

Abstract
The most commonly studied laboratory rodents possess a specialized form of fat called brown adipose tissue (BAT) that generates heat to help maintain body temperature in cold environments. In humans, BAT is abundant during embryonic and early postnatal development, but is absent or present in relatively small amounts in adults where it is located in paracervical and supraclavicular regions. BAT cells can `burn' fatty acid energy substrates to generate heat because they possess large numbers of mitochondria in which oxidative phosphorylation is uncoupled from ATP production as a result of a transmembrane proton leak mediated by uncoupling protein 1 (UCP1). Studies of rodents in which BAT levels are either increased or decreased have revealed a role for BAT in protection against diet-induced obesity. Data suggest that individuals with low levels of BAT are prone to obesity, insulin resistance and cardiovascular disease, whereas those with higher levels of BAT maintain lower body weights and exhibit superior health as they age. BAT levels decrease during aging, and dietary energy restriction increases BAT activity and protects multiple organ systems including the nervous system against age-related dysfunction and degeneration. Future studies in which the effects of specific manipulations of BAT levels and thermogenic activity on disease processes in animal models (diabetes, cardiovascular disease, cancers, neurodegenerative diseases) are determined will establish if and how BAT affects the development and progression of age-related diseases. Data from animal studies suggest that BAT and mitochondrial uncoupling can be targeted for interventions to prevent and treat obesity and age-related diseases. Examples include: diet and lifestyle changes; specific regimens of mild intermittent stress; drugs that stimulate BAT formation and activity; induction of brown adipose cell progenitors in muscle and other tissues; and transplantation of brown adipose cells.
Introduction
Most body fat is white/yellow in color, is concentrated in the abdomen and subcutaneous regions, and serves the function of storing energy in the form of fatty acids which can be mobilized under conditions where dietary energy intake and liver glycogen levels are low (Lafontan and Girard, 2008). In contrast to white adipose tissue, brown adipose tissue (BAT) is relatively sparse and is located in small `pockets' in the thorax (Figure 1a). Evolutionary considerations and experimental evidence indicates that brown fat serves the important function of generating heat to maintain body temperature of homeotherms in cold environments (Jastroch et al., 2008). Smaller mammals (rats, mice, hamsters, etc.) contain relatively greater amounts of BAT compared to larger mammals including humans. BAT consists of brown adipocytes and adipocyte progenitor cells, and is highly vascularized (Figure 1b). Brown adipocytes contain very high numbers of mitochondria and smaller amounts of lipid droplets, in contrast to white adipocytes which typically have relatively few mitochondria and huge numbers of lipid droplets. As summarized below and recently reviewed elsewhere (Wijers et al., 2009), emerging evidence suggests that in addition to its role in non-shivering thermogenesis BAT may consume enough energy to impact body fat and the risk for obesity and diabetes.
Figure 1.

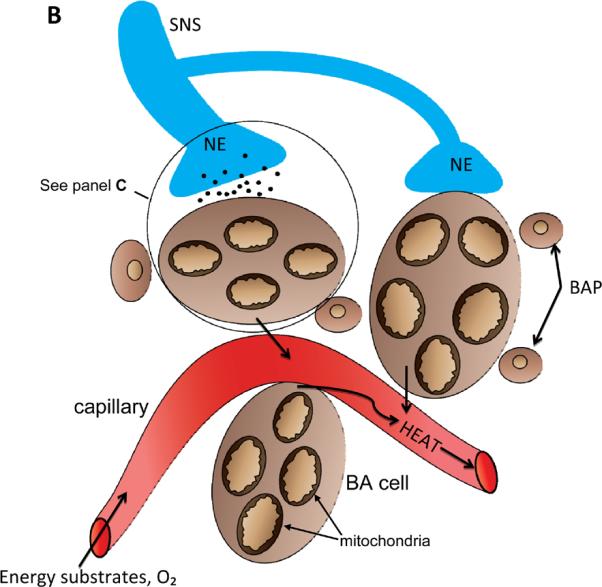
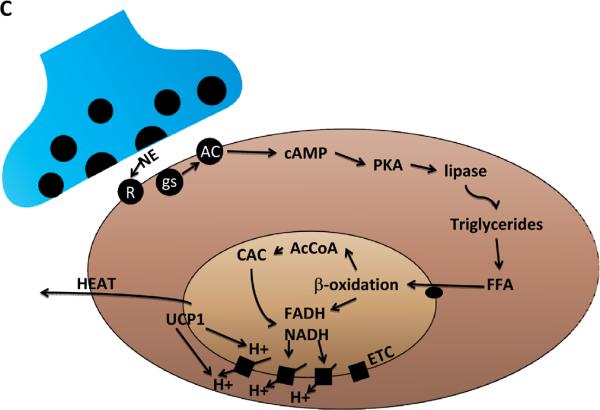
BAT: its location, cellular composition and thermogenic regulation. A. In humans brown adipose tissue (BAT) is located in interscapular and supraclavicular regions of the thorax, and is innervated by sympathetic nervous system (SNS) neurons that originate in the hypothalamus (HT) (Bamshad et al., 1999). Cold temperatures and excessive food intake activate the SNS resulting in increased BAT thermogenic activity. B. Cellular features of BAT. The principal cell of BAT is the brown adipose (BA) cell which contains large numbers of mitochondria that utilize energy substrates and O2 and generate heat which is transferred to circulating blood. BAT also contains brown adipose progenitors (BAP) which are capable of dividing and differentiation into BA cells. BAT is innervated by axons from sympathetic neurons that employ the neurotransmitter norepinephrine (NE). C. Thermogenic signal transduction in brown adipocytes. NE released from SNS presynaptic terminals activates adrenergic receptors (R) on the surface of BA cells, resulting in the activation of the GTP-binding protein gs which, in turn, activates adenylate cyclase (AC) to generate cyclic adenosine monophosphate (cAMP). Cyclic AMP then activates cAMP-dependent protein kinase (PKA) which then phosphorylates and thereby activates lipases that hydrolyze triacylglycerols to generate free fatty acids (FFA). The FFA are then transferred into mitochondria where they undergo β-oxidation to generate acetyl Co A (AcCoA) which enters the citric acid cycle (CAC). As a result of b-oxidation and the CAC the energy substrates FADH (flavanine adenine dinucleotide) and NADH (nicotinamide adenine dinucleotide) are generated. BA cells express high amounts of uncoupling protein 1 (UCP1) which, when activated by the pathway just described, mediates a proton leak that uncouples the electron transport chain resulting in heat production.
The Mechanism by Which Brown Adipocytes Consume Energy and Generate Heat
In most cells the production of ATP in mitochondria is an efficient process wherein the hydrogen ion (proton) gradient across the inner mitochondrial membrane is tightly coupled to the electron transport chain and ATP production, such that energy substrates (glucose and fatty acids) are not “wasted” and very little heat is produced (Brand, 2005; Mattson et al., 2008). In response to cold temperatures and other factors that activate the sympathetic nervous system (SNS), lipolysis occurs in the brown adipocytes as the result of activation of hormone-sensitive lipase and inactivation of perilipin (which normally covers and protects the fatty acids) (Ducharme and Bickel, 2008). The free fatty acids (FFA) are then bound to fatty acid binding proteins (FABP) and transferred into the mitochondria by the activation/carnitine shuttle system (Figure 1c). The FFA then undergo β-oxidation and, together with the citric acid cycle, the reduced electron carriers FADH and NADH are formed and are oxidized by the electron transport chain (ETC). As a result, protons are pumped across the mitochondrial membrane to form the H+ concentration gradient that allows UCP1 to drive protons back into the mitochondrial matrix in a process that generates heat (Rousset et al., 2004).
UCP1 (also known as thermogenin) is a 32 kDa member of the mitochondrial carrier protein family (Dulloo and Samee, 2001). UCP1 in as integral membrane protein located in the outer mitochondrial membrane; it has six transmembrane domains with both the N- and C-terminal ends of the protein located on the cytosolic side of the membrane. Regions of UCP1 that are conserved in other UCPs (UCP2, UCP3 and UCP4) include a region involved in binding GDP and conserved proline residues in three transmembrane domains. In addition, there are two domains that are unique to UCP1, one in the central loop and another in the carboxy-terminal region on the cytosolic side of the membrane. In the presence of cytosolic nucleotides (GDP) and with low levels of FFA, UCP1 activity is low. FFA activate UCP1 and thermogensis because mitochondria lacking UCP1 are an order of magnitude less sensitive to FFA than wild type mitochondria and non-shivering thermogenesis is severely impaired or absent in UCP1-deficient mice (Shabalina et al., 2002). It is as yet unclear exactly how FFA activate UCP1, but they may function as cofactors, as allosteric regulators or as proton shuttles.
The Importance of the Location, Vascular Supply and Innervation of BAT
In the most commonly studied laboratory rodents (rats, mice, hamsters and gerbils) BAT is located in identifiable clusters (e.g., interscapular, surrounding the kidneys and aorta, and intercostal), and brown adipocytes may also be distributed diffusely within white adipose tissue (Cannon and Nedergaard, 2004). BAT is highly vascularized (Figure 1b), which maximizes the transfer of heat to the tissues perfused by the blood that passes through the BAT including the vital organs of the thorax and nervous system (Smith, 1964). The vascularization of BAT is increased in response to cold exposure by a mechanism involving stimulation of angiogenesis by SNS activation (Asano et al., 1997). Vascular endothelial cell growth factor is believed to mediate angiogenesis in BAT (Tonello et al., 1999; Fredriksson et al., 2005). In addition, it has been reported that insulin can stimulate capillary growth in BAT in rats exposed to high temperatures (Yamashita et al., 1992).
BAT is innervated by axons of the SNS which use norepinephrine (NE) as a neurotransmitter (Trayhurn and Ashwell, 1987; Cannon and Nedergaard, 2004). By activating β- or α-adrenergic receptors, NE controls the processes of thermogenesis of differentiated brown adipocytes, and the generation of new brown adipocytes from progenitor cells within the BAT. Activation of β3 adrenoreceptors by norepinephrine induces thermogenesis in BAT; cold temperatures and overeating are examples of stimuli that can activate the SNS and BAT β3 receptors. The events that occur upon β3 receptor activation that result in increased thermogenesis include activation of the GTP-binding protein Gs, resulting in adenylate cyclase activation and cyclic AMP production (Figure 3). The cyclic AMP-dependent protein kinase (PKA) is then activated and phosphorylates several protein substrates that are believed to mediate the changes that increase mitochondrial uncoupling; these substrates include the transcription factor CREB (cyclic AMP response element binding protein), the protein phosphatase DARPP, and proteins involved in fatty acid hydrolysis. Collectively, these and other, yet-to-be identified, PKA substrates induce the expression of UCP1, increase UCP1 activity and provide energy substrates for thermogenesis.
In addition, to the acute effects of norepinephrine receptor activation on brown adipocyte lipolysis and mitochondrial uncoupling, several growth factors may mediate long-term changes in BAT cellular composition. For example, basic fibroblast growth factor promotes the enlargement of BAT during long-term acclimation to cold temperatures (Yamashita et al., 1994), in part by stimulating the proliferation of brown adipose progenitor cells (Konishi et al., 2000). Insulin-like growth factor 1 can also induce the proliferation of pre-adipocytes in BAT (Porras et al., 1998). Norepinephrine stimulates the production of some of the growth factors that increase the production and growth of brown adipocytes (Garcia and Obregon, 1997).
While the function of BAT located in discrete depots in the thorax is well-established, recent findings suggests that brown adipocyte progenitor cells, and possibly differentiated brown adipocytes, exist as scattered individual cells and small cell clusters distributed diffusely within skeletal muscle tissue (Crisan et al., 2008; Wijers et al., 2008). Studies of embryonic development have revealed surprising evidence that brown adipocytes arise from a myocte progenitor, whereas white fat cells arise from a distinct cell lineage (Timmons et al., 2007; Seale et al., 2008). However, overexpression of the zinc-finger protein PRDM16 in white fat depots induces the formation of brown adipocytes, whereas depletion of PRDM16 (using small interfering RNAs) in brown fat cells causes them to stop expressing UCP1 and lose their thermogenic capability (Seale et al., 2007). The presence of brown adipocyte progenitor cells in skeletal muscle, and the ability to induce brown adipocyte differentiation in vivo, suggest the possibility of a therapeutic approach for obesity based on brown adipocyte-mediated energy expenditure.
Evidence that BAT Thermogenesis can Prevent Obesity
From an evolutionary perspective it is believed that BAT is an adaptation to cold climates that allows maintenance of body temperature to support the temperature-dependent physiology of homeotherms. On the other hand, expending energy to generate heat comes at the expense of energy needed to maintain body mass that can be critical for survival during extended periods of food scarcity. It would therefore be expected that species and individuals with low amounts of BAT would be prone to excessive weight gain under conditions of plentiful food supplies. In support of such a possibility in humans, a genetic association study found that a polymorphism in the UCP1 promoter that reduces UCP1 gene expression is associated with a higher BMI and abdominal obesity (Sramkova et al., 2007). It will be of interest to compare levels of BAT in individuals who maintain a low body weight with those who are either obese or predisposed (as the result of genetic or environmental factors) to obesity.
Studies of animals lacking BAT or UCP1 have clearly demonstrated the ability of BAT thermogenesis to protect against diet-induced obesity. For example, a laboratory in Germany generated mice in which diphtheria toxin A-chain was expressed under the control of a UCP1 promoter, resulting in the selective destruction of brown adipocytes (Hamann et al., 1998). The BAT-deficient mice were obese and exhibited reduced energy expenditure and insulin resistance; they were hypersensitive to severe obesity when maintained on a high fat diet. Mice lacking UCP1 exhibit a marked attenuation of SNS-induced fatty acid utilization and non-shivering thermogenesis, which is particularly striking and leads to obesity when the mice are fed a high fat diet (Kontani et al., 2005). However, data in the latter study showed that when UCP1 deficient mice are maintained on a normal diet at usual warm housing temperature they do not develop obesity and appear to live a normal lifespan. In another study, noradrenergic input to BAT was eliminated in mice by disruption of the dopamine β-hydroxylase gene (Thomas and Palmiter, 1997). The mice were hyperphagic and exhibited extreme sensitivity to cold temperatures, but did not become obese because their basal metabolic rate was elevated. In addition, whereas treatment of wild type mice with a β3-adrenergic agonist (CL-316,243) increased whole body oxygen consumption and brown fat temperature, it failed to do so in UCP1-deficient mice (Inokuma et al., 2006). Moreover, growth hormone signaling-deficient long-lived dwarf mice exhibit increased levels of BAT and UCP1, and have a lower BMI, compared to their wild-type counterparts (Li et al., 2003).
Studies in which inter-cellular signaling mechanisms involved in the generation and differentiation of brown adipocytes have been manipulated further support a role for UCP1 in preventing obesity. Bone morphogenic protein 7 (BMP7) can stimulate the differentiation of brown adipose progenitor cells, and multipotent mesenchymal stem cells into mature brown adipocytes that express relatively high levels of PGC1α and UCP1 (Tseng et al., 2008). Mouse embryos lacking BMP7 exhibit negligible brown fat and little or no UCP1. Overexpression of BMP7 in mice results in an increased BAT, increased energy expenditure and reduced body weight. Cytokines may also influence BAT. For example, treatment of rats with interleukin-15 resulted in increased expression of genes encoding UCP1, PPARδ, fatty acid translocase and fatty acid transport protein in BAT (Almendro et al., 2008).
One of the most interesting animal studies, with major implications for the ongoing dramatic increase in childhood obesity, involved a model in which rat pups gained weight excessively as the result of rearing in small litters (Xiao et al., 2007). Compared with rats raised in a normal litter size (8 pups per litter), rats raised in a small litter (3 pups per litter) exhibited excess weight gain, reduced thermogenic capacity and lower levels of UCP1 as adults. BAT from small litter size rats was less active and less responsive to cold compared to BAT from normal litter size rats. Consistent with reduced BAT thermogenic activity, the overweight pups exhibited reduced expression of several lipid lipases, and reduced SNS responsiveness to isoproterenol. Collectively, the findings in this study suggest that over-nourished young animals possess relatively low amounts of BAT and impaired SNS activity relative to young animals with a lower dietary energy intake. The resulting deficit in BAT/thermogenic capacity may contribute to a lifelong propensity for obesity.
Dietary Energy Intake, BAT and Aging
Dietary energy restriction has been conclusively shown to increase the average and maximum lifespan of a range of mammals including non-human primates, and data from epidemiological and clinical studies support similar benefits of dietary energy restriction in humans (Berner and Stern, 2004; Redman and Ravussin, 2009). Considerable evidence suggests that dietary energy restriction during adult life reduces the risk for development of diabetes, cardiovascular disease, neurodegenerative disorders and many types of cancer (Fontana, 2008; Howell et al., 2009). Two major mechanisms have been proposed for the “anti-aging” effects of dietary energy restriction: 1) reduced production of mitochondrial free radicals (Hunt et al., 2006); and 2) increased production of cellular stress resistance proteins (Mattson, 2008).
In addition to its beneficial effects on the cardiovascular and musculoskeletal systems, dietary energy restriction has been shown to improve the functionality, and resistance to aging and disease, of the nervous system. For example, life long dietary energy restriction ameliorates the age-related decline in cognitive function in rodents (Komatsu et al., 2008). Moreover, long-term (months) dietary energy restriction protects neurons in the brain against dysfunction and degeneration in animal models of stroke, Alzheimer's disease, Parkinson's disease and Huntington's disease (Bruce-Keller et al., 1999; Yu and Mattson, 1999; Maswood et al., 2004; Duan et al., 2003; Halagappa et al., 2007; Arumugam et al., 2009).
While dietary energy restriction has profound beneficial effects on health and longevity, increased energy expenditure by regular exercise confers similar, albeit typically less pronounced health benefits (Lanza et al., 2008). Exercise reduces the risk for diabetes and cardiovascular disease, and so may extend average lifespan, particularly in individuals genetically predisposed to the insulin resistance syndrome (Lakka and Laaksonen, 2007). Providing rats or mice access to a running wheel increases their resistance to impaired neurogenesis, synaptic dysfunction and cognitive impairment as they grow old (van Praag, 2008). Exercise imposes a transient stress on muscle and cardiovascular cells, as well as on cells in other organs including the brain. This cellular stress activates adaptive stress response pathways that result in increased production of proteins that protect cells against stress including growth factors (IGF-1 and BDNF, for example), protein chaperones and antioxidant enzymes (Berg and Bang, 2004; Marini et al., 2007; Stranahan et al., 2009). Factors that promote angiogenesis, including VEGF, are also increased in muscle and brain tissues in response to exercise (Prior et al., 2004; Ding et al., 2006).
BAT and UCP1 may be regulated by molecular mechanisms believed to play fundamental roles in responses of cells and organisms to changes in energy intake and expenditure. Sirtuins (SIRT1 – SIRT7) are a family of histone deacetylases, several of which appear to mediate cellular responses to energetic and oxidative stress. SIRT1, an NAD+-dependent deacetylase, has been shown to extend longevity in yeast, worms and flies (Guarente and Picard, 2005), and recent findings suggest that activation of SIRT1 may counteract the adverse effects of obesity on health in mice (Baur et al., 2006). Treatment of mice with SRT1720, a specific chemical activator of SIRT1, resulted in enhanced lipid oxidation in BAT, skeletal muscle and liver cells, and protected the mice against diet-induced insulin resistance and obesity (Feige et al., 2008). The latter study provided evidence that SIRT1 activation controls fatty acid oxidation by inducing a genetic network involving PGC-1α, FOXO1, p53 and AMPK. The mitochondria-associated histone deacetylase SIRT3 may play an important role in the regulation of mitochondrial energy metabolism and thermogenesis in BAT. Overexpression of SIRT3 results in increased expression of UCP1, while inhibition of SIRT3 results in decreased UCP1 expression (Shi et al., 2005). The pathway downstream of SIRT3 that promotes mitochondrial uncoupling involves PGC1α and the transcription factor CREB. Interestingly, SIRT3 levels are reduced in BAT from several lines of genetically obese mice (Shi et al., 2005), suggesting a role for impaired SIRT3-mediated signaling in obesity.
The possibility that BAT can protect the body against the aging process is suggested by several additional findings. BAT in humans is responsive to cold temperatures, and this responsiveness is diminished during aging. Thus, when exposed to a temperature of 19°C for 2 hours, 17 of 32 young subjects and only 2 of 24 older subjects exhibited an increased in radiolabeled glucose uptake into supraclavicular and paraspinal adipose tissue (Saito et al., 2009). Interestingly, the activity of this presumptive BAT was inversely related to body mass index (BMI) and visceral fat mass, consistent with both an increased need for non-shivering thermogenesis in skinny individuals and the notion that energy expended by BAT can contribute to maintenance of a low BMI and visceral fat mass. Moreover, the findings of Saito et al. (2009) suggest that depletion of BAT may contribute to the increased susceptibility of elderly individuals to hypothermia.
Animal studies support roles for diminished BAT activity in aging. Lifelong dietary energy restriction, which retards aging, attenuates the age-related decline in mitochondrial mass, cyclooxygenase activity and uncoupling levels in BAT of rats (Valle et al., 2008). BAT-mediated non-shivering thermogenesis induced by either cold or norepinephrine is significantly decreased in old compared to young rats, with the effect of age on BAT activity being greater in males than in females (McDonald and Horwitz, 1999). The diminished non-shivering thermogenesis during aging is apparently the result of changes in the brown adipose cells themselves, rather than an effect of age on the SNS. In light of the inverse relationship between levels of brown and white adipose tissue, the decline in BAT during aging may contribute to increased visceral adiposity which typically occurs during aging. Data in the study of McDonald and Horwitz (1999) further suggest that a loss of functional BAT late in life may predispose the elderly to hypothermia. Changes in circulating levels of sex and thyroid hormones may contribute to the age-related decline in BAT. For example, a study of young, middle-age and old rats showed that BAT atrophy occurs late in life and is correlated with a decrease in triiodothyronine levels, but not with changes in sex steroids (Valle et al., 2008).
The evidence that BAT can protect against multiple age-related diseases is substantial, albeit mostly circumstantial. The argument is as follows (Figure 2): 1) individuals with low BAT levels are prone to excess accumulation of white adipose tissue and weight gain (Hansen and Kristiansen, 2006); 2) visceral adiposity and obesity are strongly associated with the risk for several major age-related diseases including type 2 diabetes, cardiovascular disease, stroke and some cancers (Phillips and Prins, 2008); 3) dietary energy restriction, which increases levels of BAT, protects against diabetes, cardiovascular disease, stroke, cancers (Varady and Hellerstein, 2007; Stranahan and Mattson, 2008) and possibly neurodegenerative disorders such as Alzheimer's disease (Maalouf et al., 2009); and 4) BAT removal promotes white adipose tissue growth and weight gain (Stephens et al., 1981; Stern et al., 1984).
Figure 2.
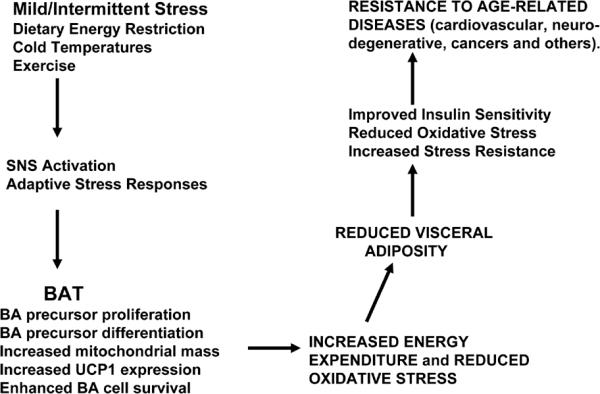
Possible mechanisms by which BAT and mitochondrial uncoupling can protect against obesity and age-related diseases. See text for discussion.
While preventing excessive weight gain and visceral adiposity is likely a major mechanism by which BAT activity might protect against age-related disease, another mechanism may involve enhancement of adaptive responses to stress. Environmental manipulations that increase BAT mass/activity have also been shown to increase cellular stress resistance in several different organ systems. When male rats were subjected to a daily 3 hour immobilization stress for 1–8 weeks, they exhibited lower body weights and improved cold tolerance compared to a non-stress control group of rats (Kuroshima et al., 1984). Non-shivering thermogenesis was increased in the stressed rats, and this was associated with an increase in BAT content and increased numbers of mitochondria in brown adipocytes. Interestingly, plasma insulin levels were decreased in response to repeated daily mild stress, suggesting a beneficial effect of the intermittent stress. It therefore appears that moderate intermittent stress can stimulate BAT growth and thermogenic activity, and this response to stress may be attenuated in old age.
Can BAT be Targeted for the Prevention and Treatment of Age-Related Diseases in Humans?
Compared to adult rodents which exhibit considerable BAT, adult humans possess very little BAT. Although functionally BAT has not been detected using radiotracer imaging methods in the majority of human subjects examined, evidence was obtained suggesting that at least a small percentage of adult humans possess BAT (Nedergaard et al., 2007; Cypess et al., 2009; van Marken et al., 2009; Virtanen et al., 2009). A histological analysis of neck adipose tissue samples obtained from patients undergoing thyroid surgery revealed the presence of islands of UCP1 immunoreactive brown adipocytes that were innervated by SNS axons (Zingaretti et al., 2009). In addition, cells exhibiting characteristics of brown adipocyte precursors were present adjacent to capillaries in the presumptive BAT. The latter study further showed that neck BAT is present in greater amounts in younger and leaner subjects compared to older overweight subjects. Dietary regimens that reduce overall energy intake (e.g., controlled daily calorie restriction and alternate day calorie restriction or fasting) would therefore be expected to increase levels of BAT while decreasing levels of white adipose cells. While there are undoubtedly BAT-independent mechanisms by which dietary energy restriction improves health and extends lifespan, BAT-mediated thermogenic energy expenditure may contribute to the “anti-aging” effect of dietary energy restriction (Fried et al., 1983). For animals living in cold climates, an increase in BAT levels during times of limited food supply may provide a mechanism for maintenance of body temperature.
Two approaches to increase BAT thermogenic potential are to increase the mass of BAT (i.e., numbers of brown adipocytes) and to increase the thermogenic capacity of individual brown adipocytes. Studies using animal models have shown that both approaches are possible. For example, Kajimura et al. (2009) reported the intriguing finding that myoblasts (muscle cell precursor cells) and fibroblasts can be induced to form brown adipocytes by overexpression of the zinc finger protein PRDM16 and the transcription regulator C/EBP-β. Moreover, they showed that the brown adipocytes generated from fibroblasts can be successfully transplanted into adult mice where they exhibit thermogenic activity. The latter study provides experimental evidence to support the possibility of taking fibroblasts from a person, transducing them into brown adipocytes in culture, and then transplanting them back into the same person (an approach expected to eliminate immune rejection of the transplanted cells).
The uncoupling/thermogenic activity of BAT can be increased by intermittent exposures to cold temperatures and by dietary energy restriction. Applications of “cold temperature therapy” to humans have, to my knowledge, not been applied to disorders such as diabetes and obesity that would be expected to benefit from increased BAT activity. While dietary energy restriction and regular exercise can cure obesity, compliance with such practical approaches is typically low, and so efforts have been made to develop pharmacological interventions that suppress appetite or increase energy expenditure. One such approach has been to activate the SNS with drugs such as ephedrine (Astrup, 1986). However, while clearly increasing energy expenditure, long-term activation of the SNS has numerous adverse side effects on the cardiovascular, nervous and other organ systems (Haller and Benowitz, 2000).
The discoveries of BAT and mitochondrial uncoupling proteins have important implications for novel therapeutic interventions for diseases of aging that may extend to most types of cells in the body. Mitochondrial uncoupling not only consumes energy to generate heat, it also reduces the production of potentially damaging reactive oxygen species. Studies of experimental models of ischemic stroke (Mattiasson et al., 2003; Vincent et al., 2004; Liu et al., 2006) and Parkinson's disease (Conti et al., 2005) have provided evidence that uncoupling proteins (UCP2 and UCP4) can protect neurons against dysfunction and degeneration. UCP4, a neuron-specific uncoupling protein, has been shown to protect neurons by decreasing free radical production and stabilizing cellular calcium homeostasis (Chan et al., 2006). UCP4 activity can also induce an adaptive shift in energy metabolism that helps sustain neurons under conditions of metabolic compromise (Liu et al., 2006). Interestingly, dietary energy restriction up-regulates UCP4 expression in the brain, which may contribute to the neuroprotective effects of low energy diets (Liu et al., 2006). Mild uncoupling in neurons might also be accomplished by treatments with uncoupling agents such as 2, 4-dinitrophenol (DNP) and n-trifluoromethoxycarbonycyanidephenylhydrazone (FCCP) which have been reported to lessen brain damage and improve functional outcomes in animal models of stroke and traumatic brain injury (Korde et al., 2005; Pandya et al., 2007).
Activation of thyroid hormone receptors in brown adipose cells and their precursors may be another approach to increase BAT cell mass and thermogenesis. As evidence, treatment with a thyroid receptor β-selective agonist resulted in increased resistance to diet-induced obesity in rats (Amorim et al., 2009). Rats treated with the thyroid receptor agonist exhibited increased energy expenditure, reduced adiposity and triglyceride levels, and increased insulin sensitivity.
Fasting induces the expression of mitochondrial uncoupling proteins and PPAR-γ in BAT of rats (Kageyama et al., 2003). Several studies have demonstrated the ability of PPAR-γ (peroxisome proliferator-activated receptor γ) agonists to stimulate both the production and thermogenic activity of brown adipose cells. For example, PPAR-γ agonists suppressed visceral white adipose genes while inducing the expression of brown adipose genes (including UCP1) in white adipocytes (Vernochet et al., 2009). Another study demonstrated the ability of the thiazolidinedione darglitazone (a PPAR-γ agonist) to induce the proliferation of BAT in rats and monkeys (Aleo et al., 2003), although major adverse side effects also occurred. Petrovic et al. (2008) reported that treatment with a PPAR-γ agonist caused brown adipose progenitor cells to differentiate into thermogenically competent, SNS responsive, brown adipocytes. Therefore, it may be possible to develop more selective and potent PPAR-γ agonists that stimulate BAT growth and thermogenic activity.
Diet and lifestyle changes, specific regimens of mild intermittent stress, drugs that stimulate BAT formation and activity, induction of brown adipose cell progenitors in muscle and other tissues, and transplantation of brown adipose cells are all potential means of increasing BAT mass and thermogenic activity. In addition, stimulation of UCP expression in non-adipose cells (neurons, muscle cells, hepatocytes and others) may prove an effective means of protecting those cells against severe stress and the adversities of aging. However, while studies of such approaches in animal models of obesity and age-related diseases appear promising, their efficacy and safety in human subjects remains to be established.
References
- Aleo MD, Lundeen GR, Blackwell DK, Smith WM, Coleman GL, Stadnicki SW, Kluwe WM. Mechanism and implications of brown adipose tissue proliferation in rats and monkeys treated with the thiazolidinedione darglitazone, a potent peroxisome proliferator-activated receptor-gamma agonist. J Pharmacol Exp Ther. 2003;305:1173–1182. doi: 10.1124/jpet.102.042648. [DOI] [PubMed] [Google Scholar]
- Almendro V, Fuster G, Busquets S, Ametller E, Figueras M, Argilés JM, López-Soriano FJ. Effects of IL-15 on rat brown adipose tissue: uncoupling proteins and PPARs. Obesity (Silver Spring) 2008;16:285–289. doi: 10.1038/oby.2007.47. [DOI] [PubMed] [Google Scholar]
- Amorim BS, Ueta CB, Freitas BC, Nassif RJ, Gouveia CH, Christoffolete MA, Moriscot AS, Lancelloti CL, Llimona F, Barbeiro HV, de Souza HP, Catanozi S, Passarelli M, Aoki MS, Bianco AC, Ribeiro MO. A TRbeta-selective agonist confers resistance to diet-induced obesity. J Endocrinol. 2009;203:291–299. doi: 10.1677/JOE-08-0539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arumugam TV, Phillips TM, Cheng A, Morrell CH, Mattson MP, Wan R. Age and energy intake interact to modify cell stress pathways and stroke outcome. Ann. Neurol. 2009 doi: 10.1002/ana.21798. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano A, Morimatsu M, Nikami H, Yoshida T, Saito M. Adrenergic activation of vascular endothelial growth factor mRNA expression in rat brown adipose tissue: implication in cold-induced angiogenesis. Biochem J. 1997;328:179–183. doi: 10.1042/bj3280179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astrup A. Thermogenesis in human brown adipose tissue and skeletal muscle induced by sympathomimetic stimulation. Acta Endocrinol Suppl (Copenh) 1986;278:1–32. [PubMed] [Google Scholar]
- Bamshad M, Song CK, Bartness TJ. CNS origins of the sympathetic nervous system outflow to brown adipose tissue. Am J Physiol. 1999;276:R1569–1578. doi: 10.1152/ajpregu.1999.276.6.R1569. [DOI] [PubMed] [Google Scholar]
- Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, Pistell PJ, Poosala S, Becker KG, Boss O, Gwinn D, Wang M, Ramaswamy S, Fishbein KW, Spencer RG, Lakatta EG, Le Couteur D, Shaw RJ, Navas P, Puigserver P, Ingram DK, de Cabo R, Sinclair DA. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg U, Bang P. Exercise and circulating insulin-like growth factor I. Horm Res. 2004;62(Suppl 1):50–58. doi: 10.1159/000080759. [DOI] [PubMed] [Google Scholar]
- Berner YN, Stern F. Energy restriction controls aging through neuroendocrine signal transduction. Ageing Res Rev. 2004;3:189–198. doi: 10.1016/j.arr.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Brand MD. The efficiency and plasticity of mitochondrial energy transduction. Biochem Soc Trans. 2005;33:897–904. doi: 10.1042/BST0330897. [DOI] [PubMed] [Google Scholar]
- Bruce-Keller AJ, Umberger G, McFall R, Mattson MP. Food restriction reduces brain damage and improves behavioral outcome following excitotoxic and metabolic insults. Ann Neurol. 1999;45:8–15. [PubMed] [Google Scholar]
- Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. 2004;84:277–359. doi: 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- Chan SL, Liu D, Kyriazis GA, Bagsiyao P, Ouyang X, Mattson MP. Mitochondrial uncoupling protein-4 regulates calcium homeostasis and sensitivity to store depletion-induced apoptosis in neural cells. J Biol Chem. 2006;281:37391–37403. doi: 10.1074/jbc.M605552200. [DOI] [PubMed] [Google Scholar]
- Conti B, Sugama S, Lucero J, Winsky-Sommerer R, Wirz SA, Maher P, Andrews Z, Barr AM, Morale MC, Paneda C, Pemberton J, Gaidarova S, Behrens MM, Beal F, Sanna PP, Horvath T, Bartfai T. Uncoupling protein 2 protects dopaminergic neurons from acute 1,2,3,6-methyl-phenyl-tetrahydropyridine toxicity. J Neurochem. 2005;93:493–501. doi: 10.1111/j.1471-4159.2005.03052.x. [DOI] [PubMed] [Google Scholar]
- Crisan M, Casteilla L, Lehr L, Carmona M, Paoloni-Giacobino A, Yap S, Sun B, Léger B, Logar A, Pénicaud L, Schrauwen P, Cameron-Smith D, Russell AP, Péault B, Giacobino JP. A reservoir of brown adipocyte progenitors in human skeletal muscle. Stem Cells. 2008;26:2425–2433. doi: 10.1634/stemcells.2008-0325. [DOI] [PubMed] [Google Scholar]
- Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, Kuo FC, Palmer EL, Tseng YH, Doria A, Kolodny GM, Kahn CR. Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 2009;360:1509–1517. doi: 10.1056/NEJMoa0810780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding YH, Li J, Zhou Y, Rafols JA, Clark JC, Ding Y. Cerebral angiogenesis and expression of angiogenic factors in aging rats after exercise. Curr Neurovasc Res. 2006;3:15–23. doi: 10.2174/156720206775541787. [DOI] [PubMed] [Google Scholar]
- Duan W, Guo Z, Jiang H, Ware M, Li XJ, Mattson MP. Dietary restriction normalizes glucose metabolism and BDNF levels, slows disease progression, and increases survival in huntingtin mutant mice. Proc Natl Acad Sci USA. 2003;100:2911–2916. doi: 10.1073/pnas.0536856100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducharme NA, Bickel PE. Lipid droplets in lipogenesis and lipolysis. Endocrinology. 2008;149:942–949. doi: 10.1210/en.2007-1713. [DOI] [PubMed] [Google Scholar]
- Dulloo AG, Samec S. Uncoupling proteins: their roles in adaptive thermogenesis and substrate metabolism reconsidered. Br J Nutr. 2001;86:123–139. doi: 10.1079/bjn2001412. [DOI] [PubMed] [Google Scholar]
- Feige JN, Lagouge M, Canto C, Strehle A, Houten SM, Milne JC, Lambert PD, Mataki C, Elliott PJ, Auwerx J. Specific SIRT1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab. 2008;8:347–358. doi: 10.1016/j.cmet.2008.08.017. [DOI] [PubMed] [Google Scholar]
- Fontana L. Calorie restriction and cardiometabolic health. Eur J Cardiovasc Prev Rehabil. 2008;15:3–9. doi: 10.1097/HJR.0b013e3282f17bd4. [DOI] [PubMed] [Google Scholar]
- Fredriksson JM, Nikami H, Nedergaard J. Cold-induced expression of the VEGF gene in brown adipose tissue is independent of thermogenic oxygen consumption. FEBS Lett. 2005;579:5680–5684. doi: 10.1016/j.febslet.2005.09.044. [DOI] [PubMed] [Google Scholar]
- Fried SK, Hill JO, Nickel M, DiGirolamo M. Novel regulation of lipoprotein lipase activity in rat brown adipose tissue: effects of fasting and caloric restriction during refeeding. J Nutr. 1983;113:1870–1874. doi: 10.1093/jn/113.9.1870. [DOI] [PubMed] [Google Scholar]
- Garcia B, Obregon MJ. Norepinephrine potentiates the mitogenic effect of growth factors in quiescent brown preadipocytes: relationship with uncoupling protein messenger ribonucleic acid expression. Endocrinology. 1997;138:4227–4233. doi: 10.1210/endo.138.10.5455. [DOI] [PubMed] [Google Scholar]
- Guarente L, Picard F. Calorie restriction--the SIR2 connection. Cell. 2005;120:473–482. doi: 10.1016/j.cell.2005.01.029. [DOI] [PubMed] [Google Scholar]
- Halagappa VK, Guo Z, Pearson M, Matsuoka Y, Cutler RG, Laferla FM, Mattson MP. Intermittent fasting and caloric restriction ameliorate age-related behavioral deficits in the triple-transgenic mouse model of Alzheimer's disease. Neurobiol Dis. 2007;26:212–220. doi: 10.1016/j.nbd.2006.12.019. [DOI] [PubMed] [Google Scholar]
- Haller CA, Benowitz NL. Adverse cardiovascular and central nervous system events associated with dietary supplements containing ephedra alkaloids. N Engl J Med. 2000;343:1833–1838. doi: 10.1056/NEJM200012213432502. [DOI] [PubMed] [Google Scholar]
- Hamann A, Flier JS, Lowell BB. Obesity after genetic ablation of brown adipose tissue. Z Ernahrungswiss. 1998;37(Suppl 1):1–7. [PubMed] [Google Scholar]
- Hansen JB, Kristiansen K. Regulatory circuits controlling white versus brown adipocyte differentiation. Biochem J. 2006;398:153–168. doi: 10.1042/BJ20060402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell A, Chapman M, Harvie M. Energy restriction for breast cancer prevention. Recent Results Cancer Res. 2009;181:97–111. doi: 10.1007/978-3-540-69297-3_11. [DOI] [PubMed] [Google Scholar]
- Hunt ND, Hyun DH, Allard JS, Minor RK, Mattson MP, Ingram DK, de Cabo R. Bioenergetics of aging and calorie restriction. Ageing Res Rev. 2006;5:125–143. doi: 10.1016/j.arr.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Inokuma K, Okamatsu-Ogura Y, Omachi A, Matsushita Y, Kimura K, Yamashita H, Saito M. Indispensable role of mitochondrial UCP1 for antiobesity effect of beta3-adrenergic stimulation. Am J Physiol Endocrinol Metab. 2006;290:E1014–1021. doi: 10.1152/ajpendo.00105.2005. [DOI] [PubMed] [Google Scholar]
- Jastroch M, Withers KW, Taudien S, Frappell PB, Helwig M, Fromme T, Hirschberg V, Heldmaier G, McAllan BM, Firth BT, Burmester T, Platzer M, Klingenspor M. Marsupial uncoupling protein 1 sheds light on the evolution of mammalian nonshivering thermogenesis. Physiol Genomics. 2008;32:161–169. doi: 10.1152/physiolgenomics.00183.2007. [DOI] [PubMed] [Google Scholar]
- Kageyama H, Osaka T, Kageyama A, Kawada T, Hirano T, Oka J, Miura M, Namba Y, Ricquier D, Shioda S, Inoue S. Fasting increases gene expressions of uncoupling proteins and peroxisome proliferator-activated receptor-gamma in brown adipose tissue of ventromedial hypothalamus-lesioned rats. Life Sci. 2003;72:3035–3046. doi: 10.1016/s0024-3205(03)00225-x. [DOI] [PubMed] [Google Scholar]
- Kajimura S, Seale P, Kubota K, Lunsford E, Frangioni JV, Gygi SP, Spiegelman BM. Initiation of myoblast to brown fat switch by a PRDM16-C/EBP-beta transcriptional complex. Nature. 2009;460:1154–1158. doi: 10.1038/nature08262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu T, Chiba T, Yamaza H, Yamashita K, Shimada A, Hoshiyama Y, Henmi T, Ohtani H, Higami Y, de Cabo R, Ingram DK, Shimokawa I. Manipulation of caloric content but not diet composition, attenuates the deficit in learning and memory of senescence-accelerated mouse strain P8. Exp Gerontol. 2008;43:339–346. doi: 10.1016/j.exger.2008.01.008. [DOI] [PubMed] [Google Scholar]
- Konishi M, Mikami T, Yamasaki M, Miyake A, Itoh N. Fibroblast growth factor-16 is a growth factor for embryonic brown adipocytes. J Biol Chem. 2000;275:12119–12122. doi: 10.1074/jbc.275.16.12119. [DOI] [PubMed] [Google Scholar]
- Kontani Y, Wang Y, Kimura K, Inokuma KI, Saito M, Suzuki-Miura T, Wang Z, Sato Y, Mori N, Yamashita H. UCP1 deficiency increases susceptibility to diet-induced obesity with age. Aging Cell. 2005;4:147–155. doi: 10.1111/j.1474-9726.2005.00157.x. [DOI] [PubMed] [Google Scholar]
- Korde AS, Pettigrew LC, Craddock SD, Maragos WF. The mitochondrial uncoupler 2,4-dinitrophenol attenuates tissue damage and improves mitochondrial homeostasis following transient focal cerebral ischemia. J Neurochem. 2005;94:1676–1684. doi: 10.1111/j.1471-4159.2005.03328.x. [DOI] [PubMed] [Google Scholar]
- Kuroshima A, Habara Y, Uehara A, Murazumi K, Yahata T, Ohno T. Cross adaption between stress and cold in rats. Pflugers Arch. 1984;402:402–408. doi: 10.1007/BF00583941. [DOI] [PubMed] [Google Scholar]
- Lafontan M, Girard J. Impact of visceral adipose tissue on liver metabolism. Part I: heterogeneity of adipose tissue and functional properties of visceral adipose tissue. Diabetes Metab. 2008;34:317–327. doi: 10.1016/j.diabet.2008.04.001. [DOI] [PubMed] [Google Scholar]
- Lakka TA, Laaksonen DE. Physical activity in prevention and treatment of the metabolic syndrome. Appl Physiol Nutr Metab. 2007;32:76–88. doi: 10.1139/h06-113. [DOI] [PubMed] [Google Scholar]
- Lanza IR, Short DK, Short KR, Raghavakaimal S, Basu R, Joyner MJ, McConnell JP, Nair KS. Endurance exercise as a countermeasure for aging. Diabetes. 2008;57:2933–2942. doi: 10.2337/db08-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Knapp JR, Kopchick JJ. Enlargement of interscapular brown adipose tissue in growth hormone antagonist transgenic and in growth hormone receptor gene-disrupted dwarf mice. Exp Biol Med (Maywood) 2003;228:207–215. doi: 10.1177/153537020322800212. [DOI] [PubMed] [Google Scholar]
- Liu D, Chan SL, de Souza-Pinto NC, Slevin JR, Wersto RP, Zhan M, Mustafa K, de Cabo R, Mattson MP. Mitochondrial UCP4 mediates an adaptive shift in energy metabolism and increases the resistance of neurons to metabolic and oxidative stress. Neuromolecular Med. 2006;8:389–414. doi: 10.1385/NMM:8:3:389. [DOI] [PubMed] [Google Scholar]
- Maalouf M, Rho JM, Mattson MP. The neuroprotective properties of calorie restriction, the ketogenic diet, and ketone bodies. Brain Res Rev. 2009;59:293–315. doi: 10.1016/j.brainresrev.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marini M, Lapalombella R, Margonato V, Ronchi R, Samaja M, Scapin C, Gorza L, Maraldi T, Carinci P, Ventura C, Veicsteinas A. Mild exercise training, cardioprotection and stress genes profile. Eur J Appl Physiol. 2007;99:503–510. doi: 10.1007/s00421-006-0369-4. [DOI] [PubMed] [Google Scholar]
- Maswood N, Young J, Tilmont E, Zhang Z, Gash DM, Gerhardt GA, Grondin R, Roth GS, Mattison J, Lane MA, Carson RE, Cohen RM, Mouton PR, Quigley C, Mattson MP, Ingram DK. Caloric restriction increases neurotrophic factor levels and attenuates neurochemical and behavioral deficits in a primate model of Parkinson's disease. Proc Natl Acad Sci USA. 2004;101:18171–18176. doi: 10.1073/pnas.0405831102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattiasson G, Shamloo M, Gido G, Mathi K, Tomasevic G, Yi S, Warden CH, Castilho RF, Melcher T, Gonzalez-Zulueta M, Nikolich K, Wieloch T. Uncoupling protein-2 prevents neuronal death and diminishes brain dysfunction after stroke and brain trauma. Nat Med. 2003;9:1062–1068. doi: 10.1038/nm903. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Dietary factors, hormesis and health. Ageing Res Rev. 2008;7:43–48. doi: 10.1016/j.arr.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Gleichmann M, Cheng A. Mitochondria in neuroplasticity and neurological disorders. Neuron. 2008;60:748–766. doi: 10.1016/j.neuron.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald RB, Horwitz BA. Brown adipose tissue thermogenesis during aging and senescence. J Bioenerg Biomembr. 1999;31:507–516. doi: 10.1023/a:1005404708710. [DOI] [PubMed] [Google Scholar]
- Nedergaard J, Bengtsson T, Cannon B. Unexpected evidence for active brown adipose tissue in adult humans. Am J Physiol Endocrinol Metab. 2007;293:E444–452. doi: 10.1152/ajpendo.00691.2006. [DOI] [PubMed] [Google Scholar]
- Pandya JD, Pauly JR, Nukala VN, Sebastian AH, Day KM, Korde AS, Maragos WF, Hall ED, Sullivan PG. Post-Injury Administration of Mitochondrial Uncouplers Increases Tissue Sparing and Improves Behavioral Outcome following Traumatic Brain Injury in Rodents. J Neurotrauma. 2007;24:798–811. doi: 10.1089/neu.2006.3673. [DOI] [PubMed] [Google Scholar]
- Petrovic N, Shabalina IG, Timmons JA, Cannon B, Nedergaard J. Thermogenically competent nonadrenergic recruitment in brown preadipocytes by a PPARgamma agonist. Am J Physiol Endocrinol Metab. 2008;295:E287–296. doi: 10.1152/ajpendo.00035.2008. [DOI] [PubMed] [Google Scholar]
- Phillips LK, Prins JB. The link between abdominal obesity and the metabolic syndrome. Curr Hypertens Rep. 2008;10:156–164. doi: 10.1007/s11906-008-0029-7. [DOI] [PubMed] [Google Scholar]
- Porras A, Alvarez AM, Valladares A, Benito M. p42/p44 mitogen-activated protein kinases activation is required for the insulin-like growth factor-I/insulin induced proliferation, but inhibits differentiation, in rat fetal brown adipocytes. Mol Endocrinol. 1998;12:825–834. doi: 10.1210/mend.12.6.0122. [DOI] [PubMed] [Google Scholar]
- Prior BM, Yang HT, Terjung RL. What makes vessels grow with exercise training? J Appl Physiol. 2004;97:1119–1128. doi: 10.1152/japplphysiol.00035.2004. [DOI] [PubMed] [Google Scholar]
- Redman LM, Ravussin E. Endocrine alterations in response to calorie restriction in humans. Mol Cell Endocrinol. 2009;299:129–136. doi: 10.1016/j.mce.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset S, Alves-Guerra MC, Mozo J, Miroux B, Cassard-Doulcier AM, Bouillaud F, Ricquier D. The biology of mitochondrial uncoupling proteins. Diabetes. 2004;53(Suppl 1):S130–135. doi: 10.2337/diabetes.53.2007.s130. [DOI] [PubMed] [Google Scholar]
- Saito M, Okamatsu-Ogura Y, Matsushita M, Watanabe K, Yoneshiro T, Nio-Kobayashi J, Iwanaga T, Miyagawa M, Kameya T, Nakada K, Kawai Y, Tsujisaki M. High incidence of metabolically active brown adipose tissue in healthy adult humans: effects of cold exposure and adiposity. Diabetes. 2009;58:1526–1531. doi: 10.2337/db09-0530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seale P, Kajimura S, Yang W, Chin S, Rohas LM, Uldry M, Tavernier G, Langin D, Spiegelman BM. Transcriptional control of brown fat determination by PRDM16. Cell Metab. 2007;6:38–54. doi: 10.1016/j.cmet.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S, Scimè A, Devarakonda S, Conroe HM, Erdjument-Bromage H, Tempst P, Rudnicki MA, Beier DR, Spiegelman BM. PRDM16 controls a brown fat/skeletal muscle switch. Nature. 2008;454:961–967. doi: 10.1038/nature07182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabalina IG, Jacobsson A, Cannon B, Nedergaard J. Fatty acid-induced uncoupling in brown-fat mitochondria from wild-type and UCP1-ablated mice. Biochim Biophys Acta. 2002;(Suppl 12):262–262. [Google Scholar]
- Shi T, Wang F, Stieren E, Tong Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J Biol Chem. 2005;280:13560–13567. doi: 10.1074/jbc.M414670200. [DOI] [PubMed] [Google Scholar]
- Smith RE. Thermoregulatory and adaptive behavior of brown adipose tissue. Science. 1964;146:1686–1689. doi: 10.1126/science.146.3652.1686. [DOI] [PubMed] [Google Scholar]
- Sramkova D, Krejbichova S, Vcelak J, Vankova M, Samalikova P, Hill M, Kvasnickova H, Dvorakova K, Vondra K, Hainer V, Bendlova B. The UCP1 gene polymorphism A-3826G in relation to DM2 and body composition in Czech population. Exp Clin Endocrinol Diabetes. 2007;115:303–307. doi: 10.1055/s-2007-977732. [DOI] [PubMed] [Google Scholar]
- Stephens DN, Nash SC, Proffitt C. Dietary obesity in adult and weanling rats following removal of interscapular brown adipose tissue. Pflugers Arch. 1981;392:7–12. doi: 10.1007/BF00584574. [DOI] [PubMed] [Google Scholar]
- Stern JS, Inokuchi T, Castonguay TW, Wickler SJ, Horwitz BA. Scapular brown fat removal enhances development of adiposity in cold-exposed obese Zucker rats. Am J Physiol. 1984;247:R918–926. doi: 10.1152/ajpregu.1984.247.5.R918. [DOI] [PubMed] [Google Scholar]
- Stranahan AM, Mattson MP. Impact of energy intake and expenditure on neuronal plasticity. Neuromolecular Med. 2008;10:209–218. doi: 10.1007/s12017-008-8043-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranahan AM, Lee K, Martin B, Maudsley S, Golden E, Cutler RG, Mattson MP. Voluntary exercise and caloric restriction enhance hippocampal dendritic spine density and BDNF levels in diabetic mice. Hippocampus. 2009;19:951–961. doi: 10.1002/hipo.20577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas SA, Palmiter RD. Thermoregulatory and metabolic phenotypes of mice lacking noradrenaline and adrenaline. Nature. 1997;387:94–97. doi: 10.1038/387094a0. [DOI] [PubMed] [Google Scholar]
- Timmons JA, Wennmalm K, Larsson O, Walden TB, Lassmann T, Petrovic N, Hamilton DL, Gimeno RE, Wahlestedt C, Baar K, Nedergaard J, Cannon B. Myogenic gene expression signature establishes that brown and white adipocytes originate from distinct cell lineages. Proc Natl Acad Sci U S A. 2007;104:4401–4406. doi: 10.1073/pnas.0610615104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonello C, Giordano A, Cozzi V, Cinti S, Stock MJ, Carruba MO, Nisoli E. Role of sympathetic activity in controlling the expression of vascular endothelial growth factor in brown fat cells of lean and genetically obese rats. FEBS Lett. 1999;442:167–172. doi: 10.1016/s0014-5793(98)01627-5. [DOI] [PubMed] [Google Scholar]
- Trayhurn P, Ashwell M. Control of white and brown adipose tissues by the autonomic nervous system. Proc Nutr Soc. 1987;46:135–142. doi: 10.1079/pns19870017. [DOI] [PubMed] [Google Scholar]
- Tseng YH, Kokkotou E, Schulz TJ, Huang TL, Winnay JN, Taniguchi CM, Tran TT, Suzuki R, Espinoza DO, Yamamoto Y, Ahrens MJ, Dudley AT, Norris AW, Kulkarni RN, Kahn CR. New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature. 2008;454:1000–1004. doi: 10.1038/nature07221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valle A, Guevara R, García-Palmer FJ, Roca P, Oliver J. Caloric restriction retards the age-related decline in mitochondrial function of brown adipose tissue. Rejuvenation Res. 2008;11:597–604. doi: 10.1089/rej.2007.0626. [DOI] [PubMed] [Google Scholar]
- van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JM, Kemerink GJ, Bouvy ND, Schrauwen P, Teule GJ. Cold-activated brown adipose tissue in healthy men. N Engl J Med. 2009;360:1500–1508. doi: 10.1056/NEJMoa0808718. [DOI] [PubMed] [Google Scholar]
- van Praag H. Neurogenesis and exercise: past and future directions. Neuromolecular Med. 2008;10:128–140. doi: 10.1007/s12017-008-8028-z. [DOI] [PubMed] [Google Scholar]
- Varady KA, Hellerstein MK. Alternate-day fasting and chronic disease prevention: a review of human and animal trials. Am J Clin Nutr. 2007;86:7–13. doi: 10.1093/ajcn/86.1.7. [DOI] [PubMed] [Google Scholar]
- Vernochet C, Peres SB, Davis KE, McDonald ME, Qiang L, Wang H, Scherer PE, Farmer SR. C/EBPalpha and the corepressors CtBP1 and CtBP2 regulate repression of select visceral white adipose genes during induction of the brown phenotype in white adipocytes by peroxisome proliferator-activated receptor gamma agonists. Mol Cell Biol. 2009;29:4714–4728. doi: 10.1128/MCB.01899-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent AM, Olzmann JA, Brownlee M, Sivitz WI, Russell JW. Uncoupling proteins prevent glucose-induced neuronal oxidative stress and programmed cell death. Diabetes. 2004;53:726–734. doi: 10.2337/diabetes.53.3.726. [DOI] [PubMed] [Google Scholar]
- Virtanen KA, Lidell ME, Orava J, Heglind M, Westergren R, Niemi T, Taittonen M, Laine J, Savisto NJ, Enerbäck S, Nuutila P. Functional brown adipose tissue in healthy adults. N Engl J Med. 2009;360:1518–1525. doi: 10.1056/NEJMoa0808949. [DOI] [PubMed] [Google Scholar]
- Wijers SL, Schrauwen P, Saris WH, van Marken Lichtenbelt WD. Human skeletal muscle mitochondrial uncoupling is associated with cold induced adaptive thermogenesis. PLoS One. 2008 Mar 12. 2008;3(3):e1777. doi: 10.1371/journal.pone.0001777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijers SL, Saris WH, van Marken Lichtenbelt WD. Recent advances in adaptive thermogenesis: potential implications for the treatment of obesity. Obes Rev. 2009;10:218–226. doi: 10.1111/j.1467-789X.2008.00538.x. [DOI] [PubMed] [Google Scholar]
- Xiao XQ, Williams SM, Grayson BE, Glavas MM, Cowley MA, Smith MS, Grove KL. Excess weight gain during the early postnatal period is associated with permanent reprogramming of brown adipose tissue adaptive thermogenesis. Endocrinology. 2007;148:4150–4159. doi: 10.1210/en.2007-0373. [DOI] [PubMed] [Google Scholar]
- Yamashita H, Sato N, Yamamoto M, Sato Y, Nakanishi K, Suzuki M, Habara Y, Ohno H. Insulin administration induces capillary growth in brown adipose tissue of heat-exposed rats. Comp Biochem Physiol Comp Physiol. 1992;103:673–678. doi: 10.1016/0300-9629(92)90165-m. [DOI] [PubMed] [Google Scholar]
- Yamashita H, Sato Y, Kizaki T, Oh-ishi S, Nagasawa J, Ohno H. Basic fibroblast growth factor (bFGF) contributes to the enlargement of brown adipose tissue during cold acclimation. Pflugers Arch. 1994;428:352–356. doi: 10.1007/BF00724518. [DOI] [PubMed] [Google Scholar]
- Yu ZF, Mattson MP. Dietary restriction and 2-deoxyglucose administration reduce focal ischemic brain damage and improve behavioral outcome: evidence for a preconditioning mechanism. J Neurosci Res. 1999;57:830–839. [PubMed] [Google Scholar]
- Zingaretti MC, Crosta F, Vitali A, Guerrieri M, Frontini A, Cannon B, Nedergaard J, Cinti S. The presence of UCP1 demonstrates that metabolically active adipose tissue in the neck of adult humans truly represents brown adipose tissue. FASEB J. 2009;23:3113–3120. doi: 10.1096/fj.09-133546. [DOI] [PubMed] [Google Scholar]