

Abstract
A series of thiadiazole derivatives has been designed as potential allosteric, substrate competitive inhibitors of the protein kinase JNK. We report on the synthesis, characterization and evaluation of a series of compounds that resulted in the identification of potent and selective JNK inhibitors targeting its JIP-1 docking site.
The c-Jun N-terminal kinases (JNKs) were initially described in the early 1990s as a family of serine/threonine protein kinases, activated by a range of stress stimuli and able to phosphorylate the N-terminal transactivation domain of the c-Jun transcription factor.1 Three distinct genes encoding JNKs have been identified as JNK-1, JNK-2, and JNK-3, and at least 10 different splicing isoforms exist in mammalian cells.2-4 The three JNK isoforms share more than 90% amino acid sequence identity and the ATP pocket is highly conserved (>98% identities). These proteins are activated in response to cellular stresses such as heat shock, irradiation, hypoxia, chemotoxins, and peroxides. They are also activated in response to various cytokines and participate in the onset of apoptosis.5,6 It has been reported that up-regulation of JNK activity is associated with a number of disease states such as type- 2 diabetes, obesity, cancer, inflammation, and stroke.1-3 Therefore, JNK inhibitors are expected to be effective therapeutic agents against a variety of diseases.
JNKs bind to substrates and scaffold proteins, such as JIP-1, that contain a D-domain, as defined by the consensus sequence R/KXXXXLXL.7,8 A peptide corresponding to the D-domain of JIP-1 (aa 153-163; pep-JIP1), inhibits JNK activity in vitro and displays noteworthy selectivity with little inhibition of the closely related Erk and p38 MAPKs.9-12 Recent in vivo data, generated for studies focusing on pep-JIP1 fused to the cell permeable HIV-TAT peptide, show that its administration in various mice models of insulin resistance and type-2 diabetes restores normoglycemia without causing hypoglycemia.13 Despite these encouraging data, peptide’s instability in vivo may hamper the development on novel JNK-related therapies based on such peptides.9-13
Hence, there has been considerable effort to identify small molecule JNK inhibitors over the past several years.14-22 A drug discovery program in our laboratory was initiated with the aim of identifying and characterizing small molecule JNK inhibitors as novel chemical entities targeting its JIP binding site rather then the highly conserved ATP binding site of the protein. Very recently, we have reported the identification of 5-(5-nitrothiazol-2-ylthio)-1,3,4-thiadiazol-2-amine series 20 related to compound BI-78D319(Figure 1), as initial JIP mimetic inhibitors. These compounds were discovered using a displacement assay with a biotinylated-pepJIP1 peptide and employing a DELFIA assay platform (experimental section) in a medium size screening campaign.19 In our continued interest in the development of JNK inhibitors, 18-21 we now report further structure activity relationship studies describing novel small molecules thiadiazole derivatives as JNK inhibitors targeting its JIP/substrate docking site.
Figure 1.
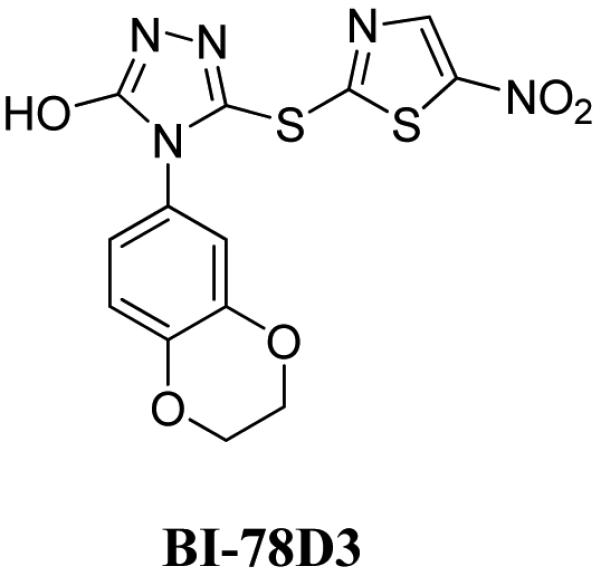
Chemical structures of pepJIP1 based tool compounds previously reported from our lab.
Recent work from our laboratory demonstrates that compound BI-78D3 (Figure 1) served as a useful tool compound for understanding the consequences of JNK substrate competitive inhibitors in vitro and in vivo; however, the compound lacked many chemical features considered desirable in a clinical candidate, including plasma stability. Moreover, it is possible that the compound may covalently bind to a surface exposed Cys residue, located at the center of the JIP binding groove.19 Hence, in our continued search for a JNK inhibitor suitable for clinical studies, we report on a thiadiazole series and focused on improved cellular potency, solubility, cell permeability and plasma stability.
The general synthetic routes utilized thiols (3, Scheme 1) that were either synthesized according to previously reported procedures, 23, 24 or that were commercially available from Enamine (Ukraine). Final compounds (4, BI-90B5 to BI-90H10, Table 1 and Supplementary material) were synthesized by nucleophilic substitution of 2-bromo-5-nitrothiazole with the corresponding thiols of thiadiazoles in the presence of NaOMe in methanol at room temperature (experimental section). Initially, we synthesized several analogs of aryl derivatives of thiadiazoles (BI-90B5, 90B7, 90D8, 90D7, 90D8, 90D10, 90E1, 90E6, 90E9, and 90F2, see in Supplementary materials) but none of these resulted in compound with appreciable inhibition of JNK in a kinase activity assay, although some compounds displayed a significant ability to displace pepJIP1 in a DELFIA assay. Our explanation is that the small nature of our test compounds may result in different binding modes along the extended JIP binding groove. We can only speculate that compounds that displace pepJIP1 by binding on the JIP surface occupied by the conserved LXL motif, in closer proximity to the ATP site, may result more effective kinase inhibitors. We therefore decided to focus on the benzylthiadiazole series. We observed that a 4-methoxybenzyl (BI-90H8) showed better activity than a simple benzyl derivative (BI-90E2). We subsequently found that compounds of the alkylthiadiazole series had improved activities in both assays although the bulkier tert-butyl (BI-90E7), cyclopropyl, and cyclohexyl groups (supplementary material) were not tolerated on 2- position of thiadiazole series suggesting steric hindrance with the target. Interestingly, substituting in the 2-position with either an ethyl group (BI-90G12) or an n-butyl group (BI-90H4) resulted in compounds with limited activity. However, when the substituent in 2- position is a 2-methoxyethyl group (BI-90H9), sec-butyl group (BI-98A10), and n-propyl group (BI-90H6) these lead to compounds with improved activity in both the pepJIP1 displacement (DELFIA) and the kinase activity (LANTHA) assays. Of these compounds, BI-90H9 showed an IC50 of 4.8 μM in the kinase assay (Figure 2A) in a substrate competitive manner and accordingly it displaced pepJIP1 with an IC50 of 158 nM (Figure 2B). Furthermore, compound BI-90H9 was found to be 20 times less active (Table 2) against p38α, a member of the MAPK family with high structural similarity to JNK, and practically inactive against the kinase Akt. These compounds were also inactive against other unrelated proteins under investigation in our laboratory, including metallo-proteases such as anthrax lethal factor, and a serin protease (furin) (Table 2), further corroborating that these compounds may selectively interfere with the JNK docking site. This selectivity is in agreement with our previous findings with compound BI-78D319 (Figure 1) and with the reported data on pepJIP1.9-13
Scheme 1.

Synthetic scheme used to prepare the thiadiazole derivatives reported in Table 1.
Table 1.
Inhibition results for thiadiazole derivatives against JNK
| Compound | ID | IC50a (μM) |
IC50b (μM) |
LEc |
|---|---|---|---|---|
| BI-90H5 | 10 | 0.47 | 0.32 | |
| BI-90H6 | 6.7 | 0.29 | 0.39 | |
| BI-90H8 | 9.1 | 0.10 | 0.23 | |
| BI-90H9 | 4.8 | 0.16 | 0.38 | |
| BI-90H10 | 5.7 | 0.10 | 0.26 | |
| BI-98A10 | 3.0 | 0.14 | 0.39 | |
| BI-90G12 | ~100 | 10 | 0.32 | |
| BI-90E2 | d | d | d | |
| BI-90E7 | d | d | d | |
| BI-90H4 | d | d | d | |
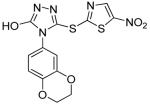 |
BI-78D3 | 0.28 | 0.50 | 0.36 |
JNK1 kinase inhibition assay (LANTHA); values are means of at least three or more experiments.
pepJIP1 displacement assay (DELFIA); values are means of at least three or more experiments.
These compounds did not show a significant inhibition in the kinase assay up to 25 μM, although showed some activity (~ 20%) in displacing pepJIP1 in DELFIA assay at 50 μM.
Figure 2. In vitro activity of compound BI-90H9.

A) Dose response curve for the in vitro inhibition of the kinase activity JNK1 by BI-90H9 using GFP-c-Jun as substrate (LANTHA assay platform). B) Dose dependent curve for the in vitro displacement of biotinylated pep-JIP1 from GST-JNK1 (DELFIA assay platform).
Table 2. Selectivity profile.
| Compound | JNK1, IC50 (μM) |
p-38α , IC50 (μM) |
Akt, IC50 (μM) |
Furin, IC50 (μM) |
LF, IC50 (μM) |
|---|---|---|---|---|---|
| BI-90H6 | 6.7 | >100 | >100 | >50 | >100 |
| BI-90H9 | 4.8 | >100 | >100 | >50 | >100 |
| BI-90H10 | 5.7 | >100 | >100 | >50 | >100 |
| BI-98A10 | 3.0 | >100 | >100 | >50 | >100 |
Previous modeling studies 18-19, supported by NMR relaxation measurements, suggest that compound BI-78D3 may bind at the JIP site with the nitrothiazole group crossing the ridge close to residue Cys163 (Figure 3A). Its benzo-dioxane group occupies the deep hydrophobic pocket along the JIP binding groove, occupied by the side chains of the essential Leu residues of the R/KXXXXLXL binding motif in pepJIP1. In such docked conformation, the compound brings the thio-ether atom in proximity to a surface exposed Cys residue in the JIP1 binding groove (Figure 3A). Previous ITC measurements with compound BI-78D3 and a single point C163S JNK2 mutant suggested a possible involvement of this residue in the binding properties of the compound19. Interestingly, docking studies with compound BI-90H9 suggest that the compound protrudes its methoxy group into the JIP binding groove occupied by the Leu residues of pepJIP1, and that the thiadiazole group can form hydrogen bonds with protein backbone atoms (Figure 3B). In agreement with this docked pose and the observed SAR, the length of the side chain at the 2-position is critical to properly locate the thiadiazole to form hydrogen bonding interactions and to fully occupy the Leu binding pocket (Figure 3B). Moreover, in this docking pose, the thioether of BI-90H9 is far from the side chain of Cys163, hence its binding properties should not be dependent on this residue. Accordingly, ITC measurements with a C163S JNK2 mutant reveal that the compound binding to this mutant with a dissociation constant Kd of 4.2 μM (Figure 2C) versus 2.8 μM obtained against wt-JNK2, whereas compound BI-78D3 showed over 50 fold reduction in binding affinity to the mutant.19
Figure 3. Molecular docking studies.

Surface representation of JNK1 (PDB code 1UKI) with docked compounds occupying the JIP binding site and the ATP site as indicated. The position of residue Cys163 is indicated. In A) the compound BI-78D3 (on the left) and an ATP mimetic compound (on the right) are reported. In B), the compound BI-90H9 (on the left) and the same ATP mimetic (on the right) are reported. Intermolecular hydrogen bonding interactions are depicted as dashed yellow lines.
In an attempt to further profile the properties of compound BI-90H9 in the context of a complex cellular milieu, we employed the cell -based LanthaScreen™ kinase assay. In this assay platform, compound BI-90H9 is able to inhibit TNF-α stimulated phosphorylation of c-Jun (IC50 = 8 μM). We also tested some other compounds on the cell based assay, among them BI-90H9 showed the most cellular activities (IC50 values for BI-90H8, and BI-98A10 were 21 μM, and 20 μM respectively). It should be noted that the cell-based system employed makes use of a GFP-c-Jun stable expression system. As a result, the levels of GFP-c-Jun in these cells are higher than endogenous levels. This could have an inflationary effect on the IC50 values obtained with this assay when testing substrate competitive compounds. Nonetheless, this finding establishes that compound BI-90H9 is able to function in a cellular context and that its activity parallels the in vitro findings
Finally, liquid chromatography/mass spectrometry bio-availability analysis (see experimental section) demonstrated that compound BI-90H9 had favorable plasma stability (68% remaining after 60 min in plasma stability analysis) and cell permeability, improving upon our previous lead molecule, BI-78D3 (Table 3). We observed that a simple n-propyl group (BI-90H6) and 3,4-dimethoxyphenethyl group (BI-90H10) in 2-position of 1,3,4-thiadiazole derivatives degraded rapidly after 1 h of incubation in rat plasma (Table 3 and supplemental data). These results suggested that a 2-methoxyethyl group on 2-position of thiadiazole (BI-90H9) is the most suitable group for improving plasma stability.
Table 3. Plasma and cell permeability data.
| Compound | Plasma stability | Cell permeability, PAMPA % Fluxa |
|---|---|---|
| BI-78D3 | 42% remaining after 60 min | 30% |
| BI-90H6 | 35% remaining after 60 min | 49.8% |
| BI-90H9 | 68% remaining after 60 min | 76.3% |
| BI-90H10 | 0% remaining after 60 min | 5.5% |
Flux values: <5% low permeation; 5-30% medium permeation; 30-100% high permeation.
In summary, compound BI-90H9 can be considered to have a good balance of potency, selectivity, solubility, cellular activity, and plasma stability. These data and the binding mode of the compound provide a solid basis for further optimizations. For example, the predicted proximity of the compound to the ATP site suggests that it may be possible to obtain bi-dentate molecules spanning both sites 18. We are currently exploring this possibility. Nonetheless, our results indicate once more that targeting the protein JIP docking site with a small molecule is a novel and promising avenue for the development of protein kinase related therapeutics.
Experimental
General
Unless otherwise indicated, all anhydrous solvents were commercially obtained and stored in Sure-seal bottles under nitrogen. All other reagents and solvents were purchased as the highest grade available and used without further purification. Thin-layer chromatography (TLC) analysis of reaction mixtures was performed using Merck silica gel 60 F254 TLC plates, and visualized using ultraviolet light. NMR spectra were recorded on Varian 300 or 500 MHz instruments. Chemical shifts (δ) are reported in parts per million (ppm) referenced to 1H (Me4Si at 0.00). Coupling constants (J) are reported in Hz throughout. Mass spectral data were acquired on Shimadzu LCMS-2010EV for low resolution, and on an Agilent ESI-TOF for either high or low resolution. Purity of all compounds was obtained in a HPLC Breeze from Waters Co. using an Atlantis T3 3μm 4.6×150 mm reverse phase column. The eluant was a linear gradient with a flow rate of 1 ml/min from 95% A and 5% B to 5% A and 95% B in 15 min followed by 5 min at 100% B (Solvent A: H2O with 0.1% TFA; Solvent B: ACN with 0.1% TFA). The compounds were detected at λ=254 nm. Purity of key compounds was established by elemental analysis as performed on a Perkin Elmer series II-2400. Combustion analysis was performed by NuMega Resonance labs, Inc., San Diego, CA.
Synthesis of N-(2-methoxyethyl)-5-(5-nitrothiazol-2-ylthio)-1,3,4-thiadiazol-2-amine (BI-90H9)
To a solution of 5-(2-methoxyethylamino)-1,3,4-thiadiazole-2-thiol (100 mg, 0.523 mmol) in MeOH (3 mL) was added MeONa (1.25 mL, 0.5 M solution in MeOH) and stirred. After 5 minutes, 2-bromo-5-nitrothiazole (119 mg, 0.575 mmol) was added to the reaction mixture and stirred at room temperature until deemed complete by TLC (16 h). The reaction mixture was acidified with 1 N HCl and the resulting precipitate was collected by filtration and washed with water (2 × 30 mL), haxanes (2 × 30 mL), and 10% ethyl acetate in hexanes (2 × 30 mL) to give a white solid. The residue was chromatographed over silica gel (70% ethyl acetate in hexane) to afford the BI- 90H9 (118 mg, 71%). 1H NMR (300 MHz, DMSO-d6) δ 3.24-3.30 (m, 2 H), 3.32 (s, 3 H), 3.46-3.57 (m, 2 H), 8.47 (t, J = 5.4 Hz, 1 H, NH), 8.75 (s, 1 H); MS m/z 341 (M+Na)+, 319 (M+H)+, 217, 171, 147, 138, 125, 106, 102, 97, 84; HRMS calcd for C8H10N5O3S3 (M+H) 319.9940, found 319.9945. Anal. calcd for C8H9N5O3S3: C, 30.08; H, 2.84; N, 21.93; S, 30.12. Found: C, 30.16; H, 2.95; N, 21.80; S, 30.01.
Following above mentioned procedure (BI-90H9) and the appropriate starting materials and reagents used; compounds (BI-90B7 to 90H10 and BI-98A10) were synthesized.
N-ethyl-5-(5-nitrothiazol-2-ylthio)-1,3,4-thiadiazol-2-amine (BI-90G12)
Yield:62% ; 1H NMR (300 MHz, DMSO-d6) δ1.20 (t, J = 6.5 Hz, 3 H), 3.31 (quintet, J = 6.5 Hz, 2 H), 8.41 (s, 1 H), 8.75 (t, J = 5.4 Hz, 1 H, NH); MS m/z 311 (M+Na)+, 289 (M+H)+, 204, 190, 138, 106, 102, 84; HRMS calcd for C7H8N5O2S3 (M+H) 289.9835, found 289.9839.
5-(5-nitrothiazol-2-ylthio)-N-((tetrahydrofuran-2-yl)methyl)-1,3,4-thiadiazol-2-amine (BI-90H5)
Yield: 41%; 1H NMR (300 MHz, DMSO-d6) δ 1.50-1.65 (m, 2H), 1.80-2.00 (m, 2H), 3.65 (q, J = 6.9 Hz, 2H), 3.78 (q, J = 7.8 Hz, 2H), 4.03 (sextet, J = 4.5 Hz, 1H), 8.48 (t, J = 6 Hz, NH), 8.74 (s, 1H); MS m/z 346 (M+H)+, 158, 147, 121, 110, 102, 100, 84; HRMS calcd for C10H12N5O3S3 ( M+H) 346.0097, found 346.0100.
5-(5-nitrothiazol-2-ylthio)-N-propyl-1,3,4-thiadiazol-2-amine (BI-90H6)
Yield: 60% ; 1H NMR (300 MHz, DMSO-d6) δ 0.93 (t, J = 7.8 Hz, 3 H), 1.61 (sextet, J = 7.2 Hz, 2 H), 3.29 (q, J = 7.2 Hz, 2 H), 8.43 (t, J = 5.4 Hz, 1H, NH), 8.75 (s, 1 H); MS m/z 325 (M+Na)+, 303 (M+H)+, 204, 190, 138, 126, 106, 102, 84; HRMS calcd for C8H10N5O2S3 (M+H) 303.9991, found 303.9996.
N-(4-methoxybenzyl)-5-(5-nitrothiazol-2-ylthio)-1,3,4-thiadiazol-2-amine (BI-90H8)
Yield: 29%; 1H NMR (300 MHz, DMSO-d6) δ 3.74 (s, 3H), 4.48 (d, J = 5.4 Hz, 2H), 6.92 (d, J = 7.8 Hz, 2H), 7.31 (d, J = 7.8 Hz, 2H), 8.74 (s, 1H), 8.81 (s, NH); MS m/z 403 (M+Na)+, 382 (M+H)+, 359, 349, 316, 185, 147, 132, 105, 100, 90, 64; HRMS calcd for C13H12N5O3S3 (M+H) 382.0097, found 382.0095.
N-(2-methoxyethyl)-5-(5-nitrothiazol-2-ylthio)-1,3,4-thiadiazol-2-amine (BI-90H9)
Yield: 71%; 1H NMR (300 MHz, DMSO-d6) δ 3.24-3.30 (m, 2 H), 3.32 (s, 3 H), 3.46-3.57 (m, 2 H), 8.47 (t, J = 5.4 Hz, 1 H, NH), 8.75 (s, 1 H); MS m/z 341 (M+Na)+, 319 (M+H)+, 217, 171, 147, 138, 125, 106, 102, 97, 84; HRMS calcd for C8H10N5O3S3 (M+H) 319.9940, found 319.9945. Anal. calcd for C8H9N5O3S3: C, 30.08; H, 2.84; N, 21.93; S, 30.12. Found: C, 30.16; H, 2.95; N, 21.80; S, 30.01.
N-(3,4-dimethoxyphenethyl)-5-(5-nitrothiazol-2-ylthio)-1,3,4-thiadiazol-2-amine (BI-90H10)
Yield: 49%; 1H NMR (300 MHz, DMSO-d6) δ 2.84 (t, J = 6.75, 2H), 3.58 (q, J = 6.0 Hz, 2H), 3.70 (s, 3H), 3.73 (s, 3 H), 6.76 (d, J = 8.7 Hz, 1H), 6.85-6.90 (m, 2H), 8.44 (t, J = 6 Hz, NH), 8.77 (s, 1H); MS m/z 448 (M+Na)+, 426 (M+H)+, HRMS calcd for C15H16N5O4S3 (M+H) 426.0359, found 426.0359
N-sec-butyl-5-(5-nitrothiazol-2-ylthio)-1,3,4-thiadiazol-2-amine (BI-98A10)
Yield: 64%; 1H NMR (300 MHz, DMSO-d6) δ 0.91 (t, J = 7.5 Hz, 3 H), 1.19 (d, J = 6.6 Hz, 3 H), 1.56 (quintet, J = 7.5 Hz, 2 H), 3.71-3.79 (m, 1 H), 8.32 (d, J = 6.2 Hz, 1 H, NH), 8.75 (s, 1 H); MS m/z 339 (M+Na)+, 318 (M+H)+, 252, 226, 158, 147, 138, 106, 84; HRMS calcd for C9H12N5O2S3 (M+H) 318.0148, found 318.0147.
Plasma Stability Assay
Briefly, each test compound solution was incubated (1 μM, 2.5% final DMSO concentration) with fresh rat plasma at 37 °C. The reactions were terminated at 0, 30, and 60 min by the addition of two volumes of methanol containing internal standard. Following protein precipitation and centrifugation, the samples were analyzed by LC-MS. The percentage of parent compound remaining at each time point relative to the 0 min sample is calculated from peak area ratios in relation to the internal standard. Compounds were run in duplicate with a positive control known to be degraded in plasma.
PAMPA Permeability Assay
PAMPA is an in vitro model of passive, transcellular permeation useful for ranking compounds based on their potential to cross cell membranes and the gastrointestinal wall. A 96-well microtiter plate completely filled with aqueous buffer solutions (pH 7.4) is covered with a microtiter filterplate in a sort of sandwich construction. The hydrophobic filter material (Durapore/ Millipore; pore size 0.22-0.45 m) of wells (sample) of the filterplate is impregnated with a 1-20% solution of phospholipid in an organic solvent (Avanti Polar Lipids). Transport studies were started by the transfer of 100-200 μL of a 20 μM stock solution on top of the filterplate in the sample and in the reference section, respectively. An equilibrium plate (compounds at the theoretical equilibrium, i.e. the resulting concentration if the donor and the acceptor solutions are combined) was also created and analyzed. This Acceptor plate and equilibrium plate concentrations were used to calculate the percentage of permeation (flux) of the compounds. The maximum DMSO content of the stock solutions was <2%. The PAMPA % flux was 76.3% for BI-90H9 indicated high permeation (Table 3). Reference compounds were included and reported as supplementary material.
DELFIA Assay (dissociation enhanced lanthanide fluoro-immuno assay)
To each well of 96-well streptavidin-coated plates (Perkin-Elmer) 100 μL of a 100 ng/ml solution of biotin-labeled pep-JIP11 (Biotin-lc-KRPKRPTTLNLF, where lc indicates a hydrocarbon chain of 6 methylene groups) was added. After 1 hr incubation and elimination of unbound biotin-pep-JIP11 by 3 washing steps, 87 μL of Eu-labeled anti-GST antibody solution (300ng/ml; 1.9 nM), 2.5 μL DMSO solution containing test compound, and 10 μL solution of GST-JNK2 for a final protein concentration of 10 nM was added. After 1 hr incubation at 0 °C, each well was washed 5 times to eliminate unbound protein and the Eu-antibody if displaced by a test compound. Subsequently, 200 μL of enhancement solution (Perkin-Elmer) was added to each well and fluorescence measured after 10 min incubation (excitation wavelength, 340 nm; emission wavelength, 615 nm). Controls include unlabeled peptide and blanks receiving no compounds. Protein and peptide solutions were prepared in DELFIA buffer (Perkin-Elmer).
In vitro Kinase Assay
The LanthaScreen™ assay platform from Invitrogen was utilized. The time-resolved fluorescence resonance energy transfer assay (TR-FRET) was performed in 384 well plates. Each well received JNK1 (35 ng/mL, JNK1 MW = 45 KDa), ATF2 (400 nM), and ATP (0.2 μM) in 50mM HEPES, 10mM MgCl 2, 1mM EGTA and 0.01% Brij-35, pH 7.5 and test compounds. The kinase reaction was performed at room temperature for 1 hr. After which, the terbium labeled antibody and EDTA were added into each well. After an additional hour incubation, the signal was measured at 520/495 nm emission ratio on a fluorescence plate reader (Victor 2, Perkin-Elmer).
Cell based assays for c-Jun phosphorylation
The cell based kinase assays for c-Jun and ATF2 phosphorylation were carried out using the LanthaScreen c-Jun (1-79) Hela (Invitrogen, Carlsbad, CA) which stably express GFP-c-Jun 1-79. Phosphorylation was determined by measuring the time resolved FRET (TR-FRET) between a terbium labeled phospho-specific antibody and the GFP-fusion protein12. The cells were plated in white tissue culture treated 384 well plates at a density of 10000 cell per well in 32 μl assay medium (Opti-MEM®, supplemented with 1% charcoal/dextran-treated FBS, 100 U/mL penicillin and 100 μg/mL streptomycin, 0.1 mM non-essential amino acids, 1 mM sodium pyruvate, 25 mM HEPES pH 7.3, and lacking phenol red). After overnight incubation, cells were pretreated for 60 min with compound (indicated concentration) followed by 30 min of stimulation with 2 ng/ml of TNF-alpha which stimulates both JNK and p38. The medium was then removed by aspiration and the cells were lysed by adding 20 μl of lysis buffer (20 mM TRIS-HCl pH 7.6, 5 mM EDTA, 1% NP-40 substitute, 5 mM NaF, 150 mM NaCl, 1:100 protease and phosphatase inhibitor mix, SIGMA P8340 and P2850 respectively). The lysis buffer included 2 nM of the terbium labeled anti-pc-Jun (pSer73) detection antibodies (Invitrogen). After allowing the assay to equilibrate for 1 h at room temperature, TR-FRET emission ratios were determined on a BMG Pherastar fluorescence plate reader (excitation at 340 nm, emission 520 nm and 490 nm; 100 μs lag time, 200 μs integration time, emission ratio = Em520 / Em 490).
Isothermal Titration Calorimetry
Titrations were done using a VP-ITC calorimeter from Microcal (Northampton, MA). C163S JNK2 and wt-JNK2 were used at 50 μM in 20 mM sodium phosphate buffer (pH 7.4), 5% DMSO, and 0.01% triton X-100. Titrants were used at 750 μM in the same buffer. Titrations were carried out at 25 °C. Data were analyzed using Microcal Origin software provided by the ITC manufacturer (Microcal, Northampton, MA).
Molecular Modeling
Molecular modeling studies were conducted on a Linux workstation and a 64 3.2-GHz CPUs Linux cluster. Docking studies were performed using the X-ray coordinates of JNK1 (PDB code 1UKH).21, 25 The complexed JIP peptide and ATO mimetic compound SP600125 were extracted from the protein structure and was used to define the binding site for docking of small molecules. The docked geometry of the indazole ATP mimetic reported in Figure 3 was simply derived from the X-ray coordinates of the parent compound, SP600125. The genetic algorithm (GA) procedure in the GOLD docking software performed flexible docking of small molecules whereas the protein structure was static.25-29 For each compound, 20 solutions were generated and subsequently ranked according to Chemscore.25 The protein surface was prepared with the program MOLCAD as implemented in Sybyl and was used to analyze the binding poses for studied small molecules. 25-29
Supplementary Material
Acknowledgments
We gratefully acknowledge financial support from the NIH (grants # DK073274 and DK080263) and Syndexa pharmaceuticals (to MP).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- (1).Manning G. The protein kinase complement of human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- (2).Manning AM, Davis RJ. Targeting JNK for therapeutic benefit: from junk to gold. Nat. Rev. Drug Discovery. 2003;2:554–565. doi: 10.1038/nrd1132. [DOI] [PubMed] [Google Scholar]
- (3).Bogoyevitch MA, Arthur PG. Inhibitors of c-Jun N-terminal kinases-JuNK no more. Biochim. Biophys. Acta. 2008;1784:76–93. doi: 10.1016/j.bbapap.2007.09.013.and references cited therein.
- (4).Gupta S, Barrett T, Whitmarsh AJ, Cavanagh J, Sluss HK, Dérijard B, Davis RJ. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996;15:2760–2770. [PMC free article] [PubMed] [Google Scholar]
- (5).Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 2001;81:807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- (6).Pearson G, Robinson R, Gibson TB, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr. Rev. 2001;22:153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- (7).Kallunki T, Deng T, Hibi M, Karin M. c-Jun can recruit JNK to phosphorylate dimerization partners via specific docking interactions. Cell. 1996;87:929–939. doi: 10.1016/s0092-8674(00)81999-6. [DOI] [PubMed] [Google Scholar]
- (8).Yang S-H, Whitmarsh AJ, Davis RJ, Sharrocks AD. Differential targeting of MAP kinases to the ETS-domain transcription factor Elk-1. EMBO J. 1998;17:1740–1749. doi: 10.1093/emboj/17.6.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Barr RK, Kendrick TS, Bogoyevitch MA. Identification of the features of a small peptide inhibitor of JNK activity. J. Biol. Chem. 2002;277:10987–10997. doi: 10.1074/jbc.M107565200. [DOI] [PubMed] [Google Scholar]
- (10).Bonny C, Oberson A, Negri S, Sauser C, Schorderet DF. Cell-permeable peptide inhibitors of JNK: novel blockers of beta-cell death. Diabetes. 2001;50:77–82. doi: 10.2337/diabetes.50.1.77. [DOI] [PubMed] [Google Scholar]
- (11).Dickens M, Roger JS, Cavanagh J, Raitano A, Xia Z, Halpern JR, Greenberg ME, Sawyers CL, Davis RJ. A cytoplasmic inhibitor of the JNK signal transduction pathways. Science. 1997;277:693–696. doi: 10.1126/science.277.5326.693. [DOI] [PubMed] [Google Scholar]
- (12).Heo Y-S, Kim S-K, Seo CI, Kim Y-K, Sung B-J, Lee HS, Lee JI, Park S-Y, Kim JH, Hwang KY, Hyun Y-L, Jeon YH, Ro S, Cho JM, Lee TG, Yang C-H. Structural basis for the selective inhibition of JNK1 by the scaffolding protein JIP1 and SP600125. EMBO J. 2004;23:2185–2195. doi: 10.1038/sj.emboj.7600212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kaneto H, Nakatani Y, Miyatsuka T, Kawamori D, Matsuoka T, Matsuhisa M, Kajimoto Y, Ichijo H, Yamasaki Y, Hori M. Possible novel therapy for diabetes with cell-permeable JNK inhibitor peptide. Nature Medicine. 2004;10:1128–1132. doi: 10.1038/nm1111. [DOI] [PubMed] [Google Scholar]
- (14).Shin Y, Chen W, Habel J, Duckett D, Ling YY, Koenig M, He Y, Vojkosvsky T, LoGrasso P, Kamenecka TM. Synthesis and SAR of piperazine amides as novel c-Jun N-terminal kinase (JNK) inhibitors. Bioorg. Med. Chem. Lett. 2009;19:3344–3347. doi: 10.1016/j.bmcl.2009.03.086.and references cited therein.
- (15).Gaillard P, Jeanclaude-Etter I, Ardissone V, Arkinstall S, Cambet Y, Camps M, Chabert C, Church D, Cirillo R, Gretener D, Halazy S, Nichols A, Szyndralewiez C, Vitte P-A, Gotteland J-P. Design and synthesis of the first generation of novel potent, selective, and in vivo active (benzothiazol-2-yl)acetonitrile inhibitors of the c-Jun N-terminal kinase. J. Med. Chem. 2005;48:4596–4607. doi: 10.1021/jm0310986. [DOI] [PubMed] [Google Scholar]
- (16).Rückle T, Biamonte M, Grippi-Vallotton T, Arkinstall S, Cambet Y, Camps M, Chabert C, Church DJ, Halazy S, Jiang X, Martinou I, Nichols A, Sauer W, Gotteland J-P. Design, synthesis, and biological activity of novel, potent, and selective (benzoylaminomethyl)thiophene sulfonamide inhibitors of c-Jun-N-terminal kinase. J. Med. Chem. 2004;47:6921–6934. doi: 10.1021/jm031112e. [DOI] [PubMed] [Google Scholar]
- (17)).Bennett BL, Sasaki DT, Murray BW, O’Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS, Manning AM, Anderson DW. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. U. S. A. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Vazquez J, De SK, Chen L-H, Riel-Mehan M, Emdadi A, Cellitti J, Stebbins JL, Rega MF, Pellecchia M. Development of paramagnetic probes for molecular recognition studies in protein kinases. J. Med. Chem. 2008;51:3460–3465. doi: 10.1021/jm800068w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Stebbins JL, De SK, Machleidt T, Becattini B, Vazquez J, Kuntzen C, Chen L-H, Cellitti JF, Riel-Mehan M, Emdadi A, Solinas G, Karin M, Pellecchia M. Identification of a new JNK inhibitor targeting JNK-JIP interaction site. Proc. Natl. Acad. Sci. U. S. A. 2008;105:16809–16813. doi: 10.1073/pnas.0805677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).De SK, Stebbins JL, Chen L-H, Riel-Mehan M, Machleidt T, Dahl R, Yuan H, Emdadi A, Barile E, Chen V, Murphy R, Pellecchia M. Design, Synthesis, and Structure-Activity Relationship of substrate competitive, selective, and in vivo active triazole and thiadiazole inhibitors of the c-Jun N-terminal kinase. J. Med. Chem. 2009;52:1943–1952. doi: 10.1021/jm801503n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Zhao H, Serby MD, Xin Z, Szczepankiewicz BG, Liu M, Kosogof C, Liu B, Nelson LTJ, Johnson EF, Wang S, Pederson T, Gum RJ, Clampit JE, Haasch DL, Abad-Zapatero C, Fry EH, Rondinone C, Trevillyan JM, Sham HL, Liu G. Discovery of potent, highly selective, and orally bioavailable pyridine carboxamide c-Jun NH2-terminal kinase inhibitors. J. Med. Chem. 2006;49:4455–4458. doi: 10.1021/jm060465l. [DOI] [PubMed] [Google Scholar]
- (22).Chen T, Kablauoi N, Little J, Timofeevski S, Tschantz WR, Chen P, Peng J, Charlton M, Stanton R, Bauer P. Identification of small-molecule inhibitors of the JIP-JNK interaction. Biochem J. 2009;420:283–294. doi: 10.1042/BJ20081899. [DOI] [PubMed] [Google Scholar]
- (23).Chu C-H, Hui X-P, Xu P-F, Zhang Z-Y, Li Z-C, Lioa R-A. Synthesis and antifungal activities of ω-(5-arylamino-1,3,4 thiadiazole-2-yl) thiol –ω-(1H-1,2,4-triazol-1yl) acetophenone. Indian J. Chem. Sec. B. 2002;41B:2436–2438. [Google Scholar]
- (24).Raphael E, Joshua CP, Koshy L. Alkali-catalyzed thermal cyclization of 1-substituted and 1,6-disubstituted 2,5-dithiobiureas. Formation of 1,3,4-trizolidine-3,5 dithiones and /or 1,3,4-thiadiazoline-5-thiones. Indian J. Chem. Sec. B. 1989;28B:635–638. [Google Scholar]
- (25).GOLD, Version, 2.1. The Cambridge Crystallographic Data Centre; 12, Union Road, Cambridge, CB2 1EZ, UK: [Google Scholar]
- (26).Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- (27).Eldridge MD, Murray CW, Auton TR, Paolini GV, Mee RP. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput.-Aided Mol. Des. 1997;11:425–455. doi: 10.1023/a:1007996124545. [DOI] [PubMed] [Google Scholar]
- (28).Teschner M, Henn C, Volhardt H, Reiling S, Brickmann J. Texture mapping: A new tool for molecular graphics. J. Mol. Graphics. 1994;12:98–105. doi: 10.1016/0263-7855(94)80074-x. [DOI] [PubMed] [Google Scholar]
- (29).Pearlman RS. Concord. Tripos International; St. Louis, Missouri, 63144, USA: distributed by. [Google Scholar]
- (30).Hopkins AL, Groom CR, Alex A. Ligand efficiency: A useful metric for lead selection. Drug Discovery Today. 2004;9:430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- (31).Kuntz ID, Chen K, Sharp KA, Kollman PA. The maximal affinity of ligands. Proc. Natl. Acad. Sci, USA. 1999;96:9997–10002. doi: 10.1073/pnas.96.18.9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.