

Abstract
We modified a series of (N)-methanocarba nucleoside 5 -uronamides to contain dialkyne groups on an extended adenine C2 substituent, as synthetic intermediates leading to potent and selective A3 adenosine receptor (AR) agonists. The proximal alkyne was intended to promote receptor recognition, and the distal alkyne reacted with azides to form triazole derivatives (click cycloaddition). Click chemistry was utilized to couple an octadiynyl A3AR agonist to azido-containing fluorescent, chemically reactive, biotinylated, and other moieties with retention of selective binding to the A3AR. A bifunctional thiol-reactive crosslinking reagent was introduced. The most potent and selective novel compound was a 1-adamantyl derivative (Ki 6.5 nM), although some of the click products had Ki values in the range of 200–400 nM. Other potent, selective derivatives (Ki at A3AR in nM) were intended as possible receptor affinity labels: 3-nitro-4-fluorophenyl (10.6), α-bromophenacyl (9.6), thiol-reactive isothiazolone (102), and arylisothiocyanate (37.5) derivatives. The maximal functional effects in inhibition of forskolin-stimulated cAMP were measured, indicating that this class of click adducts varied from partial to full A3AR agonist compared to other widely used agonists. Thus, this strategy provides a general chemical approach to linking potent and selective A3AR agonists to reporter groups of diverse structure and to carrier moieties.
Keywords: G protein-coupled receptor, purines, azide, structure activity relationship, radioligand binding
Introduction
The A3 adenosine receptor (AR), a G protein-coupled receptor (GPCR), is found in myocytes, astrocytes, neurons, neutrophils, eosinophils, and other cell types.1 Both A3AR agonists and antagonists are proposed for the treatment of cancer and inflammatory diseases.2–6 An antiischemic effect of A3AR agonists also suggests their use in protection of skeletal muscle and cardiac muscle.7
The crystallographic structure of the human (h) A2AAR has been determined, but a directly determined structure is lacking for the A3AR.8 We recently used the coordinates of the A2AAR structure as a template for homology modeling of the A3AR and have predicted specific interactions of the ligand with the receptor protein.9 Hypotheses for agonist binding at the A3AR have been supported using site-directed mutagenesis and reengineering of the putative binding site to recognize tailored nucleoside ligands (neoceptors).10 Thus, structural insights into recogntion in the A3AR binding site have been gained even in the absence of an X-ray structure. The 2 and 6 positions of the adenine ring are most amenable to chain derivatization without sterically interfering in the binding process.
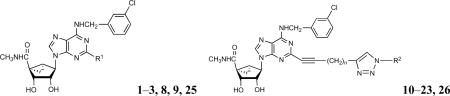
Molecular probes for the A3AR containing a rigid bicyclo[3.1.0]hexane ring system of the North conformation ((N)-methanocarba) in place of the freely twisting ribose moiety were recently reported.9,11,12 Amide-linked fluorescent 1 and biotin-containing probes 2 and 3 of high A3AR affinity and selectivity were designed using a functionalized congener approach based on stepwise chain extension designed to preserve or enhance receptor affinity (Chart 1).13
Chart 1.

Molecular probes in the (N)-methanocarba series of adenosine A3AR agonists that were previously reported (1 – 3)9 and a general formula (4) for triazole derivatives prepared in this study.
The present study introduces 2-dialkynyl groups into selective A3AR ligands of the structural class of the (N)-methanocarba-5′N-methyluronamides. The proximal alkyne was intended to promote receptor recognition.9 Earlier studies established the enhancing effect in recognition at both A2A and A3ARs of introducing an alkynyl group adjacent to the purine C2.14 The distal alkyne was shown to react selectively with alkyl or aryl azides by click cycloaddition to form triazole derivatives of the general structure 4.15 Furthermore, an optimized N6-(3-chlorobenzyl) group, which favors A3AR selectivity in human, mouse, and rat, was included in the nucleoside structures.12 Fluorescent, chemically reactive, biotinylated, and other moieties were incorporated by this chemical route, leading to selective A3AR agonists.
Results
Chemical Synthesis
We have introduced terminal alkynyl groups on the C2 position substituents of (N)-methanocarba nucleoside derivatives to serve as cross-linking sites on the A3AR agonists, such as for the introduction of reporter groups (R1 in general structure 4 of Chart 1). Prior to introduction of a terminal alkyne for coupling of the nucleosides to reporter groups, the structure activity relationship (SAR) of this series of N6-(3-chlorobenzyl)-(N)-methanocarba-5′N-methyluronamide derivatives was explored in detail.9,11 Thus, here we started with a nucleoside pharmacophore that was already optimized for activation of the A3AR. Only the adenine C2 position was structurally modified, and all modifications were intended for click chemistry.
The synthetic route to the small molecular 5′-N-methyluronamide (N)-methanocarba 2-alkynyl triazole-containing derivatives 10 – 23 is shown in Scheme 1. The synthesis of the 2′,3′-protected dialkynyl intermediates 6 and 7 was performed using a Sonogashira coupling16 on the corresponding 2-iodo intermediate 5. After deprotection of the 2′,3′-hydroxyl groups to provide nucleosides 8 and 9, compounds 10 – 23 were obtained using the Cu(I)-catalyzed 2 + 3 cyclization reaction of the terminal acetylene group with an appropriate azide.15 The reactions were generally selective for the terminal alkyne, but reaction with 4-isothiocyanatophenylazide 24 initially produced the disubstituted product 25 as the major product isolated (Scheme 2). It is well known that copper-catalyzed click reactions direct the formation of only one regioisomer, i.e. the 1,4-regioisomer is formed (out of the two possibilities).17,18 In the case of compound 25, although there is a possibility of two regioisomers, we obtained only one isomer. Based on the regiochemistry of a single click reaction, the reaction on the internal alkyne moiety was assumed to occur from its less hindered face. Use of the Cu(I)-stabilizing catalyst tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl] amine (TBTA)19 in this reaction provided the desired monosubstituted analogue 26.
Scheme 1.

Use of click reactions to couple a terminal alkynyl groups present on a functionalized chain of an adenosine analogue to various azido moieties. Reagents: A. (i) HC≡C(CH2)nC≡CH (n = 3, 4), Pd(PPh3)4, CuI, Et3N, DMF, rt; (ii) 10% TFA, MeOH, 70°C; (iii) appropriate azide, CuSO4.5H2O, sodium ascorbate, TBTA, THF/H2O or t-BuOH/H2O, rt; (iv) acetic acid N-hydroxysuccinimide ester, DMF, rt.
Scheme 2.

The unusual example of click reaction occuring at both alkynyl groups present on a functionalized chain of an adenosine analogue. When the catalyst TBTA was used, only the monosubstituted product 26 was isolated. Reagents: (i) CuSO4.5H2O, sodium ascorbate, THF/H2O, rt.; (ii) CuSO4.5H2O, sodium ascorbate, TBTA, t-BuOH/H2O, rt.
Compounds 15 and 17 were intended as models compounds for the amide coupling of amine functionalized congeners such as 16 to alkyl carboxylic carriers. Compound 18 was included in order to probe steric tolerance at the distal position.
The fluorescent squaraine-rotaxane derivative 22 was prepared from a commercial reactive azide, of undisclosed structure, which fluoresces strongly at 680 nm.20,21 Such rotaxane derivatives have the following advantages over other small fluorophores: improved chemical and photochemical stability, sharp absorption and emission bands, and stability over the pH range from 2–12. They are typically much brighter in fluorescence than the Alexa dyes.22 The elemental composition of this proprietary dye moiety, but not the full chemical structure, was revealed by the supplier, and the integrity of the synthetic product 22 was demonstrated by high resolution mass spectroscopy. The expected molecular weight of the click product of the 1:1 reaction with dialkyne 9 was obtained.
An isothiazolone azide derivative 32 designed as a bifunctional linker for thiol reactivity was synthesized as shown in Scheme 3. This served as the precursor for the adenosine derivative 19. The synthesis of isothiazolone derivatives from 3-benzoylpropionic acid was reported by Tsolomitis et al.23 An amide coupling reaction between 27 and p-toluidine formed 4-oxo-4-phenyl-N-p-tolyl-butyramide 28, which was treated with an excess of thionyl chloride to give 5-benzoyl-2-p-tolylisothiazol-3-one 29. Debenzoylation of 29 afforded 2-p-tolylisothiazol-3-one 30 in the presence of 10% aqueous sodium hydroxide in benzene, along with dithietane derivatives as by-products. Compound 30 was transformed with N-bromosuccinimide in the presence of a catalytic amount of benzoyl peroxide in carbon tetrachloride to the bromomethyl derivative 31, which upon treatment with sodium azide afforded 2-(4-azidomethylphenyl)isothiazol-3-one 32.
Scheme 3.

Synthesis of isothiazolone derivative 32 to serve as a thiol-reactive crosslinker. Reagents: (i) p-toluidine, EDC, HOBT, Et3N, 1,4-dioxane, rt, 16 h; (ii) thionyl chloride, rt, 16 h; (iii) 10% aqueous NaOH, benzene, rt, 3 d; (iv) N-bromosuccinimide, benzoyl peroxide, carbon tetrachloride, reflux, 2 h; (v) sodium azide, DMF.
Quantification of Pharmacological Activity
Binding assays at three hAR subtypes were carried out on the alkyne and triazole derivatives using standard radioligands24–26 and membrane preparations from Chinese hamster ovary (CHO) cells (A1 and A3) or human embryonic kidney (HEK293) cells (A2A) stably expressing a hAR subtype (Table 1).27,28 The previously reported molecular probes (1 – 3) were used for comparison in the biological assays.
Table 1.
Potency of a series of (N)-methanocarba adenosine derivatives at three subtypes of human ARs.
| Compd | Structure | Affinity (Ki, nM) or % inhibitiona | |||
|---|---|---|---|---|---|
| A1 | A2A | A3 | |||
| R1 | |||||
| 1b,c | C≡C(CH2)2CONH(CH2)2NH-CO-(CH2)5Cy5 | (36±3%) | 4730±1020 | 17.2±3.1 | |
| 2b,c | C≡C(CH2)2CONH(CH2)2NH-biotin | (1±1%) | (51±2%) | 36.4±5.6 | |
| 3b,c | C≡C(CH2)2CONH(CH2)2NH-CO(CH2)5NH-biotin | (12±4%) | (47±11%) | 57.7±16.2 | |
| 25e | 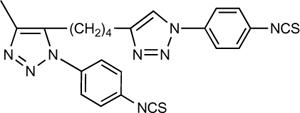 |
(4±2%) | (40±3%) | 8.8±1.3 | |
| 8 | C≡C(CH2)3C≡CH | (36±4%) | 4330±500 | 23.6±3.9 | |
| 9 | C≡C(CH2)4C≡CH | (31±2%) | 7040±1430 | 29.4±9.8 | |
| n | R2 | ||||
| 10 | 3 | 3-nitro-4-fluorophenyl | (10±3%) | (39±4%) | 26.0±8.2 |
| 11e | 4 | 3-nitro-4-fluorophenyl | (8±4%) | 6730±280 | 10.6±3.8 |
| 12 | 4 | 4-aminophenyl | (3±1%) | 5490±1150 | 87.1±13.1 |
| 13 | 4 | 4-carboxyphenyl | (0±0%) | (40±l%) | 180±23 |
| 26 | 4 | 4-isothiocyanatophenyl | (2±11%) | (28±4%) | 37.5±16.0 |
| 14e | 4 | 4-(α-Br-phenacyl) | (12±3%) | 5740±730 | 9.6±1.3 |
| 15 | 4 | (CH2)2NHCOCH3 | (12±4%) | 2440±320 | 22.3±1.6 |
| 16 | 4 | (CH2)4NH2 | (13±3%) | 1630±350 | 47.0±1.8 |
| 17 | 4 | (CH2)4NHCOCH3 | (17±1%) | 7240±510 | 89.5±12.6 |
| 18e | 4 | 1 -adamantyl | (22±2%) | 3280±700 | 6.5±0.5 |
| 19 | 4 | 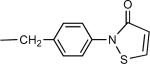 |
(15±3%) | (49±1%) | 102±25 |
| 20d | 4 | -(CH2)6NH-biotin | (0±0%) | (27±1%) | 285±54 |
| 21d | 4 | -(CH2)6CONH(CH2)2[O(CH2)]4NH-biotin | (0±0%) | (19±5%) | 235±43 |
| 22 | 4 | Rotaxane derivative | (0±0%) | (2±1%) | 239±43 |
| 23d | 4 | (CH2)6NHCO-Alexa Fluor | (0±0%) | (23±5%) | 416±45 |
All experiments were done on CHO or HEK293 (A2A only) cells stably expressing one of four subtypes of human ARs. The binding affinity for A1 A2A and A3ARs was expressed as Ki values (n = 3–5) and was determined by using agonist radioligands ([3H]36; [3H]37; or [125I]38; respectively), unless noted. A percent in parentheses refers to inhibition of radioligand binding at 10 μM.
Values from Tosh et al.9
Structure given in Chart 1.
Structure given in Scheme 1.
11, MRS5223; 14, MRS5226; 15, MRS5233; 18, MRS5224; 25, MRS5225.
The two homologous dialkyne intermediates 8 and 9 were equipotent with Ki values in A3AR binding of 24–29 nM. The selectivity of both intermediates in comparison to the A1 and A2AARs was >400 and roughly 300-fold, respectively. Following the click reaction with either aryl or alkyl azides, considerable affinity and selectivity at the A3AR were preserved. Thus, the main pharmacophore maintained its receptor recognition function in the triazole-extended series. The 3-nitro-4-fluorophenyl adducts 10 and 11 were roughly equipotent to the dialkyne precursors. The A3AR affinity of the p-substituted phenyl adducts 12, 13, 14, and 26 varied depending on the aryl substituent. The order of potency depending on the 4 position substitution was: bromoacetyl 14 > isothiocyanate 26 > amino 12 > carboxy 13. Compound 14 displayed a Ki value of 9.6 nM. The disubstituted click product 25 was highly potent in binding to the A3AR, with a Ki value of 8.8 nM. Thus, in spite of the added steric bulk, it was suprisingly 4-fold more potent than the monosubstituted isothiocyanate derivative 26.
Among adducts of alkyl azides, a short chain acetamidoalkyl derivative 15 was moderately potent with a Ki value of 22 nM, and this ethyl derivative was 4-fold more potent than a higher butyl homologue 17. Compound 15 was 109-fold selective for the A3AR in comparison to the A2AAR (Figure 1A). The free aminobutyl precursor derivative 16 displayed only moderate affinity at the A3AR. The sterically bulky 1-adamantyl adduct 18 was highly potent in binding with a Ki value of 6.5 nM at the A3AR and 500-fold selectivity in comparison to the A2AAR (Figure 1B).
Figure 1.

Inhibition of radioligand binding by (N)-methanocarba nucleoside analogues in membranes of CHO cells expressing the human A1 (■) and A2A (▴ ) and A3 (▾) ARs. Inhibition curves are shown for the agonist analogues containing a triazole group, the adamantyl derivative 18, and the amide model compound for click linkage to carriers 15. Both of the analogues shown were highly selective for the hA3AR in comparison to the A1 and A2AARs.
A thiol-reactive derivative 19 displayed only moderate affinity at the A3AR. The two derivatives of biotin 20 and 21 having unbranched chains and a fluorescent derivative of Alexa Fluor 488 23 were relatively weak in binding to the A3AR with Ki values in the range of 200 – 400 nM. The longer biotin derivative 21 contained a tetraethylene glycol spacer, which apparently did not enhance affinity. The rotaxane derivative 22 bound to the A3AR receptor with a Ki value of 239 nM.
The click products were tested in a functional assay at the A3AR (inhibition of forskolin-stimulated cAMP production29,30 in A3AR-expressing CHO cells) as shown in Table 2. The (N)-methanocarba derivatives displayed A3AR agonist properties with varying degrees of maximal inhibition of cAMP production at 10 μM. In general, the degree of inhibition was similar to or less than that of 5′-N-ethylcarboxamidoadenosine (NECA), taken as a reference standard. Therefore, some of these derivatives, e.g., 4-fluoro-3-nitrophenyl 11, bromophenacyl 14, acetamidoethyl 15, and rotaxane 22 derivatives, were highly efficacious agonists. Compounds that displayed intermediate (50–90%) efficacies were: 9, 13, 16, 17, 19, and 25. Agonists of lower efficacy (<50%) were the short dialkyne derivative 8 and the aminophenyl 12, isothiocyanate 26, and 1-adamantyl 18 triazole derivatives. The affinity of compounds 20, 21, and 23 was so low that a 10 μM test concentration in the cAMP assay was not sufficient to ensure full receptor occupancy. The corresponding efficacy values for two A3AR agonists, N6-(3-iodobenzyl)-5′-N-methylcarboxamidoadenosine (IB-MECA) and its 2-chloro analogue Cl-IB-MECA, which are currently in Phase II clinical trials, are included for comparison.1 Both are full agonists in this assay.
Table 2.
Maximal efficacy of (N)-methanocarba adenosine derivatives in a functional assay at the A3AR.
| Compound | % Inhibition of cAMP formationa at hA3 AR |
|---|---|
| IB-MECA | 99±6 |
| Cl-IB-MECA | 97 |
| 36, NECA | 100 |
| 1b | 94.4±9.6 |
| 2b | 84.5±12.0 |
| 3b | 107±18 |
| 25 | 83.6±7.2 |
| 8 | 18.3±7.6 |
| 9 | 60.2±17.0 |
| 10 | ND |
| 11 | 116±22 |
| 12 | 44.3±2.4 |
| 13 | 83.4±13.1 |
| 26 | 19.5±14.5 |
| 14 | 109±12 |
| 15 | 93.6±17.7 |
| 16 | 64.4±12.4 |
| 17 | 59.7±19.2 |
| 18 | 27.8±17.4 |
| 19 | 75.9±16.5 |
| 20 | 55.6±12.5 |
| 21 | 41.8±11.3 |
| 22 | 111±18 |
| 23 | 37.8±14.6 |
The efficacy at the human A3AR was determined by inhibition of forskolin-stimulated cyclic AMP production in AR-transfected CHO cells, as described in the text. At a concentration of 10 μM, in comparison to the maximal effect of a full agonist NECA at 10 μM. Data are expressed as mean ± standard error (n = 3). ND, not determined.
Values from Tosh et al.9
This series of (N)-methanocarba nucleosides is known to be much weaker in interaction with the A2BAR than with other subtypes.12 We have tested selected nucleosides at 10 μM for agonist activity in the stimulation of cAMP accumulation in CHO cells stably expressing the human A2BAR. The percent stimulation was: 100% (NECA), 30.2% (11), 19.4%, (15), and 36.5% (18).
Discussion
Click chemistry as a means of assembling complex ligands and for introducing structural diversity is finding increasing application in biological systems.15 With respect to the ARs, a series of triazole derivatives of adenosine was previously prepared using click chemistry, resulting in A3AR selective agonists, partial agonists, and antagonists.31 The triazole ring was directly attached at the 2 position of adenine, which caused wide variation of the receptor affinity and relative efficacy of the analogues. However, in the present study, the triazole was incorporated at a more distal position of an elongated and flexible 2-adenine substituent. This resulted in a greater degree of preservation of the agonist properties in this series than in the previous study, in which some derivatives became antagonists.31 Thus, we have used the azide/alkyne cycloaddition reaction to easily synthesize a wide range of biologically active molecules. The click reaction was intended for linking adenosine functionalized congeners to other moieties and carriers, such as reporter groups, chemically reactive groups, and macromolecular carriers. Other studies of biologically-active small molecules have used click cycloaddition reactions to incorporate reporter groups, such as fluoresecent dyes and biotin.32,33
The small molecule adducts in the present series were not uniformly potent as A3AR selective agonists. Rather, only certain derivatives displayed exceptionally high affinity at this subtype (Ki <20 nM): 14, 18, and 25. Two different lengths of dialkynyl chains were compared in binding at the A3AR. The octadiynyl derivative 9 appeared to be better suited than the shorter homologue 8 for binding to this receptor when coupled to an aryl azide (cf. 10 and 11). Therefore, the octadiynyl chain was used in subsequent derivatives. A study by Seela and coworkers also utilized octadiynyl-derivatized nucleosides that were incorporated into oligonucleotides for click reactions that took place exclusively at the distal alkynyl group.34 Compounds 10, 14, 19, and 26 were designed as potential affinity labels of the A3AR, but the chemical irreversibility of binding was not tested in the present study. The use of isothiocyanates, 3-isothiazolones, and other electrophilic derivatives to affinity label biopolymers has been described.13,35
In conclusion, the most potent and selective novel compound was a 1-adamantyl derivative (Ki 6.5 nM), which suggested the existence of a hydrophobic binding pocket in this region of the receptor. Curiously, this compound proved to be a partial agonist of the A3AR with only 27.8% of the maximal efficacy in comparison to NECA. Various other click products were in the Ki range of 200–400 nM. Other potent, selective derivatives (Ki at A3AR in nM) were intended as possible receptor affinity labels: 3-nitro-4-fluorophenyl derivative 11 (10.6), α-bromophenacyl 14 (9.6), thiol-reactive isothiazolone 19 (102), and arylisothiocyanate 26 (37.5). The maximal functional effects in inhibition of forskolin-stimulated cAMP were measured, indicating that this class of click adducts varied from partial to full A3AR agonist compared to other widely used agonists. Thus, this strategy provides a general chemical approach to linking potent and selective A3AR agonists to reporter groups of diverse structure and to carrier moieties.
One disadvantage of the standard cycloaddition reaction is that cuprous ions are required. This reactant might not be compatible with the full range of GPCR ligands desired to be coupled to carriers and is certainly not useful in cell systems. To overcome this drawback, Bertozzi and colleagues have explored cyclooctyne derivatives that are substrates for “copper-free” click chemistry.36 These groups provide greater compatibility with living systems and allow covalent coupling to azido groups on biopolymers and other biomolecules. We are currently extending these results to application of cyclooctynyl derivatives of A3AR agonists to conjugation using copper-free click chemistry.
Experimental Section
Chemical Synthesis
1H NMR spectra were obtained with a Varian Gemini 300 spectrometer. When using D2O was used as a solvent, the chemical shifts are expressed as relative ppm from HOD (4.80 ppm).
The purity of the final nucleotide derivatives were determined using a Hewlett–Packard 1100 HPLC equipped with a Zorbax Eclipse 5 mm XDB-C18 analytical column (250 × 4.6 mm; Agilent Technologies Inc, Palo Alto, CA), using a linear gradient solvent system: 5 mM TBAP (tetrabutylammonium dihydrogenphosphate)-CH3CN from 80:20 to 40:60 in 20 min with a flow rate of 1 mL/min. Peaks were detected by UV absorption (254 nm) using a diode array detector. All derivatives tested for biological activity were shown to be at least 97% pure using this analytical HPLC system.
High-resolution mass measurements were performed on a Micromass/Waters LCT Premier Electrospray Time of Flight (TOF) mass spectrometer coupled with a Waters HPLC system. Unless noted otherwise, reagents and solvents were purchased from Sigma-Aldrich (St. Louis, MO). Solutions of the nucleoside analogues in DMSO (5 mM) were prepared for biological testing and stored at −20°C.
The squaraine-rotaxane azide derivative SRfluor® 680 Azide was obtained from Molecular Targeting Technologies, Inc. (West Chester, PA). Alexa Fluor 488 azide and biotin(PEG)4 azide were purchased from Invitrogen Corp. (Carlsbad, CA). DMEM/F12 medium and 1 M Tris-HCl (pH 7.5) were purchased from Mediatech, Inc. (Herndon, VA). Unless noted otherwise, reagents and solvents were purchased from Sigma-Aldrich (St. Louis, MO).
(1′S, 2′R, 3′S, 4′S, 5′S)-4′-[6-(3-Chlorobenzylamino)-2-(1,6-heptadiynyl)-9H-purin-9-yl]-(1′S, 2′R, 3′S, 4′S, 5′S)-4′-[6-(3-Chlorobenzylamino)-2-(1,7-octadiynyl)-9H-purin-9-yl]-2′,3′-O-isopropylidenebicyclo[3.1.0]hexane-1′-carboxylic acid N-methylamide (7)
To a solution of compound 5 (440 mg, 0.73 mmol) in anhydrous DMF (12 mL), Pd(PPh3)4 (92 mg, 0.08 mmol), CuI (30.5 mg, 0.16 mmol), 1,7-octadiyne (1.0 mL, 8.01 mmol) and then triethylamine (0.22 mL, 1.6 mmol) was added. The reaction mixture was heated at 60 °C for overnight. Solvent was evaporated under vacuum and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH = 70:1) to give the compound 7 (352 mg, 83%) as foamy syrup. 1H NMR (CD3OD, 300 MHz)δ8.11 (s, 1H), 7.43 (s, 1H), 7.26–7.33 (m, 3H), 5.74 (d, J = 7.2 Hz, 1H), 5.01 (s, 1H), 4.83 (m, 1H), 2.87 (s, 3H), 2.53 (t, J = 6.9 Hz, 2H), 2.34–2.31 (m, 3H), 2.10–2.15 (m, 1H), 1.71–1.84 (m, 4H), 1.54–1.57 (m, 4H), 1.40 (t, J = 5.4 Hz, 1H), 1.29 (s, 3H). HRMS calculated for C31H34ClN6O3 (M + H)+: 573.2381; found 573.2397.
2′,3′-O-isopropylidenebicyclo[3.1.0]hexane-1′-carboxylic acid N-methylamide (6)
Compound 6 (81%) was synthesized from 5 following same procedure as for compound 7. 1H NMR (CD3OD, 300 MHz) δ 8.12 (s, 1H), 7.45 (s, 1H), 7.28–7.34 (m, 3H), 5.76 (d, J = 6.9 Hz, 1H), 5.02 (s, 1H), 4.85–4.87 (m, 1H), 2.88 (s, 3H), 2.64 (t, J = 6.9 Hz, 2H), 2.41–2.46 (m, 2H), 2.31 (t, J = 2.7, 1H) 2.12–2.17 (m, 1H), 1.85–1.94 (m, 2H), 1.56 (m, 4H), 1.40–1.44 (m, 1H). 1.30 (s, 3H). HRMS calculated for C30H32ClN6O3 (M + H)+: 559.3046; found 559.3085.
(1′S, 2′R, 3′S, 4′S, 5′S)-4′-[6-(3-Chlorobenzylamino)-2-(1,6-heptadiynyl)-9H-purin-9-yl]-2′,3′-dihydroxybicyclo[3.1.0]hexane-1′-carboxylic acid N-methylamide (8)
Compound 8 (86%) was synthesized from 6 following same procedure as for compound 9. 1H NMR (CD3OD, 300 MHz) δ 8.07 (s, 1H), 7.42 (s, 1H), 7.25–7.42 (m, 3H), 5.01 (d, J = 6.9 Hz, 1H), 4.84–4.87 (m, 1H), 3.98 (d, J = 6.6 Hz, 1H), 2.86 (s, 3H), 2.59 (t, J = 7.2 Hz, 2H), 2.36–2.44 (m, 2H), 2.28 (t, J = 2.4 Hz, 1H), 2.06–2.10 (m, 1H), 1.79–1.88 (m, 3H), 1.34–1.39 (m, 1H). HRMS calculated for C27H28ClN6O3 (M + H)+: 519.1911; found 519.1912.
(1′S, 2′R, 3′S, 4′S, 5′S)-4′-[6-(3-Chlorobenzylamino)-2-(1,7-octadiynyl)- 9H-purin-9-yl]-2′,3′-dihydroxybicyclo[3.1.0]hexane-1′-carboxylic acid N-methylamide (9)
To a solution of compound 7 (350 mg, 0.61 mmol) in methanol (7 mL), 10% trifluromethane sulfonic acid was added and heated at 70 °C for 6 h. Solvent was evaporated and the residue was purified on flash silica gel chromatography (CH2Cl2:MeOH = 40:1) to give the compound 9 (295 mg, 91%) as a syrup. 1H NMR (CD3OD, 300 MHz) δ 8.08 (s, 1H), 7.43 (s, 1H), 7.28–7.33 (m, 3H), 5.02 (d, J = 6.6 Hz, 1H), 4.80–4.82 (M, 1H), 4.00 (dd, J1 = 1.2 Hz, J2 = 5.7 Hz, 1H), 2. 87 (s, 3H), 2.51 (t, J = 6.9 Hz, 1H), 2.22–2.30 (m, 3H), 2.07–2.10 (m, 1H), 1.86 (t, J = 5.1 Hz, 1H), 1.70–1.81 (m, 4H), 1.35–1.40 (m, 1H). HRMS calculated for C28H30ClN6O3 (M + H)+: 533.2068; found 533.2082.
(1S,2R,3S,4R,5S)-4′-(6-(3-Chlorobenzylamino)-2-(6-(1-(4-fluoro-3-nitrophenyl)-1H-1,2,3-triazol-4-yl)pent-1-ynyl)-9H-purin-9-yl)-2′,3′-dihydroxybicyclo[3.1.0]hexane-1′-carboxylic acid N-methylamide (10)
Compound 10 (89%) was synthesized from 8 following same procedure as for compound 11. 1H NMR (CD3OD, 300 MHz) δ 8.52–8.56 (m, 2H), 8.18–8.22 (m, 1H), 8.08 (s, 1H), 7.59–7.65 (m, 1H), 7.41 (s, 1H), 7.25–7.32 (m, 3H), 5.04 (d, J = 5.4 Hz, 1H), 4.80–4.83 (m, 1H), 3.99 (dd, J1 = 0.9 Hz, J2 = 5.7 Hz, 1H), 3.04 (t, J = 7.5 Hz, 2H), 2.87 (s, 3H), 2.60 (t, J = 6.6 Hz, 2H), 2.07–2.17 (m, 3H), 1.86 (t, J = 5.1 Hz, 1H), 1.38–1.45 (m, 2H), 0.89–0.96 (m, 1H). HRMS calculated for C33H31ClFN10O5 (M + H)+: 701.2151; found 701.2172.
(1S,2R,3S,4R,5S)-4′-(6-(3-Chlorobenzylamino)-2-(6-(1-(4-fluoro-3-nitrophenyl)-1H-1,2,3-triazol-4-yl)hex-1-ynyl)-9H-purin-9-yl)-2′,3′-dihydroxybicyclo[3.1.0]hexane-1′-carboxylic acid N-methylamide (11)
To a mixture of compound 9 (34 mg, 0.063 mmol) and 4-fluoro-3-nitro-phenyl azide (16.2 mg, 0.088 mmol) in in THF/H2O 3:1 (2 mL), was added freshly prepared 1M sodium ascorbate (51 μL, 0.05 mmol) followed by 7.5% aqueous copper sulfate pentahydrate solution (42 μL, 0.012 mmol) and stirred for over night at room temperature. Solvent was evaporated and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH = 45:1) to give the clicked product 11 (42 mg, 94%) as a syrup. 1H NMR (CD3OD, 300 MHz) δ 8.57–8.58 (m, 1H), 8.49 (s, 1H), 8.20–8.25 (m, 1H), 8.09 (s, 1H), 7.60–7.67 (m, 1H), 7.40 (s, 1H), 7.20–7.32 (m, 3H), 5.03 (d, J = 6.6 Hz, 1H), 4.84–4.87 (m, 1H), 3.99 (d, J = 6.3 Hz, 1H), 2.91 (t, J = 7.5 Hz, 2H), 2.87 (s, 3H), 2.56 (t, J = 6.9 Hz, 2H), 1.96–2.11 (m, 3H), 1.75–1.87 (m, 3H), 1.31–1.40 (m, 2H). HRMS calculated for C34H33ClFN10O5 (M + H)+: 715.2308; found 715.2347.
(1S,2R,3S,4R,5S)-4′-(6-(3-Chlorobenzylamino)-2-(6-(1-(4-amino-phenyl)-1H-1,2,3-triazol-4-yl)hex-1-ynyl)-9H-purin-9-yl)-2′,3′-dihydroxybicyclo[3.1.0]hexane-1′-carboxylic acid N-methylamide (12)
To a mixture of compound 9 (4.46 mg, 0.008 mmol) and 4-amino-phenylazide (2 mg, 0.011 mmol) in a mixture of t-butanol (0.5 mL) and water (0.5 mL), was added TBTA (1 mg, 0.001 mmol) and freshly prepared sodium ascorbate (8.3 μL, 0.008 mmol) followed by copper sulfate (8.3 μL, 0.003 mmol). The reaction mixture was stirred at room temperature for overnight, solvent was evaporated and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH = 30:1) to give the compound 12 (4 mg, 72%) as a syrup. 1H NMR (CD3OD, 300 MHz) δ 8.08 (s, 1H), 8.07 (s, 1H), 7.23–7.41 (m, 6H), 6.74 (d, J = 8.7 Hz, 2H), 5.01 (d, J = 6.9 Hz, 1H), 4.78–4.82 (m, 1H), 3.96 (d, J = 6.0 Hz, 1H), 2.82–2.86 (m, 5H), 2.53 (t, J = 6.9 Hz, 2H), 2.06–2.16 (m, 2H), 1.93–1.98 (m, 2H), 1.71–1.87 (m, 4H), 1.29–1.39 (m, 1H). HRMS calculated for C34H35ClN10O3Na (M + Na)+: 689.2480; found 689.2465.
(1S,2R,3S,4R,5S)-4′-(6-(3-Chlorobenzylamino)-2-(6-(1-(4-carboxyl-phenyl)-1H-1,2,3-triazol-4-yl)hex-1-ynyl)-9H-purin-9-yl)-2′,3′-dihydroxybicyclo[3.1.0]hexane-1′-carboxylic acid N-methylamide (13)
Compound 13 (81%) was synthesized from 9 following same procedure as for compound 12. 1H NMR (CD3OD, 300 MHz) δ 8.39 (s, 1H), 8.09–8.14 (m, 3H), 7.81–7.84 (m, 2H), 7.22–7.44 (m, 4H), 5.04 (d, J = 6.6 Hz, 1H), 4.83–4.85 (m, 1H), 3.99 (d, J = 6.6 Hz, 1H), 2.89–2.94 (m, 5H), 2.57 (t, J = 6.6 Hz, 2H), 1.98–2.14 (m, 4H), 1.77–1.90 (m, 4H), 1.38–1.43 (m, 1H). HRMS calculated for C35H33ClN9O5 (M − H)+: 694.2293; found 694.2311.
(1S,2R,3S,4R,5S)-6-(3-Chlorobenzylamino)-4-(2-(6-(4-(2-bromoacetyl)phenyl)-1H-1,2,3-triazol-4-yl)hex-1-ynyl)-9H-purin-9-yl)-2′,3′-dihydroxybicyclo[3.1.0]hexane-1′-carboxylic acid N-methylamide (14)
Compound 14 (79%) was synthesized from 9 following same procedure as for compound 11. 1H NMR (CD3OD, 300 MHz) δ 8.43 (s, 1H), 8.33 (s, 1H), 8.07–8.15 (m, 4H), 7.21–7.42 (m, 4H), 5.01 (d, J = 6.6 Hz, 1H), 4.77–4.82 (m, 1H), 4.70 (s, 2H), 3.96 (d, J = 6.9 Hz, 1H), 2.86–2.92 (m, 5H), 2.41 (t, J = 5.1 Hz, 2H), 1.93–2.10 (m, 4H), 1.74–1.87 (m, 4H), 1.35–1.40 (m, 1H). HRMS calculated for C36H35ClBrN9O4Na (M + Na)+: 796.1738; found 796.1713.
(1S,2R,3S,4R,5S)-6-(3-Chlorobenzylamino)-(2-(6-(1-(4-acetamidoethyl)-1H-1,2,3-triazol-4-yl)hex-1-ynyl)-9H-purin-9-yl)-2′,3′-dihydroxybicyclo[3.1.0]hexane-1′-carboxylic acid N-methylamide (15)
Compound 15 (86%) was synthesized from 9 following same procedure as for compound 11. 1H NMR (CD3OD, 300 MHz) δ 8.09 (s, 1H), 7.78 (s, 1H), 7.43 (s, 1H), 7.27–7.33 (m, 3H), 5.03 (d, J = 5.1 Hz, 1H), 4.83–86 (m, 1H), 4.47 (t, J = 5.7 Hz, 2H), 4.01 (d, J = 6.6 Hz, 1H), 3.62 (t, J = 5.7 Hz, 2H), 2.87 (s, 3H), 2.79 (t, J = 7.5 Hz, 2H), 2.53 (t, J = 7.2 Hz, 2H), 2.07–2.10 (m, 1H), 1.85–1.93 (m, 5H), 1.69–1.74 (m, 2H), 1.36–1.41 (m, 1H). HRMS calculated for C32H38ClN10O4 (M + H)+: 661.2766; found 661.2751.
(1S,2R,3S,4R,5S)-6-(3-Chlorobenzylamino)-(2-(6-(1-(4-aminobutyl)-1H-1,2,3-triazol-4-yl)hex-1-ynyl)-9H-purin-9-yl)-2′,3′-dihydroxybicyclo[3.1.0]hexane-1′-carboxylic acid N-methylamide (16)
Compound 16 (73%) was synthesized from 9 following same procedure as for compound 12. 1H NMR (CD3OD, 300 MHz) δ 8.11 (s, 1H), 7.81 (s, 1H), 7.45 (s, 1H), 7.28–7.34 (m, 3H), 5.06 (d, J = 6.3 Hz, 1H), 4.80–4.83 (m, 1H), 4.42 (t, J = 6.6 Hz, 2H), 4.01 (d, J = 6.6 Hz, 1H), 2.85–2.88 (m, 5H), 2.79–2.84 (m, 4H), 2.54 (t, J = 7.2 Hz, 2H), 2.08–2.12 (m, 1H), 1.86–1.99 (m, 5H), 1.66–1.78 (m, 2H), 1.54–1.62 (m, 2H), 1.38–1.43 (m, 1H). HRMS calculated for C32H40ClN10O3 (M + H)+: 647.2973; found 647.2968.
(1S,2R,3S,4R,5S)-6-(3-Chlorobenzylamino)-(2-(6-(1-(4-acetamidobutyl)-1H-1,2,3-triazol-4-yl)hex-1-ynyl)-9H-purin-9-yl)-2′,3′-dihydroxybicyclo[3.1.0]hexane-1′-carboxylic acid N-methylamide (17)
To a solution of compound 16 (1.79 mg, 0.002 mmol) in anhydrous DMF (0.5 mL), acetic acid N-hydroxysuccinimide ester (1 mg, 0.006 mmol) was added and the mixture stirred at room temperature for overnight. Solvent was evaporated and the residue was purified in preparative TLC (CH2Cl2:MeOH = 25:1) to give the compound 17 as a syrup (1.26 mg, 66 %). 1H NMR (CD3OD, 300 MHz) δ 8.11 (s, 1H), 7.80 (s, 1H), 7.44 (s, 1H), 7.27–7.33 (m, 3H), 5.04 (d, J = 6.3 Hz, 1H), 4.83–4.85 (m, 1H), 4.39 (t, J = 6.9 Hz, 2H), 3.70 (t, J = 3.9 Hz, 4H), 3.59 (m, 4H), 2.87 (s, 3H), 2.78–2.86 (m, 2H), 2.48–2.57 (m, 2H), 1.93 (s, 3H), 1.85–1.91 (m, 2H), 1.68–1.79 (m, 2H), 1.39–1.52 (m, 2H), 1.23–1.43 (m, 2H). HRMS calculated for C34H41ClN10O4Na (M + Na)+: 711.2898; found 711.2917.
(1S,2R,3S,4R,5S)-6-(3-Chlorobenzylamino)-(2-(6-(1-(adamantyl)-1H-1,2,3-triazol-4-yl)hex-1-ynyl)-9H-purin-9-yl)-2′,3′-dihydroxybicyclo[3.1.0]hexane-1′-carboxylic acid N-methylamide (18)
Compound 18 (76%) was synthesized from 9 following same procedure as for compound 11. 1H NMR (CD3OD, 300 MHz) δ 8.09 (s, 1H), 7.81 (s, 1H), 7.44 (s, 1H), 7.25–7.33 (m, 3H), 5.03 (d, J = 6.6 Hz, 1H), 4.83–4.85 (m, 1H), 3.98 (d, J = 6.0 Hz, 1H), 2.86 (s, 3H), 2.79 (t, J = 7.2 Hz, 2H), 2.51 (t, J = 6.9 Hz, 2H), 2.16 (s, 9H), 2.06–2.10 (m, 1H), 1.68–1.95 (m, 13H), 1.36–1.40 (m, 1H). HRMS calculated for C38H45ClN9O3 (M + H)+: 710.3334; found 710.3352.
(1S,2R,3S,4R,5S)-6-(3-Chlorobenzylamino)-(2-(1-(4-3-oxoisothiazol-2(3H)-yl)benzyl-1H-1,2,3-triazol-4-yl)hex-1-ynyl)-9H-purin-9-yl)-2′,3′-dihydroxybicyclo[3.1.0]hexane-1′-carboxylic acid N-methylamide (19)
Compound 19 (81%) was synthesized from 9 following same procedure as for compound 11. 1H NMR (CD3OD, 300 MHz) δ 8.56 (d, J = 6.3 Hz, 1H), 8.08 (s, 1H), 7.83(s, 1H), 7.53–7.56 (m, 2H), 7.39–7.42 (m, 2H), 7.25–7.30 (m, 4H), 6.29 (d, J = 6.3 Hz, 1H), 5.6 (s, 2H), 5.02 (d, J = 6.9 Hz, 1H), 4.80–4.83 (m, 1H), 3.97 (d, J = 6.0 Hz, 1H), 2.86 (s, 3H), 2.80 (t, J = 7.2 Hz, 2H), 2.50 (t, J = 7.2 Hz, 2H), 2.06–2.10 (m, 1H), 1.84–1.92 (m, 4H), 1.67–1.74 (m, 2H), 1.35–1.40 (m, 1H). HRMS calculated for C38H38ClSN10O4 (M + H)+: 765.2487; found 765.2461.
(1S,2R,3S,4R,5S)-6-(3-Chlorobenzylamino)-(2-(6-(1-(6-(5-(3aS,4S,6aR)-oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)pentanamido)hexyl-1H-1,2,3-triazol-4-yl)hex-1-ynyl)-9H-purin-9-yl)-2′,3′-dihydroxybicyclo[3.1.0]hexane-1′-carboxylic acid N-methylamide (20)
Compound 20 (73%) was synthesized from 9 and the appropriate azide [ref. 38] following same procedure as for compound 12. 1H NMR (CD3OD, 300 MHz) δ 8.08 (s, 1H), 7.77 (s, 1H), 7.42 (s, 1H), 7.23–7.31 (m, 3H), 5.02 (d, J = 6.3 Hz, 1H), 4.83–4.86 (m, 1H), 4.45–4.49 (m, 1H), 4.26–4.35 (m, 3H), 3.98 (d, J = 7.2 Hz, 1H), 3.65–3.69 (m, 3H), 3.54–3.57 (m, 3H), 3.03–3.24 (m, 4H), 2.85 (s, 3H), 2.78 (t, J = 7.5 Hz, 2H), 2.51 (t, J = 7.2 Hz, 2H), 2.06–2.28 (m, 6H), 1.81–1.89 (m, 6H), 1.60–1.72 (m, 6H), 1.34–1.46 (m, 2H). HRMS calculated for C44H57ClN12O5SNa (M + Na)+: 923.3882; found 923.3887.
1S,2R,3S,4R,5S)-6-(3-Chlorobenzylamino)-(2-(6-(1-(7-oxo-7-(6-(5-(3aS,4S,6aR)-oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)pentanamido)hexylamino)heptyl)-1H-1,2,3-triazol-4-yl)hex-1-ynyl)-9H-purin-9-yl)-2′,3′-dihydroxybicyclo[3.1.0]hexane-1′-carboxylic acid N-methylamide (21)
Compound 21 (82%) was synthesized from 9 following same procedure as for compound 12. 1H NMR (CD3OD, 300 MHz) δ 8.03 (s, 1H), 7.81 (s, 1H), 4.46 (s, 1H), 7.29–7.35 (m, 3H), 5.05 (d, J = 6.5 Hz, 1H), 4.80–4.85 (m, 1H), 4.42–4.46 (m, 4H), 3.99 (d, J = 6.9 Hz, 1H), 3.79–3.85 (m, 8H), 3.56–3.62 (m, 8H), 3.15–3.26 (m, 4H), 2.89 (s, 3H), 2.23–2.57 (m, 6H), 1.03–1.91 (bm, 28H). HRMS calculated for C55H ClN78cln13O10SNa (M + Na)+: 1170.5302; found 1170.5288.
2-(6-Amino-3-imino-4,5-disulfonato-3H-xanthen-9-yl)-5-(6-(4-(6-(6-3-chlorobenzylamino)-9-((1S,2R,3S,4R,5S)-3,4-dihydroxy-5-(methylcarbamoyl)bicycle[3.1.0]hexane-2-yl)-9H-purin-2-yl)hex-5-ynyl)-1H-1,2,3-triazol-1-yl)hexylcarbamoyl)benzoate (23)
Compound 23 (76%) was synthesized from 9 following same procedure as for compound 12. 1H NMR (CD3OD, 300 MHz) δ 8.56 (s, 1H), 8.07(s, 1H), 7.83 (s, 1H), 7.26–7.43 (m, 4H), 7.09–7.12 (m, 2H), 6. 90 (d, J = 6.9 Hz, 2H), 5.75 (d, J = 6.7 Hz, 1H), 5.57 (d, J = 6.8 Hz, 1H), 4.98 (d, J = 6.6 Hz, 1H), 4.80–4.84 (m, 1H), 4.13 (t, J = 3.6 Hz, 2H), 3.98 (d, J = 7.2 Hz, 1H), 3.69–3.72 (m, 4H), 3.59–3.64 (m, 4H), 2.89 (s, 3H), 2.50–2.55 (m, 4H), 2.19–2.32 (m, 4H), 1.21–1.46 (m, 7H). HRMS calculated for C55H54ClN12O13S2 (M + H)+: 1189.3063; found 1189.3038. Fluorescence (aq. pH 7.4): λex 490 nm, λex 520 nm.
(1S,2R,3S,4R,5S)-6-(3-Chlorobenzylamino)-(2-(1,6-bis(4-isothiocyante-phenyl)-1H-bis(1,2,3-triazol-4-yl)hex-1-ynyl)-9H-purin-9-yl)-2′,3′-dihydroxybicyclo[3.1.0]hexane-1′-carboxylic acid N-methylamide (25)
To a mixture of compound 9 (17 mg, 0.031 mmol) and 4-azidophenyl isothiocyante (7.8 mg, 0.044 mmol) in in THF/H2O 3:1 (1.2 mL), was added freshly prepared 1M sodium ascorbate (26 μL, 0.025 mmol) followed by 7.5% aqueous copper sulfate pentahydrate solution (21 μL, 0.006 mmol) and stirred for 2 d at room temperature. Solvent was evaporated and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH = 20:1) to give an unusual clicked product 25 (21 mg, 75%) as a syrup. 1H NMR (CD3OD, 300 MHz) δ 8.29 (s, 1H), 8.12 (s, 1H), 7.71 (d, J = 8.7 Hz, 2H), 7.63 (d, J = 9.0 Hz, 2H), 7.50 (d, J = 6.6 Hz, 2H), 7.41 (s, 1H), 7.23–7.30 (m, 3H), 7.08 (d, J = 6.6 Hz, 2H), 5.02 (d, J = 5.2 Hz, 1H), 4.79–4.81 (m, 1H), 3.97 (d, J = 6.6 Hz, 1H), 2.83–2.91 (m, 5H), 2.55 (t, J = 6.9 Hz, 2H), 1.97–2.09 (m, 4H), 1.74–1.86 (m, 4H), 1.39–1.47 (m, 1H). HRMS calculated for C41H40ClN14O3S (M−CS)+: 843.2817; found 843.2812.
(1S,2R,3S,4R,5S)-6-(3-Chlorobenzylamino)-(2-(6-(1-(4-isothiocyante-phenyl)-1H-1,2,3-triazol-4-yl)hex-1-ynyl)-9H-purin-9-yl)-2′,3′-dihydroxybicyclo[3.1.0]hexane-1′-carboxylic acid N-methylamide (26)
To a mixture of compound 9 (4.22 mg, 0.007 mmol) and 4-isothiocyante-phenylazide (2 mg, 0.011 mmol) in a mixture of t-butanol (0.3 mL) and water (0.3 mL), was added TBTA (1 mg, 0.001 mmol) and freshly prepared sodium ascorbate (7.9 μL, 0.007 mmol) followed by copper sulfate (7.9 μL, 0.002 mmol). The reaction mixture was stirred at room temperature for overnight. The starting material and product came in same Rf value in TLC. Solvent was evaporated and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH = 25:1) to give the compound 26 (3.8 mg, 69%) as a syrup, no diclicked product was detected. 1H NMR (CD3OD, 300 MHz) δ 8.35 (s, 1H), 8.09 (s, 1H), 7.83 (d, J = 6.9 Hz, 2H), 7.40–7.43 (m, 3H), 7.24–7.32 (m, 3H), 5.02 (d, J = 6.3 Hz, 1H), 4.83–4.85 (m, 1H), 3.97 (d, J = 6.3 Hz, 1H), 2.87–2.92 (m, 5H), 2.55 (t, J = 6.9 Hz, 2H), 1.97–2.13 (m, 4H), 1.87 (t, J = 4.5 Hz, 2H), 1.74–1.83 (m, 2H), 1.38–1.41 (m, 1H). HRMS calculated for C35H34ClN10O3S (M + H)+: 709.2225; found 709.2236.
4-Oxo-4-phenyl-N-p-tolyl-butyramide(28)
EDC (347 mg, 1.81 mmol) and HOBT (254 mg, 1.88 mmol) were added to mixture of p-toluidine (129 mg, 1.20 mmol) and 3-benzoylpropionic acid 27 (218 mg, 1.21 mmol) in 1,4-dioxane (3 mL) at room temperature. It was stirred for 20 min and then added triethylamine (0.5 mL, 3.59 mmol). The reaction mixture was stirred for 20 h at room temperature and the solvent was evaporated under vacuum. The residue was purified by flash silica gel column chromatography (hexane:ethyl acetate = 3:1 to 2:1) yielded the compound 28 (252 mg, 78%). 1H-NMR (CDCl3, 300MHz) δ 2.30 (s, 3H), 2.80 (t, J = 6.3 Hz, 2H), 3.45 (t, J = 6.3 Hz, 2H), 7.10 (d, J = 8.4 Hz, 2H), 7.39 (d, J = 8.4 Hz, 2H), 7.47 (m, 2H), 7.58 (m, 1H), 7.99 (m, 1H).
5-Benzoyl-2-p-tolylisothiazol-3-one (29)
A solution of 4-oxo-4-phenyl-N-p-tolyl-butyramide 28 (252 mg, 0.944 mmol) in thionyl chloride (1 mL) was stirred for 16 h at room temperature. Excess thionyl chloride was removed under vacuum and the residue was purified by flash silica gel column chromatography (hexane:ethyl acetate = 10:1 to 4:1) to give the compound 29 (216 mg, 77%). 1H-NMR (CDCl3, 300 MHz) δ 2.40 (s, 3H), 6.82 (s, 1H), 7.28 (m, 2H), 7.54 (m, 4H), 7.71 (m, 1H), 7.96 (m, 2H). HRMS: calculated for C17H14NO2S (M+H)+ 296.0745; found 296.0762.
2-p-Tolylisothiazol-3-one (30)
Aqueous sodium hydroxide (4 mL, 10%) was added to the solution of 5-benzoyl-2-p-tolylisothiazol-3-one 29 (203 mg, 0.678 mmol) in benzene (9 mL), and the mixture was stirred for 3 d at room temperature. The reaction mixture was added water (20 mL) and extracted with ethyl acetate. The combined organic layer was dried (MgSO4), filtered and evaporated. The residue was purified by flash silica gel column chromatography (hexane:ethyl acetate = 2:1 to 1:1) and gave the compound 30 (77 mg, 59%). 1H-NMR (CDCl3, 300MHz) δ 2.38 (s, 3H), 6.32 (d, J = 6.6 Hz, 1H), 7.25 (d, J = 8.1 Hz, 2H), 7.44 (d, J = 8.1 Hz, 2H), 8.14 (d, J = 6.6 Hz, 1H). HRMS: calculated for C10H10NOS (M+H)+ 192.0483; found 192.0494.
2-(4-Bromomethylphenyl)isothiazol-3-one (31)
Catalytic amount of benzoyl peroxide (4 mg) was added to a solution of 2-p-tolylisothiazol-3-one 30 (123 mg, 0.643 mmol), and N-bromosuccinimide (121 mg, 0.673 mmol) in carbon tetrachloride (8 mL), which was refluxed for 2 h and then cooled to room temperature. Solvent was evaporated under low pressure and the residue was purified by flash silica gel column chromatography (methylene chloride:methanol = 50:1 to 20:1), afforded the compound 31 (182 mg, 93%). 1H-NMR (CDCl3, 300MHz) δ 4.50 (s, 2H), 6.33 (d, J = 6.3 Hz, 1H), 7.47 (d, J = 8.4 Hz, 2H), 7.59 (d, J = 8.4 Hz, 2H), 8.16 (d, J = 6.3 Hz, 1H). HRMS: calculated for C10H9NOSBr (M+H)+ 269.9588; found 269.9593.
2-(4-Azidomethylphenyl)isothiazol-3-one (32)
Sodium azide (6.0 mg, 0.0923 mmol) was added to a solution of 2-(4-bromomethylphenyl)isothiazol-3-one 31 (18.2 mg, 0.0674 mmol) in DMF (1 mL) and stirred for 20 h at room temperature. Water (10 mL) was added and the mixture was extracted with diethyl ether. The combined organic layer was dried (MgSO4), filtered and evaporated. The residue was purified on flash silica gel column chromatography (hexane:ethyl acetate = 4:1 to 1:1), afforded the desired compound 32 (11.7 mg, 75%). 1H-NMR (CDCl3, 300 MHz) δ 4.38 (s, 2H), 6.34 (d, J = 6.3 Hz, 1H), 7.41 (d, J = 8.4 Hz, 2H), 7.64 (d, J = 8.4 Hz, 2H), 8.17 (d, J = 6.3 Hz, 1H). HRMS: found 233.0496; calcd for C10H9N4OS (M+H)+ 233.0497.
Receptor binding and functional assays
[3H]Adenosine-5′-N-methyluronamide (36, [3H]NECA, 42.6 Ci/mmol) was obtained from Perkin Elmer. [3H](2-[p-(2-Carboxyethyl)phenyl-ethylamino]-5′-N-ethylcarboxamidoadenosine) (37, [3H]CGS21680, 40.5 Ci/mmol) and [125I]N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyluronamide (38, [125I]I-AB-MECA, 2200 Ci/mmol) were purchased from Perkin–Elmer Life and Analytical Science (Boston, MA). Test compounds were prepared as 5 mM stock solutions in DMSO and stored frozen at −20°C.
Cell Culture and Membrane Preparation
CHO cells stably expressing the recombinant hA1 and hA3Rs, and HEK-293 cells stably expressing the hA2AAR were cultured in Dulbecco's modified Eagle medium (DMEM) and F12 (1:1) supplemented with 10% fetal bovine serum, 100 units/mL penicillin, 100 μg/mL streptomycin, and 2 μmol/mL glutamine. In addition, 800 μg/mL geneticin was added to the A2A media, while 500 μg/mL hygromycin was added to the A1 and A3 media. After harvesting, cells were homogenized and suspended in PBS. Cells were then centrifuged at 240 g for 5 min, and the pellet was resuspended in 50 mM Tris-HCl buffer (pH 7.5) containing 10 mM MgCl2. The suspension was homogenized and was then ultra-centrifuged at 14,330 g for 30 min at 4 °C. The resultant pellets were resuspended in Tris buffer, incubated with adenosine deaminase (3 units/mL) for 30 min at 37 °C. The suspension was homogenized with an electric homogenizer for 10 sec, pipetted into 1 mL vials and then stored at −80 °C until the binding experiments were conducted. The protein concentration was measured using the BCA Protein Assay Kit from Pierce Biotechnology, Inc. (Rockford, IL).37
Binding assays
Into each tube in the binding assay was added 50 μL of increasing concentrations of the test ligand in Tris-HCl buffer (50 mM, pH 7.5) containing 10 mM MgCl2, 50 μL of the appropriate agonist radioligand, and finally 100 μL of membrane suspension. For the A1AR (22 μg of protein/tube) the radioligand used was [3H]36 (final concentration of 3.5 nM). For the A2AAR (20 μg/tube) the radioligand used was [3H]37 (10 nM). For the A3AR (21 μg/tube) the radioligand used was [125I]38 (0.34 nM). Nonspecific binding was determined using a final concentration of 10 μM unlabeled 36 diluted with the buffer. The mixtures were incubated at 25 °C for 60 min in a shaking water bath. Binding reactions were terminated by filtration through Brandel GF/B filters under a reduced pressure using a M-24 cell harvester (Brandel, Gaithersburg, MD). Filters were washed three times with 3 mL of 50 mM ice-cold Tris-HCl buffer (pH 7.5). Filters for A1 and A2AAR binding were placed in scintillation vials containing 5 mL of Hydrofluor scintillation buffer and counted using a Perkin Elmer Liquid Scintillation Analyzer (Tri-Carb 2810TR). Filters for A3AR binding were counted using a Packard Cobra II γ-counter. The Ki values were determined using GraphPad Prism for all assays.
Cyclic AMP accumulation assay
Intracellular cyclic AMP levels were measured with a competitive protein binding method.29,30 CHO cells that expressed the recombinant human A3AR were harvested by trypsinization. After centrifugation and resuspended in medium, cells were planted in 24-well plates in 1.0 mL medium. After 24 h, the medium was removed and cells were washed three times with 1 mL DMEM, containing 50 mM HEPES, pH 7.4. Cells were then treated with the agonist NECA and/or test compound in the presence of rolipram (10 μM) and adenosine deaminase (3 units/mL). After 45 min forskolin (10 μM) was added to the medium, and incubation was continued for an additional 15 min. The reaction was terminated by removing the supernatant, and cells were lysed upon the addition of 200 μL of 0.1 M ice-cold HCl. The cell lysate was resuspended and stored at −20°C. For determination of cyclic AMP production, protein kinase A (PKA) was incubated with [3H]cyclic AMP (2 nM) in K2HPO4/EDTA buffer (K2HPO4, 150 mM; EDTA, 10 mM), 20 μL of the cell lysate, and 30 μL 0.1 M HCl or 50 μL of cyclic AMP solution (0–16 pmol/200 μL for standard curve). Bound radioactivity was separated by rapid filtration through Whatman GF/C filters and washed once with cold buffer. Bound radioactivity was measured by liquid scintillation spectrometry.
Supplementary Material
Acknowledgements
We thank Dr. John Lloyd and Dr. Noel Whittaker (NIDDK) for mass spectral determinations. This research was supported by the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases. We thank Arunkumar Easwaran, Ph.D., Molecular Targeting Technologies, Inc. West Chester, PA for helpful discussions.
Abbreviations
- AR
adenosine receptor
- cAMP
adenosine 3′,5′-cyclic phosphate
- CHO
Chinese hamster ovary
- Cl-IB-MECA
2-chloro-N6-(3-iodobenzyl)-5′-N-methylcarboxamidoadenosine
- DMEM
Dulbecco's modified Eagle's medium
- EDC
1-ethyl-3-(3-dimethylaminopropyl) carbodiimide
- EDTA
ethylenediaminetetraacetic acid
- GPCR
G protein-coupled receptor
- HATU
2-(1H-7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate methanaminium
- HEK
human embryonic kidney
- HOBT
1-hydroxybenzotriazole
- I-AB-MECA
N6-(3-iodo-4-aminobenzyl)-5′-N-methylcarboxamidoadenosine
- IB-MECA
N6-(3-iodobenzyl)-5′-N-methylcarboxamido-adenosine
- NECA
5′-N-ethylcarboxamidoadenosine
- DMF
N,N-dimethylformamide
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HRMS
high resolution mass spectroscopy
- TBTA
tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl] amine
- TEA
triethylamine
- TLC
thin layer chromatography
Footnotes
Supporting information available: An alternate synthetic procedure the synthesis of compound 32 and NMR spectral characterization of compounds 9, 11, and 15 are available.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jacobson KA, Klutz AM, Tosh DK, Ivanov AA, Preti D, Baraldi PG. Handbook of Experimental Pharmacology. Vol. 193. Springer; 2009. p. 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gessi S, Merighi S, Varani K, Cattabriga E, Benini A, Mirandola P, Leung E, Ma c Lennan S, Feo C, Baraldi S, Borea PA. J. Cell. Physiol. 2007;211:826. doi: 10.1002/jcp.20994. [DOI] [PubMed] [Google Scholar]
- 3.Yang H, Avila MY, Peterson-Yantorno K, Coca-Prados M, Stone RA, Jacobson KA, Civan MM. Current Eye Res. 2005;30:747. doi: 10.1080/02713680590953147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hua X, Chason KD, Fredholm BB, Deshpande DA, Penn RB, Tilley SL. J Alle rgy Clin Immunol. 2008;122:107. doi: 10.1016/j.jaci.2008.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fishman P, Jacobson KA, Ochaion A, Cohen S, Bar-Yehuda S. Immun. Endoc. Metab. Agents in Med. Chem. 2007;7:298. doi: 10.2174/187152207781369878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Silverman MH, Strand V, Markovits D, Nahir M, Reitblat T, Molad Y, Rosner I, Rozenbaum M, Mader R, Adawi M, Caspi D, Tishler M, Langevitz P, Rubinow A, Friedman J, Green L, Tanay A, Ochaion A, Cohen S, Kerns WD, Cohn I, Fishman-Furman S, Farbstein M, Bar-Yehuda S, Fishman P. J. Rheumatol. 2008;35:41. [PubMed] [Google Scholar]
- 7.Zheng J, Wang R, Zambraski E, Wu D, Jacobson KA, Liang BT. Am. J. Physiol.; Heart and Circ. Physiol. 2007;293:3685. doi: 10.1152/ajpheart.00819.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jaakola V-P, Griffith MT, Hanson MA, Cherezov V, Chien EYT, Lane JR, IJzerman AP, Stevens RC. Science. 2008;322:1211. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tosh DK, Chinn M, Ivanov AA, Klutz AM, Gao ZG, Jacobson KA. J. Med. Chem. 2009;52:7580. doi: 10.1021/jm900426g. DOI: 10.1021/jm900426g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jacobson KA, Gao ZG, Liang BT. Trends Pharmacol. Sci. 2007;28:111. doi: 10.1016/j.tips.2007.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Melman A, Wang B, Joshi BV, Gao ZG, de Castro S, Heller CL, Kim SK, Jeong LS, Jacobson KA. Bioorg. Med. Chem. 2008;16:8546. doi: 10.1016/j.bmc.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Melman A, Gao ZG, Kumar D, Wan TC, Gizewski E, Auchampach JA, Jacobson KA. Bioorg. Med. Chem. Lett. 2008;18:2813. doi: 10.1016/j.bmcl.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jacobson KA. Bioconjugate Chem. 2009;20:1816. doi: 10.1021/bc9000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) Matsuda A, Shinozaki M, Yamaguchi T, Homma H, Nomoto R, Miyasaka T, Watanabe Y, Abiru T. J. Med. Chem. 1992;35:241. doi: 10.1021/jm00080a007. [DOI] [PubMed] [Google Scholar]; b) Cristalli G, Volpini R, Vittori S, Camaioni E, Monopoli A, Conti A, Dionisotti S, Zocchi C, Ongini E. J. Med. Chem. 1994;37:1720. doi: 10.1021/jm00037a024. [DOI] [PubMed] [Google Scholar]; c) Rieger JM, Brown ML, Sullivan GW, Lind en J, Macdonald TL. J. Med. Chem. 2001;44:531. doi: 10.1021/jm0003642. [DOI] [PubMed] [Google Scholar]; d) Volpini R, Costanzi S, Lambertucci C, Vittori S, Klotz KN, Cristalli G. J. Med. Chem. 2002;45:3271. doi: 10.1021/jm0109762. [DOI] [PubMed] [Google Scholar]
- 15.Kolb HC, Sharpless KB. Drug Discov. Today. 2003;8:1128. doi: 10.1016/s1359-6446(03)02933-7. [DOI] [PubMed] [Google Scholar]
- 16.Chincshilla R, Nájera C. Chem. Rev. 2007;107:874. doi: 10.1021/cr050992x. [DOI] [PubMed] [Google Scholar]
- 17.Kolb HC, Finn MG, Sharpless KB. Angew. Chem. Int. Ed. 2001;40:2004. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 18.Bock VD, Hiemstra H, van Maarseveen JH. Eur. J. Org. Chem. 2006;51:68. [Google Scholar]
- 19.Chan TR, Hilgraf R, Sharpless KB, Fokin VV. Org. Lett. 2004;6:2853. doi: 10.1021/ol0493094. [DOI] [PubMed] [Google Scholar]
- 20.Gassensmith JJ, Arunkumar E, Barr L, Baumes JM, DiVittorio KM, Johnson JR, Noll BC, Smith BD. J. Am. Chem. Soc. 2007;129:15054. doi: 10.1021/ja075567v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson JR, Fu N, Arunkumar E, Leevy WM, Gammon ST, Piwinica-Worms D, Smith BD. Angew. Chem. Int. Ed. 2007;46:5528. doi: 10.1002/anie.200701491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berlier JE, Rothe A, Buller G, Bradford J, Gray DR, Filanoski BJ, Telford WG, Yue S, Liu J, Cheung C-Y, Chang W, Hirsch JD, Beechem JM, Haugland RP, Haugland RP. J. Histochem. Cytochem. 2003;51:1699. doi: 10.1177/002215540305101214. [DOI] [PubMed] [Google Scholar]
- 23.Tsolomitis A, Sandris C. Heterocycles. 1987;25:569. [Google Scholar]
- 24.Klotz KN, Lohse MJ, Schwabe U, Cristalli G, Vittori S, Grifantini M. Naunyn Schmiedebergs Arch. Pharmacol. 1989;340:679. doi: 10.1007/BF00717744. [DOI] [PubMed] [Google Scholar]
- 25.Jarvis MF, Schutz R, Hutchison AJ, Do E, Sills MA, Williams M. J. Pharmacol. Exp. Ther. 1989;251:888. [PubMed] [Google Scholar]
- 26.Olah ME, Gallo-Rodriguez C, Jacobson KA, Stiles GL. Mol. Pharmacol. 1994;45:978. [PMC free article] [PubMed] [Google Scholar]
- 27.Englert M, Quitterer U, Klotz KN. Biochem. Pharmacol. 2002;64:61. doi: 10.1016/s0006-2952(02)01071-7. [DOI] [PubMed] [Google Scholar]
- 28.Jacobson KA, Park KS, Jiang J.-l., Kim YC, Olah ME, Stiles GL, Ji X.d. Neuropharmacology. 1997;36:1157. doi: 10.1016/s0028-3908(97)00104-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nordstedt C, Fredholm BB. Anal. Biochem. 1990;189:231. doi: 10.1016/0003-2697(90)90113-n. [DOI] [PubMed] [Google Scholar]
- 30.Post SR, Ostrom RS, Insel PA. Methods Mol. Biol. 2000;126:363. doi: 10.1385/1-59259-684-3:363. [DOI] [PubMed] [Google Scholar]
- 31.Cosyn L, Palaniappan KK, Kim SK, Duong HT, Gao ZG, Jacobson KA, Van Calenbergh S. J. Med. Chem. 2006;49:7373. doi: 10.1021/jm0608208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hsu T-L, Hanson SR, Kishikawa K, Wang S-K, Sawa M, Wong C-H. Proc. Natl. Acad. Sci USA. 2007;104:2614. doi: 10.1073/pnas.0611307104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Salic A, Mitchison TJ. Proc. Natl. Acad. Sci USA. 2008;105:2415. doi: 10.1073/pnas.0712168105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ming X, Leonard P, Heindl D, Seela F. Nucleic Acids Symposium Series. 2008;52:471. doi: 10.1093/nass/nrn239. [DOI] [PubMed] [Google Scholar]
- 35.Morley JO, Kapur AJO, Charlton MH. Int. J. Chem. Kinetics. 2007;39:254. [Google Scholar]
- 36.Codelli JA, Baskin JM, Agard NJ, Bertozzi CR. J. Am. Chem. Soc. 2008;130:11486. doi: 10.1021/ja803086r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bradford MM. Anal. Biochem. 1976;72:248. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 38.Inverarity IA, Viguier RFH, Cohen P, Hulme AN. Bioconjugate Chem. 2007;18:1593. doi: 10.1021/bc070085u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.