1002/pmic.200701049. [DOI] [PubMed] [Google Scholar]
- 61.Hodgson DM, Zingman LV, Kane GC, Perez-Terzic C, Bienengraeber M, Ozcan C, Gumina RJ, Pucar D, O'Coclain DF, Mann DL, Alekseev AE, Terzic A. Cellular remodeling in heart failure disrupts KATP channel-dependent stress tolerance. EMBO J. 2003;22:1732–42. doi: 10.1093/emboj/cdg192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gumina RJ, Pucar D, Bast PH, Hodgson DM, Kurtz CE, Dzeja PP, Miki T, Seino S, Terzic A. Knockout of Kir6.2 negates ischemic preconditioning-induced protection of myocardial energetics. Am J Physiol Heart Circ Physiol. 2003;284:H2106–13. doi: 10.1152/ajpheart.00057.2003. [DOI] [PubMed] [Google Scholar]
- 63.Terzic A, Tung RT, Kurachi Y. Nucleotide regulation of ATP-sensitive potassium channels. Cardiovasc Res. 1994;28:746–53. doi: 10.1093/cvr/28.6.746. [DOI] [PubMed] [Google Scholar]
- 64.Jahangir A, Terzic A. KATP channel therapeutics at the bedside. J Mol Cell Cardiol. 2005;39:99–112. doi: 10.1016/j.yjmcc.2005.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sattiraju S, Reyes S, Kane GC, Terzic A. KATP channel pharmacogenomics: from bench to bedside. Clin Pharmacol Ther. 2008;83:354–7. doi: 10.1038/sj.clpt.6100378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science. 2008;321:1493–5. doi: 10.1126/science.1158554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ping P. Getting to the heart of proteomics. N Engl J Med. 2009;360:532–4. doi: 10.1056/NEJMcibr0808487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mayr M, Chung YL, Mayr U, McGregor E, Troy H, Baier G, Leitges M, Dunn MJ, Griffiths JR, Xu Q. Loss of PKC-delta alters cardiac metabolism. Am J Physiol Heart Circ Physiol. 2004;287:H937–45. doi: 10.1152/ajpheart.00877.2003. [DOI] [PubMed] [Google Scholar]
- 69.Mayr M, Metzler B, Chung YL, McGregor E, Mayr U, Troy H, Hu Y, Leitges M, Pachinger O, Griffiths JR, Dunn MJ, Xu Q. Ischemic preconditioning exaggerates cardiac damage in PKC-delta null mice. Am J Physiol Heart Circ Physiol. 2004;287:H946–56. doi: 10.1152/ajpheart.00878.2003. [DOI] [PubMed] [Google Scholar]
- 70.Mayr M, Liem D, Zhang J, Li X, Avliyakulov NK, Yang JI, Young G, Vondriska TM, Ladroue C, Madhu B, Griffiths JR, Gomes A, Xu Q, Ping P. Proteomic and metabolomic analysis of cardioprotection: Interplay between protein kinase C epsilon and delta in regulating glucose metabolism of murine hearts. J Mol Cell Cardiol. 2009;46:268–77. doi: 10.1016/j.yjmcc.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dzeja PP, Redfield MM, Burnett JC, Terzic A. Failing energetics in failing hearts. Curr Cardiol Rep. 2000;2:212–7. doi: 10.1007/s11886-000-0071-9. [DOI] [PubMed] [Google Scholar]
- 72.Terzic A, Moore RL, Waldman SA. Acquired and innate cardioprotection. J Appl Physiol. 2007;103:1436–7. doi: 10.1152/japplphysiol.00834.2007. [DOI] [PubMed] [Google Scholar]
- 73.Ingwall JS. Energy metabolism in heart failure and remodelling. Cardiovasc Res. 2009;81:412–9. doi: 10.1093/cvr/cvn301. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.