

Summary
Chronic exposure to tobacco smoke, which contains over 60 tumor-initiating carcinogens, is the major risk factor for development of lung cancer, accounting for a large portion of cancer-related deaths worldwide. It is well established that tobacco smoke is a tumor initiator, but we asked whether it also acts as a tumor promoter once malignant initiation, such as caused by K-ras activation, has taken place. Here we demonstrate that repetitive exposure to tobacco smoke promotes tumor development both in carcinogen-treated mice and in transgenic mice undergoing sporadic K-ras activation in lung epithelial cells. Tumor promotion is due to induction of inflammation that results in enhanced pneumocyte proliferation and is abrogated by IKKβ ablation in myeloid cells or inactivation of JNK1.
Introduction
Currently, lung cancer is the leading cause of cancer-related mortalities in men and women, and despite extensive anti-smoking campaigns it still accounts for 15% of all new cancers and 29% of all cancer deaths in the US (Jemal et al., 2008). Amongst lung cancers, pulmonary adenocarcinoma is the predominant histological type (Jemal et al., 2008; Toh, 2009). Tobacco smoking is the major risk factor, estimated to cause 87% of lung cancer cases in the US (Hecht, 2002). Tobacco smoke (TS) contains about 4,000 chemical agents, including over 60 carcinogens: polycyclic aromatic hydrocarbons (PAHs), N-nitrosamines, such as NNK [4-(methylnitrosamino)-1-(3-pyridyl)-butanone] and aromatic amines (Hecht, 2002). Conversion of these compounds to reactive forms (metabolic activation) results in formation of DNA adducts that cause many of the genetic changes underlying lung cancer. Amongst these changes, K-ras-activating mutations are early events in the pathway leading to lung adenocarcinoma (Herbst et al., 2008). K-ras mutations occur in 30–40% of lung adenocarcinomas, but are infrequent in other lung tumor types or in lung tumors from non-smokers (Berns, 2001). TS also induces pulmonary inflammation (Vlahos et al., 2006), which is believed to play a role in progressive lung destruction in Chronic Obstructive Pulmonary Disease (COPD) (Barnes, 2008). Although chronic inflammation was suggested to contribute to tumor initiation through the production of reactive oxygen and nitrogen species that contribute to DNA damage and induction of oncogenic mutations (Hussain et al., 2003), the major effect of inflammation on tumor induction in experimental animals is exerted at the level of tumor promotion (Karin and Greten, 2005). This tumor promoting effect of inflammation, however, was so far mainly demonstrated in cancers that arise in the context of underlying infections or chronic idiotypic inflammation (Coussens and Werb, 2002; Karin et al., 2006) and its role in tumorigenesis induced by TS or other environmental irritants has not been critically evaluated. Although COPD is known to increase lung cancer risk (Tockman et al., 1987), K-ras activation in bronchial epithelial cells can cause an inflammatory response (Ji et al., 2006) and autocrine production of the chemokine CXCL-8 (IL-8) stimulates the growth of K-ras transformed lung adenocarcinoma cells (Karin, 2005; Sparmann and Bar-Sagi, 2004), the role of inflammation in smoking-induced lung tumorigenesis remains to be investigated using appropriate in vivo models. However, in none of the mouse models where TS or compounds derived from it can induce lung cancer, was TS shown to act on a different step in the tumorigenic process other than initiation (i.e. induction of oncogenic mutations).
Indeed, previous attempts to ascribe tumor promoting activity to TS in mice have failed (Witschi, 2005). In fact, mice that were first treated with a carcinogen and subsequently exposed to TS for 5 months showed a significant inhibition of tumor development rather than enhancement (Witschi et al., 1997). Following a 4-month recovery phase, TS-exposed mice exhibited the same tumor multiplicity as mice treated with carcinogen alone (Witschi et al., 1997). In NNK-treated mice, TS exposure also failed to increase lung tumor multiplicity (Finch et al., 1996). It has been recognized that in these murine models of TS exposure lung tumorigenesis is influenced by smoke-induced toxicity, manifested by weight loss, thereby necessitating the inclusion of a recovery period (Witschi, 2005). It was also noted that long term exposure desensitizes mice to TS, as maximal induction of pulmonary inflammation and cell proliferation were observed 1 to 3 weeks after initiation of TS exposure (Witschi et al., 1997). Furthermore, TS exposure can inhibit the metabolic activation of NNK and reduces the formation of NNK-induced O6-methylguanine DNA adducts (Brown et al., 1999), which strongly correlate with lung tumor yield in A/J mice (Peterson and Hecht, 1991). Taking all of these limitations into consideration, we have devised two new murine models in which intermittent, yet prolonged, exposure to mainstream TS (MTS) can successfully promote lung tumor development. We have used these models to determine how the tumor promoting effect of MTS exposure is accomplished.
Results
To better understand how exposure to TS, which contains many tumor initiators as well as irritants, promotes development of lung cancer, we sought to develop mouse models in which MTS exposure acts as a tumor promoter in addition to its established tumor initiator activity. In the first model, we used NNK as a tumor initiator and MTS as a tumor promoter. First, we optimized the initiating dose of NNK in A/J mice and found it to be 50–70 mg/kg (Figure S1A). Next, 1 week after intraperitoneal (i.p.) NNK injection into 7-week-old A/J mice, we exposed one group of mice to MTS generated by burning of 4 cigarettes (cig.)/day, 5 days per week for 1 month, followed by a 1 month rest interval and repeated this regimen 3 times. A second group was exposed to 2 cig./day, 5 days per week for 5 months, whereas the 3rd group received filtered air for the same duration, as a control (Figure 1A). All mice were given a final recovery period of 4 months. Whereas MTS-exposed mice failed to gain weight during the exposure period, mice exposed to 4 cig./day for 1 month rapidly resumed weight gain and caught up with air-exposed mice during the 1 month rest interval (Figure 1B). Yet, mice that were continuously exposed to 2 cig./day showed a sustained reduction in body weight.
Figure 1. Tobacco smoke exposure promotes development of chemically-induced lung cancer.
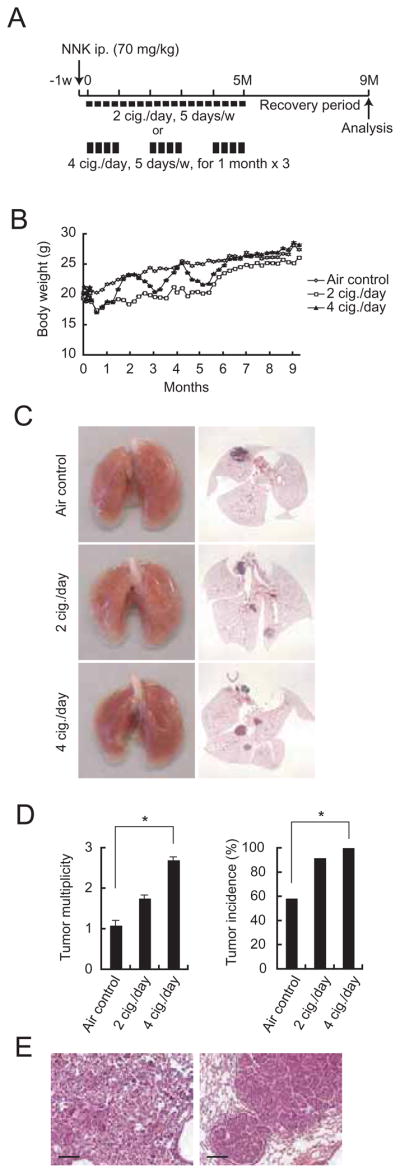
(A) Experimental protocols for promotion of NNK-induced lung cancer. 7-week-old male A/J mice were MTS-exposed using two different regimens starting 1 week after NNK injection. Tumor development was analyzed 9 months later.
(B) Effect of MTS exposure on weight gain in A/J mice. Results are means ± S.E. (air control: n = 12; 2 cig./day: n = 12; 4 cig./day: n = 13).
(C) Lung appearance (left panels) and histology (H&E stain; right panels) in A/J mice 9 months after initiation of the NNK + MTS protocol.
(D) Lung tumor multiplicity and incidence were determined by serial sectioning at 350 μm intervals. Incidence = number of tumor-bearing mice divided by the number of mice in each group. Results are means ± S.E. (n: as described in panel B). Significant difference, *P < 0.03.
(E) Histological appearance of adenoma (left panel) and adenocarcinoma with bronchial invasion (right panel) in A/J mice given NNK and 4 cig./day MTS. Scale bar = 100 μm. See also Figure S1.
We analyzed lung tumor multiplicity and incidence by microscopic examination after serial sectioning of lungs at 350 μm intervals as described (Curtin et al., 2004). Mice administered 70 mg/kg NNK and exposed to 4 cig./day exhibited significantly increased NNK-initiated lung tumor multiplicity, as well as increased tumor incidence (Figures 1C, D). Exposure to 2 cig./day MTS also increased tumor multiplicity and the effect was most pronounced when combined with the highest NNK dose that was tested 70 mg/kg (Figure S1A). Alveolar adenoma was the most common histological type in both air- and MTS-exposed mice, seen in about 70% of each cohort. About 30% of the tumors in both air- and MTS-exposed mice showed features of well-differentiated human papillary adenocarcinoma, including nuclear enlargement, prominent nucleoli, increased mitotic rate and bronchial invasion (Figure 1E and Figure S1B).
Since the most common mutation found in murine lung tumors induced by NNK is a GGT → GAT transition in codon 12 of the K-ras gene (Hecht, 1998), we developed a second model in which we investigated whether intermittent MTS exposure could promote K-ras-driven lung tumorigenesis using K-rasLA2+/− (K-rasLA2) mice, which develop lung cancer in response to sporadic activation of an oncogenic K-rasasp12 allele through a spontaneous homologous recombination event (Johnson et al., 2001). Six to eight week-old, sex-matched K-rasLA2 mice were exposed to MTS at 4 cig./day for 2 – 3 weeks followed by 2 week rest intervals on air alone, 3-times, and lung tumors were analyzed at 5 months of age (Figure 2A). In contrast to A/J mice, weight reduction in K-rasLA2 mice, which are in the C57BL6 background, was more modest and occurred only during the initial 2 cycles of MTS exposure (Figure 2B). As found in NNK-initiated mice, MTS exposure increased tumor multiplicity and maximal tumor sizes (Figures 2C, D). No tumors were found in MTS-exposed wild type (WT) mice (Figure 2C), indicating that increased tumor number in MTS-exposed K-rasLA2 mice is not due to MTS-induced tumor initiation. Similar effects were observed in male and female mice (Figures S2A, B). K-rasLA2 mice also develop thymic lymphoma due to K-rasasp12 activation in thymocytes (Johnson et al., 2001). No statistically-significant differences in the incidence of thymic lymphoma were detected between air- and MTS-exposed K-rasLA2 mice (Figure S2C), suggesting that the tumor promoting effect of MTS is limited to the primary site of exposure - the lung. Most pulmonary tumors in air- or MTS-exposed K-rasLA2 mice were alveolar adenomas (Figure 2E, Figure S2D), corresponding to a relatively early stage of lung cancer development, as reported previously (Johnson et al., 2001). Interestingly, tumors in MTS-exposed mice were more vascularized than those in air-exposed controls (Figure S2E, F). Thus, MTS exposure promotes development of both chemically- and genetically-induced lung cancer and this constitutes strong evidence that TS acts as a tumor promoter and not only as an initiator.
Figure 2. Tobacco smoke exposure promotes development of genetically-induced lung cancer.

(A) Outline of the experimental protocol in which K-rasLA2 mice were exposed to MTS and analyzed at 5 months of age.
(B) Effect of MTS exposure on weight gain in K-rasLA2 mice. Results are means ± S.E. (air control: n = 4; 4 cig./day: n = 8).
(C) Lung appearance (left side) and histology (H&E stain; right side) in 5-month-old K-rasLA2 or WT mice with or without MTS exposure.
(D) Lung tumor multiplicity and maximal tumor sizes were determined as above. Results are means ± S.E. (air control: n = 16; 4 cig./day: n = 18). Significant difference, *P < 0.002.
(E) Histological appearance of adenoma in MTS-exposed K-rasLA2 mice. Scale bar = 100 μm. See also Figure S2.
We investigated whether the tumor promoting effect of MTS exposure is due to induction of inflammation. We examined the sub-acute inflammatory pulmonary response of C57BL6 male mice exposed to MTS or air. Total cell number and numbers of various leukocyte types in bronchoalveolar lavage fluid (BALF) were significantly increased 1 and 2 weeks after MTS exposure (Figure 3A). MTS exposure induced the expression of various inflammatory cytokine and chemokine mRNAs, such as Il1β, Il6, Tnfα, Ccl3 (Mip1α), and Ccl2 (Mcp1) in the lung (Figure 3B). Induction of most of these transcripts was maximal within 2 weeks of exposure initiation and declined at later time points (Figure S3A). MTS exposure also induced secretion of TNF-α and IL-6 by lung cells (Figure 3C) and stimulated mitogen-activated protein kinases (MAPKs), including extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) (Figure 3D). Similar induction of inflammatory response genes was seen after MTS exposure of K-rasLA2 tumor-bearing mice (Figure S3B). MTS exposure modestly increased NF-κB DNA binding activity (Figure 3D) and increased cell proliferation in the lung (Figure 3E), without affecting basal rate of apoptosis (Figure S3C). Proliferating cells in the alveolar zone of MTS-exposed lungs were mainly alveolar epithelial cells and immune cells (Figure S3D). As shown below, MTS exposure also increased cell proliferation within lung adenomas. Both IL-6 and TNF-α deficient mice showed decreased MTS-enhanced lung cell proliferation (Figure S3E), indicating the importance of these inflammatory cytokines in MTS-induced cell proliferation. Lung tumors in MTS-exposed K-rasLA2 mice exhibited enhanced infiltration with IL-6-producing macrophages (Figure 3F; Figure S3F).
Figure 3. Tobacco smoke induces pulmonary inflammation and cell proliferation.

(A) Total cell number and leukocyte populations in BALF collected from C57BL6 males 24 hrs after last MTS or air exposure. Cellular composition was analyzed using cytospin preparations. Results are means ± S.E. (n = 8 for each group). Significant difference, *P < 0.02 vs. air control.
(B) Induction of inflammatory cytokine and chemokine mRNAs in lungs of MTS-exposed C57BL6 males. Lung RNA was isolated 24 hrs after last MTS exposure and analyzed by real-time PCR. Results are means ± S.E. (n = 5 for each group). Significant difference, *P < 0.03 vs. air control.
(C) Elevated cytokine secretion by lungs of MTS-exposed male mice. Fresh lungs were cut into small pieces and incubated in medium at 37°C for 48 hrs. Cytokines in culture supernatants were measured by ELISA. Results are means ± S.E. (air control: n = 7; 4 cig./day 1w: n = 9; 4 cig./day 2w: n = 9). Significant difference, *P < 0.02 vs. air control.
(D) ERK and JNK activation and NF-κB DNA binding activity in lungs of MTS-exposed mice. Lung lysates and nuclear extracts prepared 4 hrs after last MTS exposure were analyzed by immunoblotting and EMSA. Nuclear protein content was determined by immunoblotting with β-actin antibody. Shown are results from 2 representative mice per group.
(E) Cell proliferation in lungs of air- or MTS-exposed mice was determined by BrdU labeling. Results are means ± S.E. (air control: n = 7; 4 cig./day 1w: n = 5; 4 cig./day 2w: n = 8). Significant difference, *P < 4.0 × 10−5 vs. air control.
(F) Infiltration of IL-6 positive macrophages into K-rasLA2 lung tumors 2 weeks after initiation of MTS exposure. Lung sections prepared 24 hrs after last MTS exposure were analyzed by immunostaining for F4/80 (green) and IL-6 (red). Nuclei were counterstained by DAPI (blue). Scale bar = 100 μm. Results shown are for one representative mouse. For quantitation of the entire experiment see Figure S3F. See also Figure S3.
IκB kinase β (IKKβ), required for NF-κB activation, links chronic inflammation with carcinogenesis in a mouse model of colitis-associated cancer (Greten et al., 2004) and is a critical regulator of inflammatory cytokine production (Karin, 2006). To examine the contribution of IKKβ to MTS-induced sub-acute pulmonary inflammation, we used IkkβΔmye (LysM-Cre; IkkβF/F) mice that specifically lack IKKβ in the myeloid lineage (Greten et al., 2004), because myeloid cells, including alveolar macrophages, are a major source of inflammatory cytokines needed for initiation and propagation of inflammatory responses (Barnes, 2008). The efficiency of IKKβ deletion in alveolar macrophages was 80~90% (Figure 4A) and previous experiments have shown that IKKβ is not deleted in any cell type other than mature macrophages and neutrophils (Greten et al., 2004). IkkβΔmye mice showed impaired MTS-induced cell recruitment into BALF, that mostly affected neutrophils (Figure 4B), decreased induction of Il6, Tnfα, Ccl3, and Ccl2 mRNAs, as well as decreased secretion of IL-6 and TNF-α by MTS-exposed lungs, relative to MTS-exposed IkkβF/F mice (Figures 4C, D). IkkβΔmye mice also exhibited decreased MTS-induced cell proliferation (Figure 4E). We also deleted IKKβ in Clara cells which account for 70% of the airway epithelium using CC10-Cre; IkkβF/F mice (Broide et al., 2005). Clara cell-specific deletion of IKKβ had only a modest and statistically insignificant effect on MTS-induced cytokine secretion and cell proliferation (Figures S4A, B). Its effect on MTS-induced tumor promotion was therefore not explored.
Figure 4. Myeloid cell IKKβ deletion decreases MTS-induced inflammation and cell proliferation.

(A) Expression of IKKβ in alveolar macrophages of 7-week-old IkkβF/F and IkkβΔmye mice.
(B) BALF cellular composition in air- or MTS-exposed IkkβF/F and IkkβΔmye mice 24 hrs after last MTS exposure. Results are means ± S.E. (IkkβF/F air control: n = 9, IkkβF/F 4 cig./day 2w: n = 9, IkkβΔmye air control: n = 9, IkkβΔmye 4 cig./day 2w: n = 13). Significant difference, *P < 0.03.
(C) Induction of inflammatory cytokine and chemokine mRNAs in lungs of MTS-exposed IkkβF/F and IkkβΔmye mice 24 hrs after last 1 or 2 weeks MTS exposure. Results are means ± S.E. (n = 5 for each group). Significant difference, *P < 0.05.
(D) Secretion of cytokines by lungs of MTS-exposed mice was analyzed as in Fig. 3C. Results are means ± S.E. (IkkβF/F air control: n = 14; IkkβF/F 4 cig./day 2w: n = 8; IkkβΔmye air control: n = 12; IkkβΔmye 4 cig./day 2w: n = 13). Significant difference, *P < 0.04.
(E) Cell proliferation in lungs of air- or MTS-exposed mice was analyzed as in Fig. 3E. Results are means ± S.E. (n = 7 for each group). Significant difference, *P < 0.03. See also Figure S4.
To investigate the role of myeloid cell IKKβ in MTS-induced lung tumor promotion, we crossed K-rasLA2 mice with IkkβΔmye or IkkβF/F mice and exposed the hybrid strains to MTS as described above. Deletion of IKKβ in myeloid cells significantly reduced the effect of MTS exposure on lung tumor multiplicity and maximal tumor sizes (Figures 5A,B). IKKβ deletion in myeloid cells also reduced tumor angiogenesis, especially in MTS-exposed mice (Figure S2E, F). In agreement with the reduction in IL-6 production, we observed decreased MTS-induced STAT3 activation in K-rasLA2; IkkβΔmye mice (Figure S5A, B). As indicated above, IL-6 and TNF-α, whose induction in MTS-exposed lungs requires IKKβ in myeloid cells in both tumor-free (Figure 4C) and in K-rasLA2 tumor-bearing mice (Figure S3B), are essential for MTS-induced epithelial cell proliferation (Figure S3E). Consistent with these findings, IKKβ ablation in myeloid cells reduced MTS-induced proliferation of lung adenoma cells (Figure 5C). Myeloid cell IKKβ ablation, however, did not have a significant effect on apoptosis in lung adenomas, which was slightly elevated upon MTS exposure (Figure 5D). We also examined the effect of IKKβ deletion in myeloid cells on MTS-induced recruitment of various immune cell types into lungs of K-rasLA2 tumor-bearing mice. As seen in naïve mice, IKKβ ablation reduced the number of recruited neutrophils (Gr1+ cells) but did not significantly impact the MTS-induced recruitment of macrophages (F4/80+ cells) and CD4+ T cells (Figure S5C, D). The reduction in neutrophil count was supported by a marked reduction in expression of myeloperoxidase (MPO) mRNA that encodes a neutrophil-specific oxidative enzyme. MTS exposure and IKKβ deletion had no significant effect on the abundance of lung CD8+ T cells. Thus, IKKβ in myeloid cells plays a critical role in the promotion of K-rasasp12-induced lung cancer development by MTS, most likely through the induction of inflammation, angiogenesis and tumor promoting inflammatory cytokines that stimulate the proliferation of both pre-malignant and malignant cells. Our results rule out reduced apoptosis as a component of the tumor promoting effect of MTS exposure.
Figure 5. IKKβ deletion in myeloid cells inhibits MTS-induced lung tumor promotion and malignant cell proliferation in K-rasLA2 mice.

(A) Lung appearance (left) and histology (H&E stain; right) in 5-months-old K-rasLA2; IkkβF/F and K-rasLA2; IkkβΔmye mice with or without MTS exposure.
(B) Lung tumor multiplicity and maximal tumor sizes were determined as in Figure 2D in K-rasLA2; IkkβF/F and K-rasLA2; IkkβΔmye mice that were either air- or MTS-exposed. Results are means ± S.E. (K-rasLA2; IkkβF/F air control: n = 18; K-rasLA2; IkkβF/F 4 cig./day: n = 17; K-rasLA2; IkkβΔmye air control: n = 20; K-rasLA2; IkkβΔmye 4 cig./day: n = 18). Significant difference, *P < 0.02.
(C) Cell proliferation in lung adenomas from air- or MTS-exposed mice of indicated genotypes was determined by BrdU labeling. Results are means ± S.E. (n = 5–6 for each group). Significant difference, *P < 0.001.
(D) Apoptosis in lung adenomas from air- or MTS-exposed mice of indicated genotypes was determined by TUNEL staining. Results are means ± S.E. (n = 5–6 for each group). See also Figure S5.
It has been suggested that activation of MAPKs, including JNK, contributes to COPD-associated mucus overproduction and inflammation (Mercer and D’Armiento, 2006). JNK activation is also required for optimal induction of inflammatory cytokines (Karin and Gallagher, 2005). To investigate the role of JNK in MTS-induced inflammation, we analyzed the sub-acute response to MTS exposure in Jnk1−/− mice. Jnk1−/− mice showed reduced induction of Il6, Tnfα, Ccl3, and Ccl2 mRNAs, as well as decreased secretion of IL-6 and TNF-α by lung tissue upon MTS exposure, compared to WT mice (Figures 6A, B). Jnk1−/− mice also exhibited decreased induction of pneumocyte proliferation upon MTS exposure (Figure 6C).
Figure 6. Deletion of JNK1 decreases MTS-induced inflammation, cell proliferation, and lung tumor promotion.

(A) Induction of inflammatory cytokine and chemokine mRNAs in lungs of MTS-exposed Jnk1−/− mice was analyzed as in Fig. 3B. Results are means ± S.E. (n = 5 for each group). Significant difference, *P < 0.05.
(B) Secretion of cytokines in lungs of MTS-exposed Jnk1−/− mice was analyzed as in Fig. 3C. Results are means ± S.E. (WT air control: n = 12; WT 4 cig./day: n = 9; Jnk1−/− air control: n = 9; Jnk1−/− 4 cig./day: n = 9). Significant difference, *P < 0.05.
(C) JNK1 deletion decreases MTS-induced lung cell proliferation. Results are means ± S.E. (WT air control: n = 7; WT 4 cig./day: n = 8; Jnk1−/− air control: n = 10; Jnk1−/− 4 cig./day: n = 9). Significant difference, *P < 0.0003.
(D) Lung appearance (left side) and histology (H&E stain; right side) in 5-month-old K-rasLA2 or K-rasLA2; Jnk1−/− mice with or without MTS exposure.
(E) Lung tumor multiplicity and maximal tumor sizes were determined as in Fig. 2D. Results are means ± S.E. (WT air control: n = 16; WT 4 cig./day: n = 18; Jnk1−/− air control: n = 13; Jnk1−/− 4 cig./day: n = 14). Significant difference, *P < 0.002. See also Figure S6.
To examine the role of JNK1 in MTS-induced lung tumor promotion, we crossed K-rasLA2 mice with Jnk1−/− mice and generated K-rasLA2; Jnk1−/− hybrids that were exposed to MTS. Ablation of JNK1 reduced the enhancing effect of MTS on lung tumor multiplicity and maximal tumor sizes (Figures 6D, E). We also found decreased MTS-induced STAT3 activation in K-rasLA2; Jnk1−/− mice (Figure S6A, B). Therefore, JNK1 also plays a significant role in the promotion of lung inflammation and tumorigenesis subsequent to MTS exposure.
Discussion
TS exposure accounts for the majority of lung cancer-related deaths in men and women (Hecht, 1999). Despite the identification of numerous tumor-initiating carcinogens, such as NNK (Hecht, 1999), the mechanism by which prolonged TS exposure, either active or passive, enhances lung tumorigenesis is not well understood due to the paucity of proper experimental models that faithfully replicate different features of TS-induced lung tumorigenesis in a small animal species amenable to genetic analysis, i.e. the laboratory mouse. We now describe two mouse models in which TS exposure has a well-defined tumor promoting effect on both chemically (NNK)- and genetically (K-rasasp12)-induced lung cancer. We show that in both NNK-initiated A/J mice and C57BL6 mice in which lung cancer has been initiated through expression of an oncogenic K-rasasp12 allele, MTS exposure increases tumor multiplicity, incidence and size. Consistent with being a tumor promoter under our experimental conditions, MTS exposure had no discernable effect on tumor histology, although tumor size was clearly increased along with an enhanced tumor angiogenesis. Although tumor promotion may be attributed to several factors, induction of inflammation, which elevates the production of inflammatory cytokines that enhance the proliferation and survival of initiated epithelial cells, is likely to be a major tumor promoting mechanism (Karin, 2006). Here we demonstrate that induction of a low grade (sub-acute) inflammatory response is an important contributor to the tumor promoting activity of TS, leading to the enhanced proliferation of both pre-malignant and malignant pulmonary epithelial cells.
Interference with this inflammatory response through myeloid cell-specific IKKβ ablation resulted in nearly complete abrogation of MTS-induced tumor promotion, but had little effect if any on NNK- or K-rasasp12-induced tumor initiation. These findings should be contrasted with the importance of autocrine chemokine production for the growth and progression of already established and transplantable lung adenocarcinomas (Karin, 2005; Sparmann and Bar-Sagi, 2004). Although Ras-induced CXCL-8 (IL-8) produced by lung adenocarcinoma cells is a chemoattractant for myeloid cells (macrophages and neutrophils) and it and other K-ras-induced chemokines can cause severe pulmonary inflammation in mice (Ji et al., 2006), the mechanism through which such chemokines exert their tumor promoting effect was not defined (Karin, 2005). Our results suggest that one possible mechanism by which myeloid cells promote tumor growth is through the IKKβ/NF-κB-dependent production of cytokines, such as IL-6 and TNF-α, both of which stimulate the proliferation of pulmonary epithelial cells. These or other cytokines and chemokines may also enhance tumor angiogenesis.
Previous work on the tumor promoting role of inflammation has been focused on cancers that are associated with either chronic inflammatory disease, such as colitis associated cancer (Greten et al., 2004; Grivennikov et al., 2009), or persistent viral and bacterial infections, for instance hepatocellular carcinoma (Maeda et al., 2005; Pikarsky et al., 2004) and gastric cancer (Tu et al., 2008). The present work has addressed the role of inflammation in the tumorigenic effect of an environmental irritant, TS, with a major public health impact, being responsible for about 25% of all cancer related deaths in the US. Although tobacco smoking increases colonization of the airways with inflammation-causing bacteria (Saetta et al., 1994), it also delivers irritants that are capable of direct induction of inflammation, a property shared with other inhaled microparticles, such as asbestos and silica (Borm and Driscoll, 1996). Whereas chronic inflammation lasting for decades, such as in long term smokers, may lead to induction of oncogenic mutations through the production of reactive oxygen and nitrogen species (Hussain et al., 2003), it also leads to activation of NF-κB, AP-1 and other transcription factors in airway myeloid cells, resulting in elevated cytokine production (Barnes, 2008). Our work based on shorter term TS exposure in mice, suggests that activation of IKKβ and JNK signaling pathways and induction of inflammatory cytokine production in myeloid cells by TS-born irritants results in a strong tumor promoting effect that enhances the development of lung cancer, at least in mice. Although so far we have only examined the prophylactic value of anti-inflammatory intervention in TS-enhanced tumorigenesis, it is likely that appropriate anti-inflammatory therapy may slow down the development and progression of lung cancer if administered early enough and may interfere not only with tumor promotion but also with the accumulation of further genetic changes. Our findings suggest that it is also worthwhile to determine whether the great variation in the incidence of lung cancer amongst smokers is associated with genetic differences in TS-induced inflammation, as many genetic polymorphisms are known to affect the inflammatory response in the airways and other tissues (Cook et al., 2004). Identification of the long suspected, but heretofore unproven, tumor promoting effect of TS provides a sound scientific rationale to the benefits of anti-inflammatory therapy or intervention as well as for smoking cessation, which has to be practiced as early as possible after detection of the smallest primary lung tumor, if not earlier.
Experimental Procedures
Animals
IkkβF/F, LysM-Cre;IkkβF/F (referred to as IkkβΔmye), CC10-Cre;IkkβF/F, and Jnk1−/− mice were described (Broide et al., 2005; Maeda et al., 2005). K-rasLA2 mice were provided by T. Jacks (Massachusetts Institute of Technology) through the National Cancer Institute. Tnfα−/−, Il6−/−, and A/J mice were from the Jackson Laboratory. All strains, except A/J mice, used in this study were backcrossed to the C57BL6 background for more than 6 times. Mice were maintained under specific pathogen-free conditions, and experimental protocols were approved by University of California, San Diego Animal Care Program, following National Institute of Health guidelines.
NNK Treatment and Tobacco Smoke Exposure
To examine promotion of chemically-induced lung cancer, 7-week-old male A/J mice were i.p. injected with 70 mg/kg NNK (Toronto Research Chemicals) one week before initiation of MTS exposure. Mice were exposed to MTS generated by burning 3R4F reference cigarettes (College of Agriculture, Reference Cigarette Program, University of Kentucky) using a smoking machine (McChesney-Jaeger CSM-SSM Single Cigarette Machine; CH Technologies) regulated by programmable controls provided with JASPER windows 9x/2000 software over RS-232 communication ports (CH Technologies). Each smoldering cigarette was puffed for ~2 s, once every 25 s, for a total of 12 puffs/cigarette, at a flow rate of 5 L/min. Fresh smoke was diluted with filtered air and delivered to 12-port nose-only directed flow inhalation exposure system (Jaeger-NYU 12 port). The average total suspended particulate matter monitored at nose ports was 173 ± 5.3 mg/m3. In the 4 cig./day exposure group, mice were exposed to MTS from 4 cig./day for ~5 min with a 5 min break between cigarettes, 5 days/week for 1 month followed by 1 month of rest. This cycle was repeated two more times. In the 2 cig./day group, mice were exposed to MTS from 2 cig./day, administered over 5 min with 5 min break between cigarettes, 5 days/week for 5 months. Control mice were kept on filtered air. Lung tumors were analyzed 9 months after initiation of NNK plus MTS exposure. To examine promotion of genetically induced lung tumors, 6-8-week-old K-rasLA2 mutant mice were exposed to MTS at 4 cig./day, 5 days/week for 3 weeks followed by a 2 weeks rest interval on air alone, 2 times and exposed to MTS at 4 cig./day, 5 days/week for 2 more weeks. Sex-matched control K-rasLA2 mutant mice were kept on filtered air. Lung tumors were analyzed at 5 months of age. For the NNK dose optimization study, 7-week-old male A/J mice were i.p. injected with 30, 50, or 70 mg/kg NNK one week before initiation of MTS exposure. Mice were exposed to 2 cig./day MTS for 2 months and 1 cig./day for 3 months. Control mice were kept on filtered air. Lung tumors were analyzed 9 months after NNK injection. In the subacute inflammation study, 7-8-week-old male mice were exposed to 4 cig./day MTS, 5 days/week for 1 or 2 weeks. Control mice were kept on filtered air.
BrdU Labeling
To examine pulmonary cell proliferation, randomly chosen mice were i.p. injected with 100 mg/kg BrdU (Sigma) 2 hrs prior to sacrifice, and paraffin sections were stained with a BrdU in-situ detection kit (BD). 3000 to 5000 cells per lung were counted in randomly selected fields. BrdU labeling index was calculated as the percentage of labeled cells per total cells counted.
BALF Leukocyte Counts and Alveolar Macrophage Isolation
Number of total cells and leukocyte types in BALF were determined as described (Ikeda et al., 2003). Briefly, following instillation of 800 μl sterile PBS through the trachea, BALF was withdrawn, and cytospin preparations of BALF cells were prepared using Shandon Cytospin centrifuge (Shandon Lipshaw). BALF cells were visualized by Wright-Giemsa staining and percentages of leukocyte types were determined by counting 400 leukocytes in a randomly selected portion of the slide under light microscopy. BALF total leukocyte counts were performed using a hemocytometer. Alveolar macrophages were isolated as previously described (Vlahos et al., 2006).
Evaluation of Lung Tumors
To determine tumor incidence, multiplicity, and maximal sizes, whole lungs were manually inflated with and fixed in 4% paraformaldehyde for at least 24 hrs and paraffin embedded. Paraffin-embedded lungs were serially sectioned at 350 μm, and histologically examined using hematoxylin and eosin (H&E) stained sections as described (Curtin et al., 2004).
Cytokine ELISA
Fresh lung lobes were cut into small pieces and incubated in 1 ml RPMI medium (Invitrogen) at 37°C for 48 hrs. IL-1β, IL-6 and TNF-α were measured by enzyme linked immunosorbent assay (BD Biosciences, eBioscience, and R&D) using culture supernatants.
Analysis of Gene Expression
Total tissue RNA was prepared using RNeasy plus mini kit (Qiagen). Quantitative PCR was performed as described (Kim et al., 2009). RNA was reverse-transcribed using SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen). Real-time PCR was performed using SYBR green (Bio-Rad) on a Bio-Rad iQ5 machine. Expression data were normalized to β-actin mRNA amounts. Primer pairs for Il1β, Il6, Tnfα, Ccl2 and Ccl3 were:
5′-CAACCAACAAGTGATATTCTCCATG-3′/5′-GATCCACACTCTCCAGCTGCA-3′,
5′-GAGGATACCACTCCCAACAGACC-3′/5′-AAGTGCATCATCGTTGTTCATACA-3′,
5′-GACCAGGCTGTCGCTACATCA-3′/5′-CGTAGGCGATTACAGTCACGG-3′,
5′-GCAGGTCCCTGTCATGCTTC-3′/5′-TCCAGCCTACTCATTGGGATCA-3′,
5′-TGGAGCTGACACCCCGAC-3′/5′-ACGATGAATTGGCGTGGAA-3′, respectively. Primer pairs for MPO were: 5′-CACCCTCTTTGTTCGAGAGC-3′/5′-CAACACCAAGGGCAGGTAGT-3′.
Biochemical and Immunohistochemical Analyses
Nuclear proteins were isolated from lung tissues using Nuclear Extract Kit (Active Motif), and EMSA was performed using Gelshift Kit (Active Motif) and a κB site consensus oligonucleotide (Promega). Nuclear extracts were analyzed by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting with antibody to β-actin (Sigma). Total tissue lysates were prepared and analyzed by SDS-PAGE and immunoblotting with antibodies to P-JNK, ERK, P-ERK (all from Cell Signaling), JNK (Pharmingen) and IKKβ (Upstate). Apoptosis was determined using ApopTag Red In Situ Apoptosis Detection Kit (Chemicon). For immunohistochemistry, paraffin-embedded slides were deparaffinized and antigens were unmasked by incubation at 94°C for 40 min in Target Retrieval Solution S1700 (Dako). Slides were incubated with primary antibodies in 4°C overnight. Signals were detected using fluorescent microscopy, or VECTASTIN ABC Elite kit (Vector lab), DAB Substrate Kit (Vector lab) and light microscopy. The following antibodies were used: anti-IL-6-PE, anti-P-STAT3 (both from Cell Signaling), anti-F4/80-Alexa488 (Caltag), anti-CD31 anti-CD4, anti-CD8 (all three from BD Pharmingen), anti-F4/80 and anti-Gr-1 (both from eBioscience).
Statistical Analysis
Results are expressed as means ± S.E. Data were analyzed by Student’s t-test and Fisher’s exact test. P values < 0.05 were considered significant.
Supplementary Material
Acknowledgments
The authors thank Cell Signaling Technology for the gifts of antibodies. Research was supported by Grants from the NIH, including the Superfund Research Program (M.K.) and Tobacco-Related Disease Research Program (15RT-0197 to M.K. and 12RT-0071 to D.H.B). H.T. and H.O. were supported by the Japanese Respiratory Society and Kanzawa Medical Research Foundation, respectively. M.K. is an American Cancer Society research professor.
Footnotes
Supplemental Data are attached.
Significance
Tobacco smoking accounts for the majority of lung cancer-related deaths. Despite identification of numerous tumor-initiating carcinogens, it was not established whether tobacco smoke is also a tumor promoter for initiated lung epithelial cells. We now describe two murine models in which tobacco smoke exposure has a well-defined tumor promoting effect on both chemically- and genetically-initiated lung cancers. Induction of low-grade inflammation is likely to be an important contributor to the tumor promoting activity of tobacco smoke, as it depends on IKKβ activity in myeloid cells. These results provide new models and mechanistic insights for understanding tumor induction by a major human carcinogen and demonstrate that the tumorigenic activity of an environmental irritant depends on inflammation.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barnes PJ. The cytokine network in asthma and chronic obstructive pulmonary disease. J Clin Invest. 2008;118:3546–3556. doi: 10.1172/JCI36130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berns A. Cancer. Improved mouse models. Nature. 2001;410:1043–1044. doi: 10.1038/35074238. [DOI] [PubMed] [Google Scholar]
- Borm PJ, Driscoll K. Particles, inflammation and respiratory tract carcinogenesis. Toxicol Lett. 1996;88:109–113. doi: 10.1016/0378-4274(96)03725-3. [DOI] [PubMed] [Google Scholar]
- Broide DH, Lawrence T, Doherty T, Cho JY, Miller M, McElwain K, McElwain S, Karin M. Allergen-induced peribronchial fibrosis and mucus production mediated by IkappaB kinase beta-dependent genes in airway epithelium. Proc Natl Acad Sci U S A. 2005;102:17723–17728. doi: 10.1073/pnas.0509235102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown BG, Chang CJ, Ayres PH, Lee CK, Doolittle DJ. The effect of cotinine or cigarette smoke co-administration on the formation of O6-methylguanine adducts in the lung and liver of A/J mice treated with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) Toxicol Sci. 1999;47:33–39. doi: 10.1093/toxsci/47.1.33. [DOI] [PubMed] [Google Scholar]
- Cook DN, Pisetsky DS, Schwartz DA. Toll-like receptors in the pathogenesis of human disease. Nat Immunol. 2004;5:975–979. doi: 10.1038/ni1116. [DOI] [PubMed] [Google Scholar]
- Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtin GM, Higuchi MA, Ayres PH, Swauger JE, Mosberg AT. Lung tumorigenicity in A/J and rasH2 transgenic mice following mainstream tobacco smoke inhalation. Toxicol Sci. 2004;81:26–34. doi: 10.1093/toxsci/kfh175. [DOI] [PubMed] [Google Scholar]
- Finch GL, Nikula KJ, Belinsky SA, Barr EB, Stoner GD, Lechner JF. Failure of cigarette smoke to induce or promote lung cancer in the A/J mouse. Cancer Lett. 1996;99:161–167. doi: 10.1016/0304-3835(95)04059-5. [DOI] [PubMed] [Google Scholar]
- Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, Scheller J, Rose-John S, Cheroutre H, Eckmann L, Karin M. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht SS. Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chem Res Toxicol. 1998;11:559–603. doi: 10.1021/tx980005y. [DOI] [PubMed] [Google Scholar]
- Hecht SS. Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst. 1999;91:1194–1210. doi: 10.1093/jnci/91.14.1194. [DOI] [PubMed] [Google Scholar]
- Hecht SS. Cigarette smoking and lung cancer: chemical mechanisms and approaches to prevention. Lancet Oncol. 2002;3:461–469. doi: 10.1016/s1470-2045(02)00815-x. [DOI] [PubMed] [Google Scholar]
- Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359:1367–1380. doi: 10.1056/NEJMra0802714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain SP, Hofseth LJ, Harris CC. Radical causes of cancer. Nat Rev Cancer. 2003;3:276–285. doi: 10.1038/nrc1046. [DOI] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- Ji H, Houghton AM, Mariani TJ, Perera S, Kim CB, Padera R, Tonon G, McNamara K, Marconcini LA, Hezel A, et al. K-ras activation generates an inflammatory response in lung tumors. Oncogene. 2006;25:2105–2112. doi: 10.1038/sj.onc.1209237. [DOI] [PubMed] [Google Scholar]
- Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, Jacks T. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111–1116. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- Karin M. Inflammation and cancer: the long reach of Ras. Nat Med. 2005;11:20–21. doi: 10.1038/nm0105-20. [DOI] [PubMed] [Google Scholar]
- Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- Karin M, Gallagher E. From JNK to pay dirt: jun kinases, their biochemistry, physiology and clinical importance. IUBMB Life. 2005;57:283–295. doi: 10.1080/15216540500097111. [DOI] [PubMed] [Google Scholar]
- Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- Karin M, Lawrence T, Nizet V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell. 2006;124:823–835. doi: 10.1016/j.cell.2006.02.016. [DOI] [PubMed] [Google Scholar]
- Kim S, Takahashi H, Lin WW, Descargues P, Grivennikov S, Kim Y, Luo JL, Karin M. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature. 2009;457:102–106. doi: 10.1038/nature07623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Mercer BA, D’Armiento JM. Emerging role of MAP kinase pathways as therapeutic targets in COPD. Int J Chron Obstruct Pulmon Dis. 2006;1:137–150. doi: 10.2147/copd.2006.1.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson LA, Hecht SS. O6-methylguanine is a critical determinant of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone tumorigenesis in A/J mouse lung. Cancer Res. 1991;51:5557–5564. [PubMed] [Google Scholar]
- Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E, Ben-Neriah Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- Saetta M, Di Stefano A, Maestrelli P, Turato G, Ruggieri MP, Roggeri A, Calcagni P, Mapp CE, Ciaccia A, Fabbri LM. Airway eosinophilia in chronic bronchitis during exacerbations. Am J Respir Crit Care Med. 1994;150:1646–1652. doi: 10.1164/ajrccm.150.6.7952628. [DOI] [PubMed] [Google Scholar]
- Sparmann A, Bar-Sagi D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell. 2004;6:447–458. doi: 10.1016/j.ccr.2004.09.028. [DOI] [PubMed] [Google Scholar]
- Tockman MS, Anthonisen NR, Wright EC, Donithan MG. Airways obstruction and the risk for lung cancer. Ann Intern Med. 1987;106:512–518. doi: 10.7326/0003-4819-106-4-512. [DOI] [PubMed] [Google Scholar]
- Toh CK. The changing epidemiology of lung cancer. Methods Mol Biol. 2009;472:397–411. doi: 10.1007/978-1-60327-492-0_19. [DOI] [PubMed] [Google Scholar]
- Tu S, Bhagat G, Cui G, Takaishi S, Kurt-Jones EA, Rickman B, Betz KS, Penz-Oesterreicher M, Bjorkdahl O, Fox JG, Wang TC. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell. 2008;14:408–419. doi: 10.1016/j.ccr.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlahos R, Bozinovski S, Jones JE, Powell J, Gras J, Lilja A, Hansen MJ, Gualano RC, Irving L, Anderson GP. Differential protease, innate immunity, and NF-kappaB induction profiles during lung inflammation induced by subchronic cigarette smoke exposure in mice. Am J Physiol Lung Cell Mol Physiol. 2006;290:L931–945. doi: 10.1152/ajplung.00201.2005. [DOI] [PubMed] [Google Scholar]
- Witschi H. The complexities of an apparently simple lung tumor model: The A/J mouse. Exp Toxicol Pathol . 2005;57(Suppl 1):171–181. doi: 10.1016/j.etp.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Witschi H, Espiritu I, Peake JL, Wu K, Maronpot RR, Pinkerton KE. The carcinogenicity of environmental tobacco smoke. Carcinogenesis. 1997;18:575–586. doi: 10.1093/carcin/18.3.575. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.