

Abstract
The inhibition mechanism of matrix metalloproteinase 2 (MMP2) by the selective inhibitor (4-phenoxyphenylsulfonyl)methylthiirane (SB-3CT) and its oxirane analog are investigated computationally. The inhibition mechanism involves C-H deprotonation with concomitant opening of the three-membered heterocycle. SB-3CT was docked into the active site of MMP2, followed by molecular dynamics simulation to prepare the complex for combined quantum mechanics and molecular mechanics (QM/MM) calculations. QM/MM calculations with B3LYP/6-311+G(d,p) for the QM part and the AMBER force field for the MM part were used to examine the reaction of these two inhibitors in the active site of MMP2. The calculations show that the reaction barrier for transformation of SB-3CT is 1.6 kcal/mol lower than its oxirane analog, and the ring opening reaction energy of SB-3CT is 8.0 kcal/mol more exothermic than that of its oxirane analog. Calculations also show that protonation of the ring-opened product by water is thermodynamically much more favorable for the alkoxide obtained from the oxirane than for the thiolate obtained from the thiirane. A six-step partial charge fitting procedure is introduced for the QM/MM calculations to update atomic partial charges of quantum mechanics region and to ensure consistent electrostatic energies for reactants, transition states and products.
Keywords: MMP2, SB-3CT, QM/MM, ONIOM, Inhibition, RESP, Charge fitting
Matrix metalloproteinases (MMPs) are zinc-dependent endopeptidases that regulate functions of the extracellular matrix (ECM). MMPs are involved in many biological processes, such as embryonic development (1-3), tissue remodeling and repair (4-6), neurophathic pain processes (7), cancer (8-11), and other diseases (12-15). The physiological activities of the MMPs are strictly regulated by activation of the zymogen forms of the protein (pro-MMPs) (16), and by inhibition by protein tissue inhibitors of metalloproteinases (TIMPs).
Gelatinases A (MMP2) and B (MMP9) are proteolytic enzymes that digest type IV collagens (17). Uncontrolled activities of these two enzymes are implicated in tumor metastasis and angiogenesis. The structures and catalytic mechanisms of MMP2 and MMP9 have been studied extensively (18-24). Since the unregulated activities of these two enzymes have been implicated in many diseases, they are targets for selective inhibitor design (25-31). SB-3CT, one such inhibitor, selectively inhibits MMP2 with high potency and MMP9 with somewhat lower activity (32).
The key event in the inhibition of MMP2 by SB-3CT is enzyme-catalyzed ring-opening of the thiirane, giving a stable zinc-thiolate species (33). The previously proposed mechanism for MMP inhibition by SB-3CT involved nucleophilic addition of the carboxylate of the active site glutamate to a thiirane carbon (Scheme 1a). This mechanism is precedented with oxirane inhibitors of carboxypeptidase A, a structurally different but mechanistically related protease (34). However, recent experiments from the Mobashery lab indicate a different mechanism for SB-3CT (Scheme 1b) (33). In this new mechanism, the carboxylate of glutamate-404 abstracts a hydrogen from the methylene group juxtaposed between the sulfone and the thiirane. This deprotonation initiates ring opening, and also produces a thiolate capable of coordination with the zinc at the active site. This latter mechanism is supported by the observation of a primary deuterium kinetic isotope effect for the methylene group adjacent to the sulfone (33). Our previous theoretical calculations on model systems in solution revealed that the barriers of ring opening initiated by deprotonation are lower than those of the glutamate addition mechanism (35). These calculations also reproduce the observed primary kinetic isotope effect. Given these facts, the mechanism involving covalent modification of the glutamate was not pursued in current study.
Scheme 1.
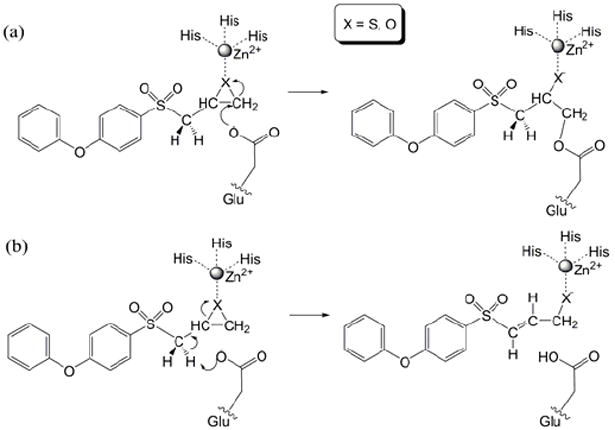
MMP2 inhibition mechanisms by SB-3CT: (a) previously proposed mechanism; (b) current mechanism.
In preparation for the present investigation, we studied the conformational preferences and stereochemical aspects of the coupled deprotonation and ring opening in solution of 2-(methylsulfonylmethyl)-thiirane and -oxirane (1 and 2 in Scheme 2) as models for SB-3CT (3) and its oxirane analog (4). Proton abstraction can occur either syn or anti to the three-membered ring, and the sulfone group exerts a comparable stereoelectronic effect in the thiirane and in the oxirane. Since no crystal structure is available for the MMP2·SB-3CT complex, the structure and stability of the complex were assessed by docking of SB-3CT into the MMP2 active site, followed by molecular dynamics studies. Then the details of the deprotonation/ring opening mechanism for inhibition were examined by combined quantum mechanics and molecular mechanics (QM/MM) methods.
Scheme 2.
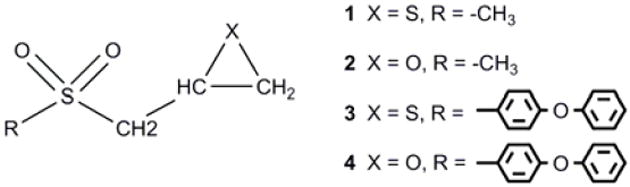
Structures of SB-3CT (3) and its analogs (1, 2, and 4).
Computational Methods
Docking and Molecular Dynamics Studies of the MMP2·SB-3CT Complex
Since the structure of the non-covalent MMP2·SB-3CT complex is not experimentally accessible, SB-3CT was docked in the active site of the crystal structure for the Ala404 mutant of MMP2 (PDB code: 1CK7) (18). Ala404 was computationally mutated to Glu404, the catalytic base in the MMP active site. The propeptide domain (residues 31 to 115) was deleted, as would be the case in the active form of MMP. The resulting MMP2 enzyme includes residues 116-449, two zinc metal ions (Zn990 and Zn991), and three calcium ions. Sybyl (Tripos 7.3) (36) was used to prepare the structures of the inhibitors. DOCK (version 5.4, UCSF) (37) was employed to dock the inhibitor in the active site, using electrostatic and van der Waals forces to score the acceptor-inhibitor interactions. The docked MMP2·SB-3CT complex was immersed in a water solvent box through energy minimization and thermodynamic equilibration procedures (using Xleap from Amber 9). AMBER force field (parm99) was used to describe the whole system, including zinc ions. The force field parameters for zinc (38) were listed in the supporting information. During these stages, position constraints were enforced for the atoms in the three histidine residues surrounding each of the zinc cations, Glu404, and SB-3CT substrate with harmonic potentials of approximately 1 Å width and force constants of 50 kcal·mol−1·Å−2. Furthermore, a distance constraint was added between Zn990 and the nitrogen atom of each histidine at the value given in the crystal structure, using a harmonic potential of width 0.2 Å and force constants of 1,000 kcal·mol−1·Å−2. A total of 2.0 ns of molecular dynamics (MD) simulation were carried out. Snapshots were extracted every 0.5 ps. The conformation of the complex was analyzed for each of the 4,000 snapshots.
QM/MM Studies of the MMP2·SB-3CT Complex
The initial structure for the QM/MM calculations of the reactant complex was prepared using the Amber software suite, version 9 (39). Since experiments show that (R)-SB-3CT is slightly more active than (S)-SB-3CT in terms of kon (40), the R stereoisomer was chosen for the QM/MM studies. The selected MMP2·(R)-SB-3CT complex from the docking and MD simulations (approximately 66,000 atoms) was subjected to geometry optimization using the Sander program from Amber 9 (approximately 12,000 conjugate gradient steps). During this optimization, a constraint was applied to the distance between an oxygen atom of the carboxylate group of Glu404 and a hydrogen atom of the CH2 group adjacent to the thiirane ring of SB-3CT to ensure that the optimized geometry would be appropriate for the abstraction step. The Amber optimized geometry was used as a starting point for the QM/MM calculation. The active site Zn2+, His403, His407, His413, Glu404, and the SB-3CT inhibitor were defined as core residues. The water molecules that are either within 3 Å of protein or within 12 Å of the core residues were kept for the QM/MM calculations. All other water molecules were deleted to facilitate the computation. The final system subjected to QM/MM calculations comprised approximately 8,800 atoms. In the QM/MM calculations, all residues within 6 Å of the core residues were allowed to move without any constraints, while all other residues were frozen. The oxirane optimizations started from the thiirane optimized geometries. Some care is needed to ensure that the hydrogen bonding pattern in the solvent water molecules is the same for the reactant complex and the transition state so that minor changes in the solvent do not overwhelm differences in the barrier heights.
A two-layer ONIOM method (41-50) was used for the QM/MM study of the inhibition mechanism of SB-3CT and its oxirane analog. The zinc ion, the three imidazole rings from His403, His407 and His413, the CH2CO2− part of the Glu404 side chain, the thiirane or oxirane with the SO2CH2 group, and two water molecules coordinating with zinc in the MD simulation were included in the QM region (49 atoms). The QM part of the system was described at the B3LYP/6-31G(d) level of density functional theory. Comparison with benchmark calculations at the CBS-QB3 level for the deprotonation/ring opening reactions in the gas phase showed that B3LYP performed significantly better than HF or MP2 calculations (35). The MM part of the system was described using the parm96 parameters of the Amber force field (51). A mechanical embedding scheme was used for geometry optimization (electrostatic interactions between the QM and MM regions are handled by MM in this approach). Transition state searches used the quadratically coupled QM/MM geometry optimizer (48) implemented in the development version of the Gaussian package (52). The optimizer explicitly calculates the transition vector, which through the quadratic coupling between the regions is not restricted to the QM region of the system. The molecular mechanics contributions to the second derivative matrix are always evaluated analytically, which makes the optimization procedure quite reliable. This method has been applied previously to calculate transition states in other enzymatic systems (53-55). The final QM/MM energies reported are based on electronic embedding (49) single point calculations at the ONIOM(B3LYP/6-311+G(d,p):AMBER) level of theory using the ONIOM(B3LYP/6-31G(d):AMBER) optimized geometries. All ONIOM calculations were carried out with the development version of Gaussian (52).
The Resp (Restrained ElectroStatic Potential) program (56, 57) was used to fit partial charges to the electrostatic potential (ESP) grid generated by gas phase calculation. In this study, Glu404 accepts a proton from the inhibitor. This changes the partial charge distribution of substrate and Glu404 significantly. Therefore, using fixed partial charges for the substrate and protein along reaction path could reduce the accuracy of the ONIOM energies. To address this issue, we developed a six-step procedure for fitting a consistent set of partial charges for the reactant, TS and product: [1] A preliminary set of partial charges was obtained for the substrate in the gas phase using the Resp procedure. [2] The reactant, TS and product were optimized by ONIOM calculations using mechanical embedding with the partial charges. [3] The QM atoms of reactant, TS and product from the ONIOM geometry optimizations in the second step were used to obtain an improved set of partial charges using the Resp procedure, and the hydrogen atoms added to cap the dangling bonds are constrained to have zero charges. [4] The reactant, TS and product were optimized in the active site by ONIOM calculations using mechanical embedding with the improved charges. [5] Step 3 and 4 are repeated until convergence is achieved. The convergence criterion is that the total ONIOM energies difference between the last two round optimizations is less than 0.1 kcal/mol. [6] The converged geometries and charges from the last step are used for single point calculations with electronic embedding. This typically required 4~6 rounds of optimization to obtain partial charges appropriate for the reactants, TSs and products in their optimal geometries in the active site. With this method, the total charge on the QM atoms is conserved along the reaction path and the changes in the electrostatic interaction energy in going from reactants to TSs to products are reproduced properly. This procedure was followed in the present work since the charge distribution was suspected to be critical for reliable results. For other studies, the dependence of the results may not be as sensitive to the charges, and iterating may not be necessary.
Results and Discussion
Docking and Molecular Dynamics Studies
Both the (R)- and (S)-SB-3CT enantiomers are effective inhibitors of this enzyme (40). Since the (S)-SB-3CT enantiomer is the slightly less reactive, (R)-SB-3CT was chosen for the initial computational study. (R)-SB-3CT was docked into the active site before being subjected to MD simulation. The MMP2·SB-3CT complex remains stable during the 2 ns MD simulation. The key electrostatic interactions that stabilize SB-3CT in the MMP2 active site involve the attraction between zinc and the sulfur of the thiirane, a hydrogen bond between one oxygen of the sulfone and the amide hydrogen of Leu191, and hydrophobic interactions between the phenoxyphenyl ring and the residues of the S1’ pocket (58-60).
The MD trajectory of MMP2·(R)-SB-3CT was analyzed to select a representative conformer for the QM/MM calculations. The most populated ranges are 2.5 ± 0.1 Å for distance between Zn and thiirane S (d1), and 1.9 ± 0.1 Å for distance between Leu191 amide H and one oxygen of the sulfone group (d2). The conformer with both d1 and d2 within the most populated ranges and the shortest distance between the Glu404 oxygen and the pro-S hydrogen of (R)-SB-3CT (d3) was chosen as a starting point for QM/MM calculations. The chosen conformer has d1 = 2.53 Å, d2 = 1.84 Å and d3 = 2.26 Å (Fig. 1).
Fig. 1.
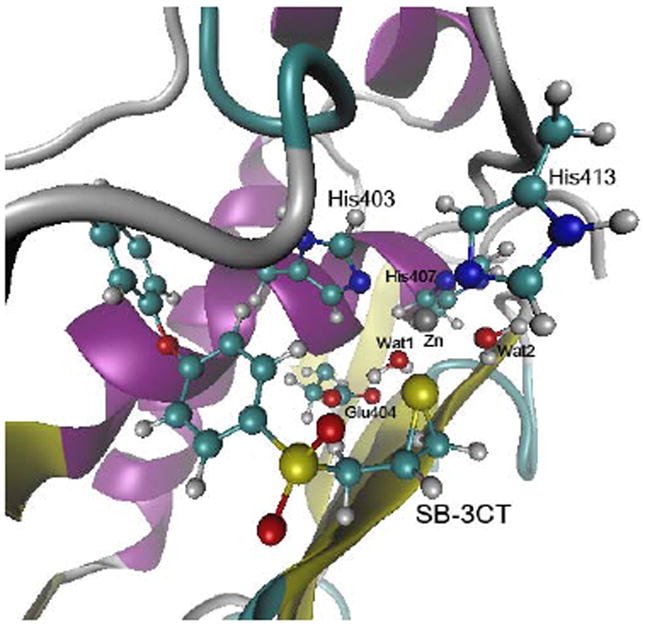
Structure of the MMP2•(R)-SB-3CT complex from the MD simulation. The distance between zinc and thiirane sulfur is 2.53 Å, which is close to the most populated value in the MD simulation. Residues are shown in ball-and-stick representation with atom colored according to atom types (H, C, N, O, S, Zn, shown in white, cyan, blue, red, yellow, and grey, respectively). The same color scheme is used in all of the figures.
(S)-SB-3CT was also subjected to MD simulation as comparison. An initial structure for the (S)-enantiomer bound in the active site of MMP2 was generated from the MD optimized (R)-enantiomer structure by inversion of configuration, and subjected to 2 ns MD simulation. The same constraints as used for the (R)-enantiomer were employed in the MD simulation of the (S)-enantiomer. The resulting conformations maintain the coordination between the zinc cation and the thiirane ring sulfur (d1 = 2.40 Å), and between a sulfone oxygen and the Leu191 amide hydrogen (d2 = 1.80 Å), as observed in the (R)-enantiomer. A conformation having a distance of 2.36 Å (d3) between the Glu404 and the pro-R hydrogen of the methylene carbon was extracted (Fig. S1). Of the distance between the oxygen and hydrogen atoms, the molecular dynamics samplings showed similar distributions over the 2 ns MD simulation for both the (R)- and (S)-enantiomers, but did indicate diastereomic preference of the glutamate for the pro-S hydrogen in the (R)-enantiomer and for the pro-R hydrogen in the (S)-enantiomer. The differences in the conformations of the (R)- and (S)-enantiomers are primarily (1) rotation of thiirane ring along C-C bond and (2) rotation of the phenoxyphenyl ether rings along the adjacent CS bond (Fig. 1; the (S) enantionmer is shown in Supporting Information, Fig. S1).
QM/MM Studies
The docking study and the subsequent MD samplings generated a reliable structure of MMP2 and SB-3CT complex. Conformations with a short distance between the hydrogen of the alpha methylene and Glu404 were sampled for the initiation of the deprotonation reaction. However, the ring opening reaction coupled with deprotonation cannot be studied by molecular mechanics level of theory used in docking and MD. Therefore, QM/MM methods were used to carry out further calculations.
(a) Water or Hydroxide in the Active Site
The MD study of MMP2·SB-3CT shows two water molecules coordinated with the zinc in the active site (Fig. 1). One water molecule is located between the zinc and Glu404 (Wat1) and the second is in contact with bulk solvent (Wat2). The QM/MM calculations show spontaneous proton transfer from Wat1 to the Glu404 carboxylate, giving a hydroxide anion coordinated to the zinc (Fig. 2; geometry parameters are given in Fig. S2). A similar proton migration from a thiol group to glutamate, to produce thiolate as a zinc ligand in pro-MMP9, was observed in the previous QM/MM study on the activation of pro-MMP9 (16). The zinc is coordinated with hydroxide and the three histidines. Neither SB-3CT nor the carboxylate side chain of Glu404 are coordinated with the zinc in this structure (5.13 and 5.08 Å). It is noteworthy that in the same study of MMP9 (16), a water molecule enters the active site, inserting itself between Glu402 and zinc during the activation process of pro-MMP9. Zinc has tetrahedral coordination with a water molecule and three histidines, and the water forms a strong hydrogen bond with the glutamate.
Fig. 2.
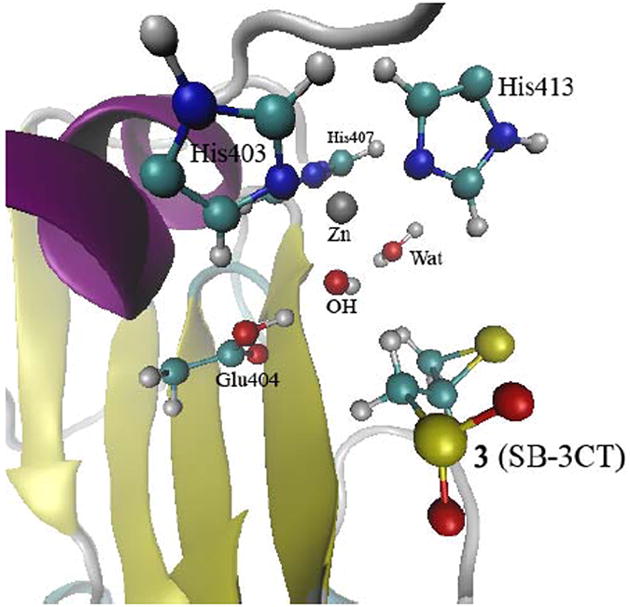
QM/MM calculations of the complex of 3 and MMP2 with two water molecules in the active site optimized at the ONIOM(B3LYP/6-31G(d):AMBER) level of theory (see Fig. S2 in the Supporting Information for details).
Since Glu404 needs to be in a deprotonated state to initiate the reaction with the inhibitor, additional QM/MM calculations were carried out with hydroxide bound to the zinc and Glu404 deprotonated (Fig. 3 and Fig. S3). The proton abstraction/ring opening energies are quite endothermic: 15.8 and 6.6 kcal/mol for the thiirane 3 and oxirane 4, respectively. The hydroxide coordinates with zinc in both the reactant and product structures. Glu404 forms a hydrogen bond with the hydroxide, and coordinates with zinc in the reactants. However, Glu404 moves away from zinc in the products. Neither the thiirane nor oxirane coordinates with zinc in the reactants. The overall reactions are endothermic because the ring-opening products do not coordinate with the zinc.
Fig. 3.
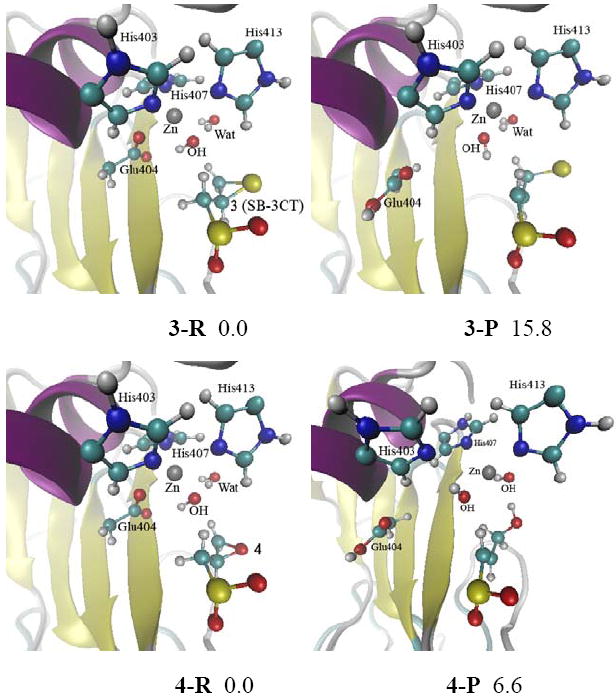
Reactant and product for 3 and 4 in the MMP2 active site optimized at the ONIOM(B3LYP/6-31G(d):AMBER) level of theory (see Fig. S3 in the Supporting Information for details). Energies (in kcal/mol) were calculated at ONIOM(B3LYP/6-311+G(d,p):AMBER) using an electronic embedding scheme with the reactant complexes as reference states.
These QM/MM calculations show that if a water molecule or hydroxide anion is in the active site shielding zinc from Glu404, the inhibitor cannot interact with the zinc, and the pathways are endothermic and not thermodynamically feasible. Therefore, subsequent QM/MM calculations were carried out without a water molecule between zinc and Glu404.
(b) Deprotonation and Ring Opening of the Inhibitor
In the initial structure for further QM/MM calculations, Glu404 and three histidines coordinate with zinc (see Fig. 4). The water molecule (Wat2) is not coordinated to the zinc, but is open to the solvent. The orientation and key intermolecular contacts between (R)-SB-3CT and the MMP2 active site closely resemble the crystallographic structures seen for other MMP inhibitors (58). Particularly, the phenoxyphenyl side chain is located as expected in the S1’ pocket, and the customary strong (1.9 Å) hydrogen bond from the backbone NH of Leu191 to the pro-S oxygen of the sulfone is preserved (Fig. 4). As is also expected, the second oxygen of the sulfone is solvent-exposed. In the QM/MM optimized reactant complex of SB-3CT, the zinc is coordinated with the three histidines, the Glu404 carboxylate, and the thiirane sulfur. No crystal structure is available for SB-3CT bound to a matrix metalloproteinase; however, this coordination agrees with the modeled complex structure between MMP9 and SB-3CT (58). These observations support the absence of a water molecule between Glu404 and zinc ion. Of particular interest to the deprotonation mechanism is the conformation of the bound SB-3CT. In addition to the intermolecular interactions enumerated above, a stereoelectronic effect governs the orientation of the phenylsulfone segment, wherein the π-orbital of the aryl carbon bonded to the sulfur of the sulfone strongly prefers to bisect the two sulfone oxygens (Fig. 4) (61, 62).
Fig. 4.
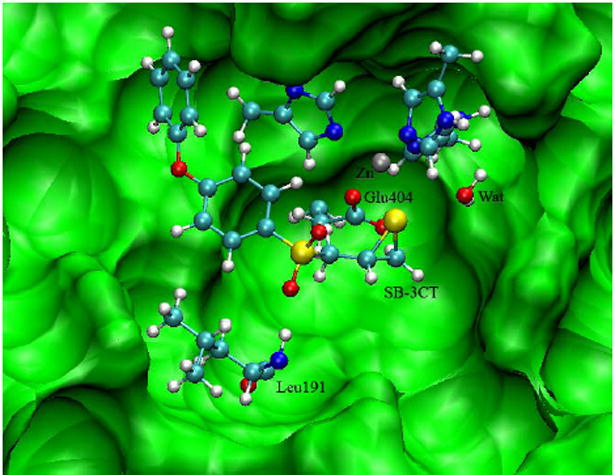
MMP2•(R)-SB-3CT complex structure from QM/MM calculation (the same geometry as 3-R in Fig. 5).
The reactant, transition state, and product structures for the inhibition of MMP2 by (R)-SB-3CT are shown in the top row of Fig. 5 and selected geometrical parameters are given in Fig. S4. Those for the oxirane analog are shown in the bottom row of these Figures. In the reactant complex of SB-3CT (3-R), an oxygen from the carboxylate group of Glu404 and the sulfur from the thiirane coordinate with the zinc at 1.99 Å and 2.91 Å, respectively, while the water is not coordinated (3.58 Å). For the oxirane reactant, 4-R, the interaction with the active site is very similar to the thiirane reactant. The QM/MM energies with electronic embedding are given in Table 1 and plotted in Fig. 6.
Fig. 5.
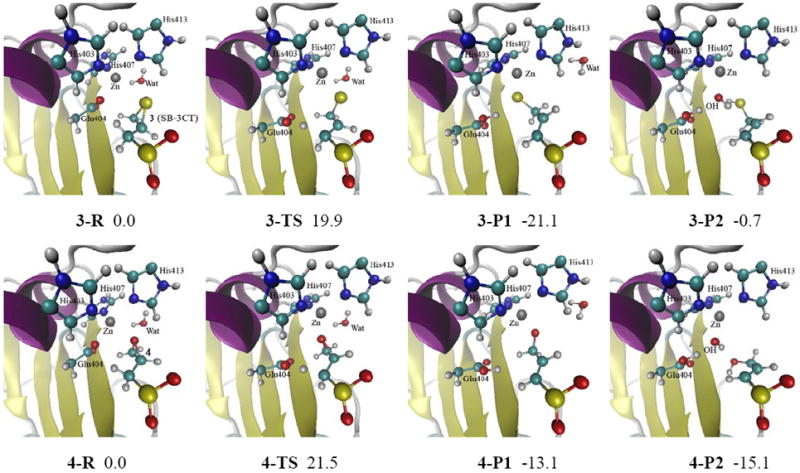
Reactants, transition states and products for SB-3CT (3) and its oxirane analog (4) in the MMP2 active site optimized at ONIOM(B3LYP/6-31G(d):AMBER) level of theory (see Fig. S4 in the Supporting Information for details). Energies (in kcal/mol) were calculated at ONIOM(B3LYP/6-311+G(d,p):AMBER) using electronic embedding with the reactant complexes used as reference states. 3-P1 and 4-P1 are unprotonated ring opening products. In 3-P2 and 4-P2, the ring opening products are protonated by water molecule, and the resulting hydroxide anion coordinates with the zinc.
Table 1.
QM/MM calculations of the energetics for the ring-opening reactions of inhibitions in the active site of MMP2a
| Inhibitor | Barrier Height | Reaction Enthalpy |
|
|---|---|---|---|
| P1 (unprotonated product) | P2 (protonated product) | ||
| SB3-CT (3)b | 19.9 | -21.1 | -0.7 |
| Oxirane Analog (4) b | 21.5 | -13.1 | -15.1 |
ONIOM(B3LYP/6-311+G(d,p):AMBER)//ONIOM(B3LYP/6-31G(d):AMBER) with electronic embedding; energies in kcal/mol.
See Fig. 5.
Fig. 6.
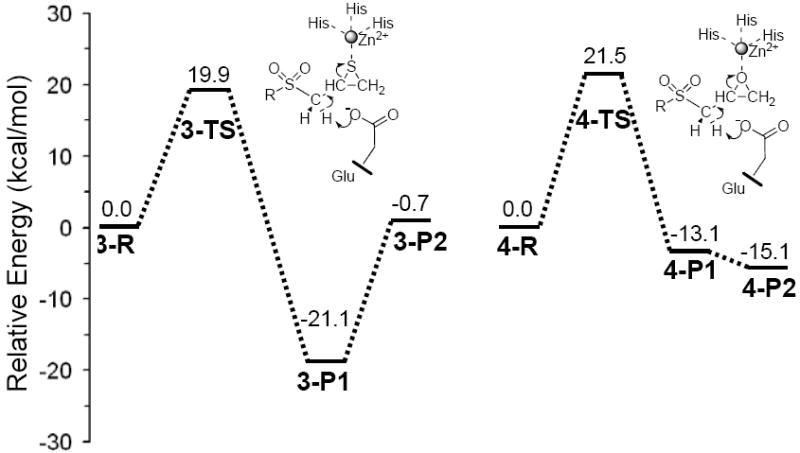
Energy profiles for SB-3CT (3) and its oxirane analog (4) in the MMP2 active site. Relative energies (in kcal/mol) were calculated at ONIOM(B3LYP/6-311+G(d,p):AMBER) using electronic embedding with the reactant complexes used as reference states.
In the QM/MM calculations of the thiirane transition state, 3-TS in Fig. 5 and Fig. S4, the transferring proton is 1.14 Å from the acceptor oxygen, and 1.57 Å from the donor carbon. The breaking C–S bond of the thiirane is elongated to 2.03 Å in 3-TS. The Glu404 side chain has moved away from zinc in order to abstract the proton. One oxygen of the sulfone accepts a hydrogen bond from the backbone NH of Leu191, as shown in Fig. 4. In the TS of the oxirane analog (4-TS), the proton transfer is earlier along the path, and the ring opening is similar to the thiirane system. The thiirane sulfur and oxirane oxygen are strongly coordinated to the zinc, but the glutamate is not.
Although the bound conformation of (R)-SB-3CT in the reactant complex shows a near-staggered conformation with respect to the mechanistically critical C-C dihedral angle for the ring opening reaction, the spatial placement of Glu404 implicates a syn relationship for proton abstraction and concomitant thiirane opening. In separate study, the model molecule of SB-3CT (1) reacting with acetate as the Brønsted base in solution, the C-S bond dihedral in the syn-elimination TS is −34° (35). In the MMP2 QM/MM TS (Fig. 5 and Fig. S4, 3-TS), this dihedral angle is 45°. To estimate the energetic cost imposed by this dihedral angle difference, the syn-elimination reactant complex and TS of 1 were recomputed by constraining the syn-elimination dihedral angle to the 45° seen in QM/MM structures. The two transition states computed in methanol solution (syn dihedral of −34° and 45°) are compared in Fig. 7. The enthalpy of TS with dihedral angle constrain is 4.1 kcal/mol higher than the optimal syn-elimination TS in methanol.
Fig. 7.
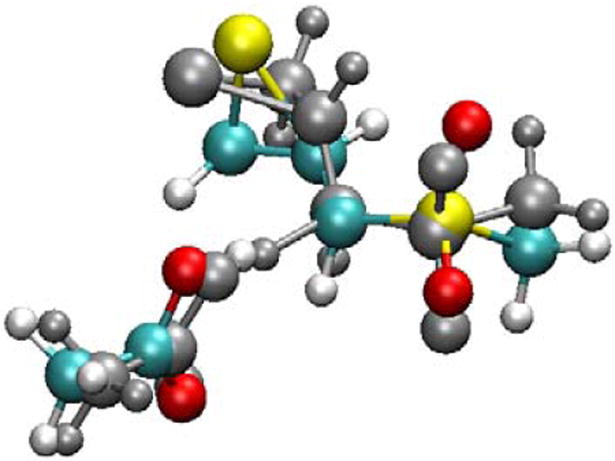
Comparison between the TS for syn-elimination in solution (structure in gray, dihedral angle -34°) and TS for syn-elimination with the same dihedral angle as in the MMP2 active site (structure in color, dihedral angle as 45°).
In the products, 3-P1 and 4-P1, the thiolate sulfur and the alkoxide oxygen remain tightly coordinated to the zinc. The protonated Glu404 stays away from zinc. As in the transition states, the zinc has tetrahedral coordination and the water molecule does not interact with the zinc. Comparing the structures of reactant complex and TS of SB-3CT (3) in the MMP2 active site (Fig. 5 and Fig. S4, 3-R and 3-TS, respectively), it is apparent that Glu404 moves a considerable distance from the zinc toward the inhibitor from reactant complex to TS. Clearly, the carboxylate of Glu404 must disengage from the zinc. The energy surface between the TS and the reactant and product complexes of MMP2·SB-3CT and its oxirane analog were examined by starting near the TS and minimizing the energy in the reactant and product directions. The energies along these optimization paths confirm that no additional barriers are encountered between the TS and the reactant or the product complexes for both 3 and 4 (see Fig. S5 and S6 in the Supporting Information).
The barriers for the thiirane and oxirane ring opening at the active site of MMP2 are 19.9 and 21.5 kcal/mol, respectively, and the corresponding reaction energies are −21.1 and −13.1 kcal/mol. These values indicate a role for the enzyme in this base-mediated elimination reaction in stabilization of the ring opening product, therefore, yielding much more favorable reaction energies for the inhibitors in the active site compared to the model systems in solution (35). The results show that the reactions of both 3 and 4 within the MMP2 active site are kinetically feasible and thermodynamically favorable.
The deprotonation-initiated ring opening TSs identified for 3 and 4 at MMP2 active site closely resemble the TSs of model systems in solution (35). The calculated KIE for the thiirane model system (kH/kD = 5.0) is in good agreement with experimental results (kH/kD = 5.0) (35). The evaluation of a kinetic isotope effect in a QM/MM calculation would necessitate the computation of a full set of vibrational frequencies or extensive sampling of the potential energy surface for the reactant and the transition state. Such calculations are not practical at this time, but may become feasible in the future.
When complexed to MMP2, the reaction of thiirane 3 is calculated to be about 8 kcal/mol more exothermic than the reaction of oxirane 4. The difference in exothermicity of 3 and 4 shows that the ring opening reaction of SB-3CT is thermodynamically more favorable than its oxirane analog. In the present computational model, the ring opening barrier of 4 is higher than 3 by 1.6 kcal/mol. This would indicate that 3 is only a factor of 15 more reactive than is 4. However, calculations in aqueous solution using a polarizable continuum model yield 6 kcal/mol for the difference in the barrier heights (35). One of the limitations of QM/MM calculations at the present state of the art is the inability to carry out extensive sampling. Since the active site is open to the solvent, fluctuations in hydrogen bonding to the solvent could add an uncertainty of ± 2 kcal/mol or more to the difference in barrier heights obtained by local optimization. Comparison with the calculations in solution suggests that the difference in the barrier may be larger than 1.6 kcal/mol. The oxirane analog is known to be a weak, linearly competitive inhibitor of MMP2 that binds poorly to the active site (conceivably, because the affinity of the oxirane oxygen for the active site zinc ion is significantly lower that than that for the sulfur of the thiirane; hence it is unable to displace hydroxide from the zinc – see section (a) above). The combination of a lower population of oxirane in the active site in the zinc coordination pose due to poorer binding and a higher barrier to ring opening may be sufficient to account for the fact that no turnover was seen in early experiments with MMP2 and the oxirane analog of SB-3CT (32). Further calculations were carried out to search for additional differences between these two reaction paths.
(c) Product Protonation by Water
Since a substantial portion of the active site of MMP2 is exposed to solvent, proton equilibration between product and solvent is expected, and is anticipated to strongly affect the stability of the product–protein complex. Since the zinc binding site is accessible to solvent, proton transfers from a water molecule near the active site to the thiirane-derived thiolate (and to the oxirane-derived alkoxide) could be energetically favorable events. Further calculations evaluated the energetics of proton transfer to the ring opening products of 3 and 4 (Fig. 5, 3-P1 and 4-P1, respectively) from an adjacent water molecule. This proton transfer leads to 3-P2 and 4-P2 in Fig. 5 and Fig. S4. The QM water (Wat2) in ONIOM calculations above was used as proton donor in these calculations, since Wat2 is close to the zinc ion and the three-membered ring in reactant complex and transition state (3-R, 3-TS, 4-R and 4-TS in Fig. 5). This proton migration produces a hydroxide anion which coordinates with zinc in both 3-P2 and 4-P2. The coordination with the zinc is much weaker for the protonated ring-opening product than for the alkoxide of the unprotonated ring-opening product 4-P1 or the thiolate of 3-P1. The computational results (P2 in Table 1 and Fig. 6) show that after proton transfer from water to the thiolate, the product complex 3-P2 is close to thermoneutral compared to the reactant complex 3-R, and is much less favorable than 3-P1. On the other hand for the oxirane system, the product complex after proton transfer to the alkoxide, 4-P2, is lower in energy and will be favored over the unprotonated product complex, 4-P1.
Conclusions
In this study, computational methods were used to examine the inhibition of MMP2 by SB-3CT (3) and its oxirane analog (4). The mechanism involves deprotonation of the inhibitor by a glutamate in the active site, opening of the thiirane ring and binding of the thiolate product to the zinc ion in the active site. Docking and MD simulation were used to prepare the inhibitors in the active site of MMP2. Since standard molecular mechanics force field cannot handle potential energy surfaces for chemical reactions and may not be optimally parameterized for interactions of metal ions, QM/MM methods were used to study the reaction of SB-3CT and its oxirane analog. The barrier for the deprotonation and ring opening reaction of 3 at MMP2 active site is 19.9 kcal/mol, and is lower than that for 4 by 1.6 kcal/mol. The ring-opening reaction of 3, (−21.1 kcal/mol), is significantly more exothermic than 4 (−13.1 kcal/mol). In reactant complexes of 3 and 4, both inhibitor and the glutamate are coordinated with the zinc. The reaction is not feasible if a water molecule is bound between the zinc and the glutamate. In the transition state, the glutamate moves away from the zinc to abstract a proton from the inhibitor. The inhibitor begins to interact with the zinc to facilitate the deprotonation and ring opening. In the products, zinc is coordinated with the thiolate or alkoxide formed by the ring opening of the inhibitor. Additional calculations show that alkoxide product from the ring opening of 4 is more easily protonated by a water molecule in the active site than is the thiolate from ring opening of 3.
Supplementary Material
Acknowledgments
We thank Wayne State University for generous allocations of computer time on its computational grid.
The abbreviations used are
- MMP2
matrix metalloproteinase 2
- SB-3CT
(4-phenoxyphenylsulfonyl)methylthiirane
- QM/MM
combined quantum mechanics and molecular mechanics
- ECM
extracellular matrix
- TIMPS
tissue inhibitors of metalloproteinases
- RESP
Restrained ElectroStatic Potential
- B3LYP
Becke 3-Parameter exchange, Lee, Yang and Parr correlation functional
- CBS
complete basis set
- HF
Hatree-Fock theory
- MP2
second-order Møller-Plesset perturbation theory
- MD
molecular dynamics
- ns
nanosecond
- ps
picosecond
Footnotes
This work is supported at Wayne State University by the National Science Foundation (CHE0512144) and at the University of Notre Dame by the National Institutes of Health (CA122417).
Supporting Information Available
MMP2·(R)-SB-3CT and MMP2·(S)-SB-3CT complexes from the MD simulation, QM/MM geometry of MMP2·(R)-SB-3CT complex with two waters at the active site, QM/MM geometries for ring opening reaction of 3 and 4 with and without hydroxide anion at the active site, QM/MM input files for thiirane and oxirane reactants, TSs and products, QM/MM energetics with mechanical and electronic embedding, energy profiles along the minimization paths from the TSs to reactants and products, and force fields parameters for zinc ions. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lei H, Furth EE, Kalluri R, Wakenell P, Kallen CB, Jeffrey JJ, Leboy PS, Strauss JF., 3rd Induction of matrix metalloproteinases and collagenolysis in chick embryonic membranes before hatching. Biol Reprod. 1999;60:183–189. doi: 10.1095/biolreprod60.1.183. [DOI] [PubMed] [Google Scholar]
- 2.Liu C-H, Wu P-S. Characterization of matrix metalloproteinase expressed by human embryonic kidney cells. Biotechnol Lett. 2006;28:1725–1730. doi: 10.1007/s10529-006-9147-y. [DOI] [PubMed] [Google Scholar]
- 3.Shi Y-B, Fu L, Hasebe T, Ishizuya-Oka A. Regulation of extracellular matrix remodeling and cell fate determination by matrix metalloproteinase stromelysin-3 during thyroid hormone-dependent post-embryonic development. Pharmacol Ther. 2007;116:391–400. doi: 10.1016/j.pharmthera.2007.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Woessner JF., Jr Matrix metalloproteinases and their inhibitors in connective tissue remodeling. FASEB J. 1991;5:2145–2154. [PubMed] [Google Scholar]
- 5.Smith MF, Ricke WA, Bakke LJ, Dow MP, Smith GW. Ovarian tissue remodeling: role of matrix metalloproteinases and their inhibitors. Mol Cell Endocrinol. 2002;191:45–56. doi: 10.1016/s0303-7207(02)00054-0. [DOI] [PubMed] [Google Scholar]
- 6.Homandberg GA, Ummadi V, Kang H. Hyaluronan enhances cartilage repair through low grade tissue remodeling involving cytokines and matrix metalloproteinases. Inflammation Res. 2004;53:534–543. doi: 10.1007/s00011-004-1292-y. [DOI] [PubMed] [Google Scholar]
- 7.Kawasaki Y, Xu Z-Z, Wang X, Park JY, Zhuang Z-Y, Tan P-H, Gao Y-J, Roy K, Corfas G, Lo EH, Ji R-R. Distinct roles of matrix metalloproteases in the early- and late-phase development of neuropathic pain. Nat Med. 2008;14:331–336. doi: 10.1038/nm1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coussens LM, Werb Z. Matrix metalloproteinases and the development of cancer. Chem Biol. 1996;3:895–904. doi: 10.1016/s1074-5521(96)90178-7. [DOI] [PubMed] [Google Scholar]
- 9.Vihinen P, Kähäri VM. Matrix metalloproteinases in cancer: prognostic markers and therapeutic targets. Int J Cancer. 2002;99:157–166. doi: 10.1002/ijc.10329. [DOI] [PubMed] [Google Scholar]
- 10.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 11.Noël A, Jost M, Maquoi E. Matrix metalloproteinases at cancer tumor-host interface. Semin Cell Dev Biol. 2008;19:52–60. doi: 10.1016/j.semcdb.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 12.Dollery CM, McEwan JR, Henney AM. Matrix metalloproteinases and cardiovascular disease. Circ Res. 1995;77:863–868. doi: 10.1161/01.res.77.5.863. [DOI] [PubMed] [Google Scholar]
- 13.Luft FC. Matrix metalloproteinases and their regulators are cardiovascular therapeutic targets. J Mol Med. 2004;82:781–783. doi: 10.1007/s00109-004-0609-1. [DOI] [PubMed] [Google Scholar]
- 14.Janssens S, Lijnen HR. What has been learned about the cardiovascular effects of matrix metalloproteinases from mouse models? Cardiovasc Res. 2006;69:585–594. doi: 10.1016/j.cardiores.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 15.Chow AK, Cena J, Schulz R. Acute actions and novel targets of matrix metalloproteinases in the heart and vasculature. Br J Pharmacol. 2007;152:189–205. doi: 10.1038/sj.bjp.0707344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosenblum G, Meroueh S, Toth M, Fisher J, Fridman R, Mobashery S, Sagi I. Molecular structures and dynamics of the stepwise activation mechanism of a matrix metalloproteinase zymogen: challenging the cysteine switch dogma. J Am Chem Soc. 2007;129:13566–13574. doi: 10.1021/ja073941l. [DOI] [PubMed] [Google Scholar]
- 17.Liotta LA, Tryggvason K, Garbisa S, Robey PG, Abe S. Partial purification and characterization of a neutral protease which cleaves type IV collagen. Biochemistry. 1981;20:100–104. doi: 10.1021/bi00504a017. [DOI] [PubMed] [Google Scholar]
- 18.Morgunova E, Tuuttila A, Bergmann U, Isupov M, Lindqvist Y, Schneider G, Tryggvason K. Structure of human pro-matrix metalloproteinase-2: activation mechanism revealed. Science. 1999;284:1667–1670. doi: 10.1126/science.284.5420.1667. [DOI] [PubMed] [Google Scholar]
- 19.Elkins PA, Ho YS, Smith WW, Janson CA, D’Alessio KJ, McQueney MS, Cummings MD, Romanic AM. Structure of the C-terminally truncated human ProMMP9, a gelatin-binding matrix metalloproteinase. Acta Crystallogr Sect D: Biol Crystallogr. 2002;D58:1182–1192. doi: 10.1107/s0907444902007849. [DOI] [PubMed] [Google Scholar]
- 20.Feng Y, Likos JJ, Zhu L, Woodward H, Munie G, McDonald JJ, Stevens AM, Howard CP, De Crescenzo GA, Welsch D, Shieh H-S, Stallings WC. Solution structure and backbone dynamics of the catalytic domain of matrix metalloproteinase-2 complexed with a hydroxamic acid inhibitor. Biochim Biophys Acta Proteins Proteomics. 2002;1598:10–23. doi: 10.1016/s0167-4838(02)00307-2. [DOI] [PubMed] [Google Scholar]
- 21.Lukacova V, Zhang Y, Kroll D, Raha S, Comez D, Balaz S. A comparison of the binding sites of matrix metalloproteinases and tumor necrosis factor-alpha converting enzyme: implications for selectivity. J Med Chem. 2005;48:2361–2370. doi: 10.1021/jm0491703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y, Lukacova V, Reindl K, Balaz S. Quantitative characterization of binding of small molecules to extracellular matrix. J Biochem Biophys Methods. 2006;67:107–122. doi: 10.1016/j.jbbm.2006.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Diaz N, Suarez D, Sordo T. Quantum chemical study on the coordination environment of the catalytic zinc ion in matrix metalloproteinases. J Phys Chem B. 2006;110:24222–24230. doi: 10.1021/jp0656882. [DOI] [PubMed] [Google Scholar]
- 24.Diaz N, Suarez D. Molecular dynamics simulations of the active matrix metalloproteinase-2: Positioning of the N-terminal fragment and binding of a small peptide substrate. Proteins: Struct Funct Bioinf. 2008;72:50–61. doi: 10.1002/prot.21894. [DOI] [PubMed] [Google Scholar]
- 25.Lukacova V, Zhang Y, Mackov M, Baricic P, Raha S, Calvo J, Balaz S. Similarity of binding sites of human matrix metalloproteinases. J Biol Chem. 2004;279:14194–14200. doi: 10.1074/jbc.M313474200. [DOI] [PubMed] [Google Scholar]
- 26.Ikejiri M, Bernardo MM, Bonfil RD, Toth M, Chang M, Fridman R, Mobashery S. Potent Mechanism-based Inhibitors for Matrix Metalloproteinases. J Biol Chem. 2005;280:33992–34002. doi: 10.1074/jbc.M504303200. [DOI] [PubMed] [Google Scholar]
- 27.Fisher JF, Mobashery S. Recent advances in MMP inhibitor design. Cancer Metastasis Rev. 2006;25:115–136. doi: 10.1007/s10555-006-7894-9. [DOI] [PubMed] [Google Scholar]
- 28.Khandelwal A, Balaz S. Improved estimation of ligand-macromolecule binding affinities by linear response approach using a combination of multi-mode MD simulation and QM/MM methods. J Comput-Aided Mol Des. 2007;21:131–137. doi: 10.1007/s10822-007-9104-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khandelwal A, Balaz S. QM/MM linear response method distinguishes ligand affinities for closely related metalloproteins. Proteins: Struct Funct Bioinf. 2007;69:326–339. doi: 10.1002/prot.21500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gupta SP. Quantitative Structure-Activity Relationship Studies on Zinc-Containing Metalloproteinase Inhibitors. Chem Rev. 2007;107:3042–3087. doi: 10.1021/cr030448t. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Y, Lukacova V, Bartus V, Nie X, Sun G, Manivannan E, Ghorpade S, Jin X, Manyem S, Sibi M, Cook G, Balaz S. Binding of matrix metalloproteinase inhibitors to extracellular matrix: 3D-QSAR analysis. Chem Biol Drug Des. 2008;72:237–248. doi: 10.1111/j.1747-0285.2008.00710.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown S, Bernardo MM, Li Z-H, Kotra LP, Tanaka Y, Fridman R, Mobashery S. Potent and Selective Mechanism-Based Inhibition of Gelatinases. J Am Chem Soc. 2000;122:6799–6800. [Google Scholar]
- 33.Forbes C, Shi Q, Fisher JF, Lee M, Hesek D, Llarrull LI, Toth M, Gossing M, Fridman R, Mobashery S. Active Site Ring-opening of a Thiirane Moiety and Picomolar Inhibition of Gelatinases. Chem Biol Drug Des. doi: 10.1111/j.1747-0285.2009.00881.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim D, Mobashery S. Mechanism-based inhibition of zinc proteases. Curr Med Chem. 2001;8:959–965. doi: 10.2174/0929867013372805. [DOI] [PubMed] [Google Scholar]
- 35.Tao P, Fisher JF, Mobashery S, Schlegel HB. DFT Studies of the Ring-Opening Mechanism of SB-3CT, a Potent Inhibitor of Matrix Metalloproteinase 2. Org Lett. 2009;11:2559–2562. doi: 10.1021/ol9008393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.SYBYL 7.3. Tripos Inc.; 1699 South Hanley Rd., St. Louis, MO, 63144: [Google Scholar]
- 37.Ewing TJA, Kuntz ID. Critical evaluation of search algorithms for automated molecular docking and database screening. J Comput Chem. 1997;18:1175–1189. [Google Scholar]
- 38.Hoops SC, Anderson KW, Merz KM. Force-field design for metalloproteins. J Am Chem Soc. 1991;113:8262–8270. [Google Scholar]
- 39.Case DA, Cheatham TE, III, Darden T, Gohlke H, Luo R, Merz KM, Jr, Onufriev A, Simmerling C, Wang B, Woods RJ. The amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee M, Bernardo MM, Meroueh SO, Brown S, Fridman R, Mobashery S. Synthesis of chiral 2-(4-phenoxyphenylsulfonylmethyl)thiiranes as selective gelatinase inhibitors. Org Lett. 2005;7:4463–4465. doi: 10.1021/ol0517269. [DOI] [PubMed] [Google Scholar]
- 41.Maseras F, Morokuma K. IMOMM: a new integrated ab initio + molecular mechanics geometry optimization scheme of equilibrium structures and transition states. J Comput Chem. 1995;16:1170–1179. [Google Scholar]
- 42.Svensson M, Humbel S, Froese RDJ, Matsubara T, Sieber S, Morokuma K. ONIOM: A multilayered integrated MO+MM method for geometry optimizations and single point energy predictions. A test for Diels-Alder reactions and Pt(P(t-Bu)(3))(2)+H-2 oxidative addition. J Phys Chem. 1996;100:19357–19363. [Google Scholar]
- 43.Humbel S, Sieber S, Morokuma K. The IMOMO method: Integration of different levels of molecular orbital approximations for geometry optimization of large systems: Test for n-butane conformation and S(N)2 reaction: RCl+Cl. J Chem Phys. 1996;105:1959–1967. [Google Scholar]
- 44.Dapprich S, Komaromi I, Byun KS, Morokuma K, Frisch MJ. A new ONIOM implementation in Gaussian98. Part I. The calculation of energies, gradients, vibrational frequencies and electric field derivatives. THEOCHEM. 1999;461-462:1–21. [Google Scholar]
- 45.Vreven T, Morokuma K. On the application of the IMOMO (integrated molecular orbital plus molecular orbital) method. J Comput Chem. 2000;21:1419–1432. [Google Scholar]
- 46.Morokuma K, Musaev DG, Vreven T, Basch H, Torrent M, Khoroshun DV. Model studies of the structures, reactivities, and reaction mechanisms of metalloenzymes. IBM J Res & Dev. 2001;45:367–395. [Google Scholar]
- 47.Vreven T, Morokuma K, Farkas O, Schlegel HB, Frisch MJ. Geometry optimization with QM/MM, ONIOM, and other combined methods. I. Microiterations and constraints. J Comput Chem. 2003;24:760–769. doi: 10.1002/jcc.10156. [DOI] [PubMed] [Google Scholar]
- 48.Vreven T, Frisch MJ, Kudin KN, Schlegel HB, Morokuma K. Geometry optimization with QM/MM methods II: Explicit quadratic coupling. Mol Phys. 2006;104:701–714. [Google Scholar]
- 49.Vreven T, Byun KS, Komaromi I, Dapprich S, Montgomery JA, Jr, Morokuma K, Frisch MJ. Combining Quantum Mechanics Methods with Molecular Mechanics Methods in ONIOM. J Chem Theory Comput. 2006;2:815–826. doi: 10.1021/ct050289g. [DOI] [PubMed] [Google Scholar]
- 50.Bearpark MJ, Ogliaro F, Vreven T, Boggio-Pasqua M, Frisch MJ, Larkin SM, Morrison M, Robb MA. CASSCF calculations for photoinduced processes in large molecules: Choosing when to use the RASSCF, ONIOM and MMVB approximations. J Photochem Photobiol A. 2007;190:207–227. [Google Scholar]
- 51.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 52.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Scalmani G, Mennucci B, Barone V, Petersson GA, Caricato M, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Li X, Hratchian HP, Peralta JE, Izmaylov AF, Kudin KN, Heyd JJ, Brothers E, Staroverov V, Zheng G, Kobayashi R, Normand J, Sonnenberg JL, Iyengar SS, Tomasi J, Cossi M, Rega N, Burant JC, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Chen W, Wong MW, Pople JA. Gaussian DV, Revision F.02. Gaussian, Inc.; Wallingford, CT: 2007. [Google Scholar]
- 53.Prabhakar R, Vreven T, Frisch MJ, Morokuma K, Musaev DG. Is the protein surrounding the active site critical for hydrogen peroxide reduction by selenoprotein glutathione peroxidase? An ONIOM study. J Phys Chem B. 2006;110:13608–13613. doi: 10.1021/jp0619181. [DOI] [PubMed] [Google Scholar]
- 54.Kwiecien RA, Khavrutskii IV, Musaev DG, Morokuma K, Banerjee R, Paneth P. Computational insights into the mechanism of radical generation in B-12-dependent methylmalonyl-CoA mutase. J Am Chem Soc. 2006;128:1287–1292. doi: 10.1021/ja056333j. [DOI] [PubMed] [Google Scholar]
- 55.Lundberg M, Kawatsu T, Vreven T, Frisch MJ, Morokuma K. Transition States in a Protein Environment - ONIOM QM:MM Modeling of Isopenicillin N Synthesis. J Chem Theory Comput. 2009;5:222–234. doi: 10.1021/ct800457g. [DOI] [PubMed] [Google Scholar]
- 56.Bayly CI, Cieplak P, Cornell W, Kollman PA. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J Phys Chem. 1993;97:10269–10280. [Google Scholar]
- 57.Cornell WD, Cieplak P, Bayly CI, Kollmann PA. Application of RESP charges to calculate conformational energies, hydrogen bond energies, and free energies of solvation. J Am Chem Soc. 1993;115:9620–9631. [Google Scholar]
- 58.Tochowicz A, Maskos K, Huber R, Oltenfreiter R, Dive V, Yiotakis A, Zanda M, Bode W, Goettig P. Crystal Structures of MMP-9 Complexes with Five Inhibitors: Contribution of the Flexible Arg424 Side-chain to Selectivity. J Mol Biol. 2007;371:989–1006. doi: 10.1016/j.jmb.2007.05.068. [DOI] [PubMed] [Google Scholar]
- 59.Rosenblum G, Meroueh SO, Kleifeld O, Brown S, Singson SP, Fridman R, Mobashery S, Sagi I. Structural Basis for Potent Slow Binding Inhibition of Human Matrix Metalloproteinase-2 (MMP-2) J Biol Chem. 2003;278:27009–27015. doi: 10.1074/jbc.M301139200. [DOI] [PubMed] [Google Scholar]
- 60.Dhanaraj V, Williams MG, Ye QZ, Molina F, Johnson LL, Ortwine DF, Pavlovsky A, Rubin JR, Skeean RW, White AD, Humblet C, Hupe DJ, Blundell TL. X-ray structure of gelatinase A catalytic domain complexed with a hydroxamate inhibitor. Croat Chem Acta. 1999;72:575–591. [Google Scholar]
- 61.Hof F, Schutz A, Fah C, Meyer S, Bur D, Liu J, Goldberg DE, Diederich F. Starving the malaria parasite: Inhibitors active against the aspartic proteases plasmepsins I, II, and IV. Angew Chem Int Ed. 2006;45:2138–2141. doi: 10.1002/anie.200504119. [DOI] [PubMed] [Google Scholar]
- 62.Lee M, Hesek D, Shi Q, Noll B, Fisher J, Chang M, Mobashery S. Conformational analyses of thiirane-based gelatinase inhibitors. Bioorg Med Chem Lett. 2008;18:3064–3067. doi: 10.1016/j.bmcl.2007.11.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.