

Abstract
The metabolism of cancer cells is altered to support rapid proliferation. Pharmacological activators of a tumor cell specific pyruvate kinase isozyme (PKM2) may be an approach for altering the classic Warburg effect characteristic of aberrant metabolism in cancer cells yielding a novel anti-proliferation strategy. In this manuscript we detail the discovery of a series of substituted N,N′-diarylsulfonamides as activators of PKM2. The synthesis of numerous analogues and the evaluation of structure activity relationships are presented as well as assessments of mechanism and selectivity. Several agents are found that have good potencies and appropriate solubility for use as chemical probes of PKM2 including 55 (AC50 = 43 nM, maximum response = 84%; solubility = 7.3 μg/mL), 56 (AC50 = 99 nM, maximum response = 84%; solubility = 5.7 μg/mL) and 58 (AC50 = 38 nM, maximum response = 82%; solubility = 51.2 μg/mL). The small molecules described here represent first-in-class activators of PKM2
Keywords: Warburg effect, pyruvate kinase, cellular metabolism, high-throughput screening, small molecule activators
Introduction
Altered metabolism was among the earliest observed differences between cancer and healthy tissues. Normally, the cellular process of glycolysis converts glucose to pyruvate which is subsequently converted to acetyl-CoA for entry into the Krebs cycle and ATP production. Under anaerobic conditions pyruvate is converted to lactate and ATP generation is limited (Figure 1A). In seminal work, Otto Warburg reported that cancer cells have increased rates of lactate production even in the presence of oxygen (Figure 1B).1,2 This altered metabolism is thought to give tumor cells a selective growth advantage relative to normal cells.3,4 Overall, this change in metabolism in cancer cells is referred to as the Warburg effect and allows cells to meet the metabolic requirements of cell proliferation.
Figure 1.

A. The fate of glucose metabolism to pyruvate in anaerobic and aerobic environments in differentiated, healthy tissue. B. The fate of glucose metabolism to pyruvate in rapidly proliferating tissue and tumors. PKM2 is activated by FBP and inhibited by tyrosine-phosphorylated proteins derived from growth factor signaling.
Pyruvate kinase operates at the final step of glycolysis catalyzing the transfer of a phosphate group from phosphoenolpyruvate (PEP) to ADP, yielding one molecule of ATP and one molecule of pyruvate. Humans have two pyruvate kinase (PK) genes that each produce two different isozymes due to alternative splicing.5,6,7 The Pyk L/R gene produces two mRNAs with defined variations in the first exon which are expressed in selective tissues including the liver (PKL form) and red blood cells (PKR form).8 The Pyk M gene also produces two mRNAs that leads to the expression of the M1 isozyme (PKM1) which is found in most normal adult tissues. However, in fetal tissue, alternative splicing results in the M2 isozyme (PKM2) with the splicing change occurring at a site required for the binding and allosteric activation of PKM2 by fructose-1,6-bis-phosphate (FBP).9–11 An early genetic observation associated with tumorigenesis recognized that normal adult PK isozymes are replaced by PKM2 in tumor cells.12–14 All tumors and cancer cell lines studied to date show exclusive expression of the PKM2 isoform.15,16 PKM2 has also been found in some normal adult tissues that require high rates of nucleic acid synthesis.
PKM2 exists in equilibrium between a low activity T-state and a high activity R-state. The active forms of PKM2, PKL and PKR are tetrameric and are allosterically activated by FBP through binding at the aforementioned flexible loop region near the dimer-dimer interface of the tetramer. The allosteric regulation of PK isoforms by FBP provides an important feedback mechanism that activates PKM2 when this early stage glycolytic intermediate accumulates to sufficient levels. It has been proposed that the low activity T-state may represent a dimer form of PKM2 which shows weak affinity for PEP while formation of the R-state involves formation of the tetrameric form exhibiting increased affinity for this substrate. In tumor cells, PKM2 has been reported to exist primarily in its low activity, dimeric form.16 The binding of FBP to the flexible loop region of PKM2 is thought to stabilize the tetrameric form of the enzyme leading to activation. In contrast, PKM1 has a high affinity for PEP in its native state and is not activated by FBP.10 Functionally, the down-regulation of PKM2 activity in cancer cells is thought to aid in shunting key glycolytic intermediates toward pathways where they, in turn, are utilized as precursors for lipid, amino acid and nucleic acid synthesis. Rapidly proliferating cells such as undifferentiated embryonic tissue and cancer cells require increased amounts of these fundamental cellular building blocks. Thus, the down-regulation of PKM2 activity provides a purposeful divergence from catabolic metabolism aimed at energy production toward an anabolic state aimed at providing the needed resources for rapid cellular construction.
Recent work has shown that replacement of PKM2 with PKM1 in cancer cells can relieve the Warburg effect and promote oxidative cellular metabolism characteristic of differentiated cells.17 Further, an assessment of growth in vivo of human lung cancer cells engineered to express only PKM1 showed delayed tumor formation in nude mouse xenografts. The tumors that did form in this model were shown to have re-expressed PKM2. In a related study, it was demonstrated that PKM2 has high affinity for binding phosphotyrosine peptides.18 Protein tyrosine phosphorylation is increased in tumor cells as a consequence of oncogenic intracellular signaling events that also drive uptake of excess nutrients. Binding of PKM2 to tyrosine phosphorylated proteins catalyzes the release of FBP from the enzyme and further down-regulates PKM2 activity to promote entry of glycolytic intermediates into biosynthetic pathways. Taken as a whole, these data suggest a therapeutic strategy whereby activation of PKM2 may restore normal cellular metabolism to a state characteristic of normal differentiated cells. We describe here the development of a PKM2 assay capable of identifying inhibitors and activators, the quantitative high-throughput screening (qHTS) of a large chemical library, and the identification and exploration of the structure activity relationships (SAR) of substituted N,N′-diarylsulfonamides as activators of PKM2.
Results
Pyruvate kinase-luciferase coupled assay
For the development of an assay for both activators and inhibitors of PKM2 we took advantage of a well utilized luminescent assay detection system for protein kinases.19,20 Briefly, these assays use the ATP-dependent reaction catalyzed by firefly luciferase to measure ATP depletion and have been widely applied protein kinase assays. In the present assay we applied this technology to measure ATP production catalyzed by PKM2. This provides for a robust increase in luminescent signal upon ATP product formation (see Supporting Information Table S1).21 The primary high-throughput screen was performed in 1,536-well microtiter plates using 4 uL/well assay volume. Using this assay design, a quantitative high-throughput screen (qHTS)22 of nearly 300,000 small molecules of the NIH Molecular Libraries23 Small Molecule Repository was performed.
The results of this screening effort are detailed in Figure 2. Small molecule activators of PKM2 were more prevalent than inhibitors, occurring at 0.27% for high confidence 1a, 1b and 2c concentration-response curves (CRCs; class 1 curves display two asymptotes, an inflection point, and r2 ≥0.9; subclasses 1a and 1b are differentiated by full (>80%) vs. partial (≤ 80%) response, respectively, while class 2a curves display a single left-hand asymptote and inflection point and full (>80%) response; see supporting information for complete curve class definitions).22,24 A cheminformatics analysis of the qHTS data included chemotype clustering, singleton identification, analysis of orthogonal activities, and structural considerations which included physical properties and anticipated optimization potential. Potency range and maximum response was additionally considered (a visual representation is shown in Figure 2C). Ultimately, this analysis led us to focus on two lead structures represented by the substituted thieno[3,2- b]pyrrole[3,2-d]pyridazinone 1 and substituted N,N′-diarylsulfonamide 2 shown in Figure 2D (a separate manuscript detailing the evaluation of 1 is in preparation).
Figure 2.

A. Summary of actives from PKM2 qHTS of 276,309 small molecules. B. Concentration response curve (CRC) representation of the full qHTS dataset. C. CRC’s for actives from the primary screen associated with curve classes 1a, 1b and 2a showing potency and maximum response. D. Chemical structures of the two lead activators of PKM2 substituted thieno[3,2-b]pyrrole[3,2-d]pyridazinone NCGC00031955 (1) and substituted N,N′-diarylsulfonamide NCGC00030335 (2).
Upon resynthesis the PKM2 activity of 2 was confirmed in the aforementioned PK coupled luciferase assay that monitored ATP production and a fluorescent PK-lactate dehydrogenase coupled assay that monitored pyruvate production. Good agreement between the two assays was observed. For PKM2, the AC50 in the luciferase-coupled assay was approximately 4-fold more potent than the AC50 determined in the LDH coupled assay but the efficacy of the response was approximately 1.5 to 2-fold higher in the LDH coupled assay. The LDH assay is performed at high saturating ADP concentrations and low (0.1 mM) PEP concentrations so differences that did occur between the two assays likely reflect preferences for the ADP-bound form of the enzyme. We discuss the data here using the luciferase-coupled assay but point to the LDH data for specific examples. The entire dataset for both assays are available in PubChem (see Methods for assay identifiers (AIDs)). We confirmed that 2 and all reported analogues did not activate firefly luciferase (PubChem AID: 1379) nor showed fluorescence at the wavelength used for NADH detection (data not shown). As well, the activators identified here were inactive in a qHTS of the Leishmania mexicana pyruvate kinase (PubChem AIDs: 1721, 1722) that used the identical format as the PKM2 qHTS. Therefore, 2 appears to be a genuine activator of PKM2.
Mode of action
We examined the mode of action 2 through analysis of the steady-state kinetics of PEP and ADP by the PKM2.11 As discussed, FBP is known to allosterically activate PKM2 through induction of an enzyme state with a high affinity for PEP. In the absence of activator, hPK shows low affinity for PEP (KM ~ 1.5 mM). In the presence of 2 or FBP the KM for PEP decreased nearly 10-fold to 0.26±0.08 mM or 0.1±0.02 mM, respectively, with lesser effects on Vmax (values of 245 pmols/min with or without FBP and 265 pmols/min with 2). The addition of excess PEP abolishes the activation of PKM2 by 2 or FBP. However, varying the concentration of ADP in the presence and absence of 2 shows that the steady-state kinetics for this substrate are not significantly affected (KM for ADP = 0.1 mM in either condition). Thus, our primary lead 2 lowered the KM for PEP but had no affect on the KM for ADP (Figure 3A) similar to what we observed for FBP (Figure 3B). .
Figure 3.

A. Initial velocity as a function of PEP and ADP concentration in the presence (open triangles) or absence (filled circles) of 2 (10 μM). B. Initial velocity as a function PEP and ADP concentration in the presence (open circles) or absence (filled circles) of FBP (10 μM). Vo, initial rate in pmol/min as determined in the PK-LDH coupled assay (kinetic assays were carried out at approximately 22 °C with 10 nM PKM2 using [KCl] = 200 mM, [MgCl2] = 15 mM, and with either [ADP] or [PEP] = 4.0 mM; see supporting information).
Synthesis of substituted N,N′-diarylsulfonamides and selected analogues
The synthetic strategy utilized to build various N,N′-diarylsulfonamide analogues was a straightforward sequence of coupling reaction, deprotection and a second coupling reaction as detailed in Scheme 1.25 Specifically, mono-boc protected piperazine was coupled to numerous aryl sulfonyl chlorides to provide the corresponding boc-protected N-arylsulfonamides. These intermediates were deprotected and subsequently coupled to a second aryl sulfonyl chloride to provide numerous N,N′-diarylsulfonamide analogues. All final compounds were purified by preparative scale HPLC and the yields for these procedures were typically high. The same procedure was utilized to explore alternate diamines between each aryl-sulfonamide moiety including cyclic diamines of different ring size (analogue 34), linear diamines (analogues 35-39), ring systems with an internal secondary amine and an exocyclic amine (analogues 40-47), and analogues with variously substituted piperazines (analogues 48-51) (scheme not shown). A similar procedure was utilized for the synthesis of several amide versions of these reagents (represented by compounds 22 and 33) in order to explore a comparative SAR of sulfonamide versus amide functionality (scheme not shown). We were also interested in exploring several of the related sulfone derivatives akin to the lead structure (Scheme 2). To synthesize these derivatives we treated N-boc-4-bromopiperidine with various aryl sulfides to afford the appropriately substituted thiol ethers. Oxidation to the sulfone followed by boc deprotection and a second coupling to various aryl sulfonyl chlorides to provided 4-(arylsulfonyl)-1-(arylsulfonyl)piperidine analogues (represented by analogues 21 and 32). Finally, it was of interest to explore N,N′-diarylsulfonamide analogues on the related piperazin-2-one core (Scheme 3). These derivatives were accessed through treatment of piperazin-2-one with 1 equivalent of various aryl sulfonyl chlorides which preferentially coupled to the free amine moiety. The resulting intermediate was converted to the 1,4-bis(arylsulfonyl)piperazin-2-ones by deprotonation of the amide with LHMDS followed by addition of various aryl sulfonyl chlorides to generate the desired products in good yields (represented by analogues 52 and 53).
Scheme 1.

Conditions and reagents: (i) TEA, CH2Cl2, 0 °C; (ii) TFA, CH2Cl2, 0 °C; (iii) TEA, CH2Cl2, 0 °C.
Scheme 2.

*Conditions and reagents: (i) K2CO3, DMF; (ii) mCPBA, CH2Cl2, 0 °C; (iii) TFA, CH2Cl2, 0 °C; (iv) TEA, CH2Cl2, 0 °C;
Scheme 3.

Conditions and reagents: (i) TEA, CH2Cl2, 0 °C; (ii) LHMDS, THF, −78 °C - then Ar2SO2Cl.
SAR of substituted N,N′-diarylsulfonamides and selected analogues
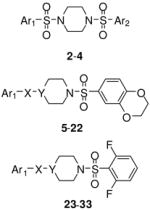
The lead N,N′-diarylsulfonamide 2 identified from the primary screen was found to possess an AC50 value versus PKM2 of 0.111 μM and maximum responses versus PKM2 (relative to activation by FBP) of 92%, respectively (Table 1). In the LDH assay, which used high saturating ADP levels and low (0.1 mM) levels of PEP, we found greater average efficacy but lower potency for 2 (AC50 values of 0.75 μM and a maximum responses of 150%). We focused our early efforts on both improving these values and further understanding of each chemotypes SAR potential and limitation. One initial focus involved symmetric versions of the N,N′-diarylsulfonamides. As such, symmetry was examined utilizing the 6-(2,3-dihydrobenzo[b][1,4]dioxine) heterocycle (analogue 3) and the 4-methoxybenzene ring (analogue 4). Each analogue had slightly diminished AC50 values (270 nM and 171 nM, respectively). From here, one aryl sulfonamide unit was held constant while exploring the SAR of the other aryl sulfonamide. Compounds 5–20 are representative examples from this strategy whereby the 6-(2,3-dihydrobenzo[b][1,4]dioxine) heterocycle remained constant. The most effective substitutions involved electron withdrawing groups in the 2- and 6-positions of the phenyl ring [for instance 2,6-difluorobenzene (analogue 10, AC50 = 65 nM, maximum response = 94%), 2,6-difluoro-4-methoxybenzene (analogue 12, AC50 = 28 nM, maximum response = 92%) and 2,6-difluoro-3-phenol (analogue 14, AC50 = 52 nM, maximum response = 95%)]. Given the improved activity observe with the di-fluoro analogues, we next chose to hold the 2,6-difluorobenzene ring constant and vary the opposite side with both substituted phenyl rings and various heterocycles (compounds 23–31). Several heterocycles were tolerated including the 7-(3,4-dihydro-2H-benzo[b][1,4]dioxepine) moiety (analogue 25, AC50 = 103 nM, maximum response = 100%) and the 6-(2-methylbenzo[d]thiazole) moiety (analogue 31, AC50 = 86 nM, maximum response = 104%). The 2-napthyl and 6-(2,2-dimethylchroman) derivatives provided significant enhancement in terms of maximum response (analogues 28 and 29, AC50 = 66 nM and 93 nM, maximum response = 138% and 119%, respectively). All amide versions of these compounds were found to be inactive in both assays (represented by derivatives 22 and 33). The sulfone derivatives that were explored showed loss of potency. Interestingly, this loss was more severe when the sulfone moiety bridged the piperidine ring system to the 6-(2,3-dihydrobenzo[b][1,4]dioxine) system relative to substituted phenyl rings (i.e. 2,6-difluorophenyl) as illustrated by analogues 21 and 32 (AC50 = 254 nM and 863 nM, maximum response = 104% and 110%, respectively).
Table 1.
SAR of selected N,N′-diarylsulfonamides with selected aryl substitutions.
| # | X | Y | Ar1 | Ar2 | hPK, M2 AC50 (μM)a | hPK, M2 M ax. Res.b | |
|---|---|---|---|---|---|---|---|
 |
2 | NA | NA | 6-(2,3-dihydrobenzo[b][1,4]dioxine) | 4-methoxybenzene | 0.111 | 92 |
| 3 | NA | NA | 6-(2,3-dihydrobenzo[b][1,4]dioxine) | 6-(2,3- dihydrobenzo[b][1,4]dioxine) | 0.270 | 90 | |
| 4 | NA | NA | 4-methoxybenzene | 4-methoxybenzene | 0.171 | 88 | |
| 5 | SO2 | N | 4-cyanobenzene | NA | 0.029 | 44 | |
| 6 | SO2 | N | 4-chlorobenzene | NA | 0.154 | 100 | |
| 7 | SO2 | N | 4-fluorobenzene | NA | 0.094 | 100 | |
| 8 | SO2 | N | 3-fluorobenzene | NA | 0.316 | 107 | |
| 9 | SO2 | N | 2-fluorobenzene | NA | 0.089 | 114 | |
| 10 | SO2 | N | 2,6-difluorobenzene | NA | 0.065 | 94 | |
| 11 | SO2 | N | 2,4,5-trifluorobenzene | NA | 0.090 | 105 | |
| 12 | SO2 | N | 2,6-difluoro-4-methoxybenzene | NA | 0.028 | 92 | |
| 13 | SO2 | N | 2,5-difluoro-4-propylbenzene | NA | 0.757 | 69 | |
| 14 | SO2 | N | 2,6-difluoro-3-phenol | NA | 0.052 | 95 | |
| 15 | SO2 | N | 2,4-difluorobenzene | NA | 0.124 | 113 | |
| 16 | SO2 | N | phenyl | NA | 0.202 | 108 | |
| 17 | SO2 | N | 3-(trifluoromethyl)benzene | NA | 0.209 | 39 | |
| 18 | SO2 | N | 3-methoxybenzene | NA | 0.113 | 90 | |
| 19 | SO2 | N | 2-pyridine | NA | 0.542 | 103 | |
| 20 | SO2 | N | 2-pyridine 1-oxide | NA | > 10 | 82 | |
| 21 | SO2 | CH | 2,6-difluorobenzene | NA | 0.254 | 104 | |
| 22 | CO | N | 2,6-difluorobenzene | NA | inactive | NA | |
| 23 | SO2 | N | 4-methoxybenzene | NA | 0.090 | 102 | |
| 24 | SO2 | N | 2,6-difluorobenzene | NA | 0.066 | 74 | |
| 25 | SO2 | N | 7-(3,4-dihydro-2H-benzo[b][1,4]dioxepine) | NA | 0.103 | 100 | |
| 26 | SO2 | N | 5-(benzo[d][1,3]dioxole) NA | NA | 0.191 | 61 | |
| 27 | SO2 | N | 7-(4-methyl-3,4-dihydro-2H-pyrido[3,2-b][1,4]oxazine) | NA | 2.71 | 94 | |
| 28 | SO2 | N | 2-naphthalene | NA | 0.066 | 138 | |
| 29 | SO2 | N | 6-(2,2-dimethylchroman) | NA | 0.093 | 119 | |
| 30 | SO2 | N | 5-(1-methyl-1H -indole) | NA | 0.387 | 91 | |
| 31 | SO2 | N | 6-(2-methylbenzo[d]thiazole) | NA | 0.086 | 104 | |
| 32 | SO2 | CH | 6-(2,3-dihydrobenzo[b][1,4]dioxine) | NA | 0.863 | 110 | |
| 33 | CO | N | 6-(2,3-dihydrobenzo[b][1,4]dioxine) | NA | inactive | NA |
AC50 values were determined utilizing the luminescent pyruvate kinase-luciferase coupled assay (ref. 22) and the data represents the results from three separate experiments. See the supporting information section for comparative values from the fluorescent pyruvate kinase-lactate dehydrogenase coupled secondary assay (ref. 27). Max Res. (Maximum Response) is% activity at 57 μM of compound. See Methods f or normalization. See supporting information section for standard deviations.
Several linear diamines and several alternate diamine core ring systems were examined. For these studies we retained the 2,6-difluorophenyl and 6-(2,3-dihydrobenzo[b][1,4]dioxine) heterocycle as the two aryl substituents to afford comparative uniformity. The results are shown in Table 2 and demonstrate that the piperazine and the related 1,4-diazepane (analogue 34, AC50 = 895 nM, maximum response = 120%) had clear advantages over other diamine moieties. Ligations with linear diamines ranging from 2- to 6-carbons in length (analogues 35-39) were found to have diminished potencies. Cis and trans versions of the cyclohexane-1,4-diamines conferred similar loss in activation potency. Interestingly, the trans version of this analogue performed significantly better than the cis version. Numerous analogues with one secondary amine contained in 4-, 5- and 6-membered rings and one exocyclic primary amine were examined (analogues 42-47) and found to be less active than the original lead compounds in both assays. In addition to the derivatives shown in Table 2, numerous bicyclic and spirocyclic diamines were examined (for instance 2,6-diazabicyclo[3.2.2]nonane and 2,7-diazaspiro[4.4]nonane) and found to be less active that the corresponding piperazine and 1,4-diazepane analogues (data not shown).
Table 2.
SAR of selected N,N′-diarylsulfonamides with divergent diamine moieties.
| # | CORE | hPK, M2 AC50 (μM)a | hPK, M2 M ax. Res.b | |
|---|---|---|---|---|
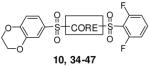 |
10 | piperazine | 0.065 | 94 |
| 34 | 1,4-diazepane | 0.866 | 120 | |
| 35 | N,N′-(ethane-1,2-diyl) | > 15 | 60 | |
| 36 | N,N′-(propane-1,3-diyl) | 3.85 | 106 | |
| 37 | N,N′-(butane-1,4-diyl) | 7.97 | 113 | |
| 38 | N,N′-(pentane-1,5-diyl) | 2.33 | 114 | |
| 39 | N,N′-(hexane-1,6-diyl) | 4.83 | 110 | |
| 40 | N,N′-((trans)-cyclohexane-1,4-diyl) | 2.11 | 91 | |
| 41 | N,N′-((cis)-cyclohexane-1,4-diyl) | > 35 | 61 | |
| 42 | 9.00 | 100 | ||
| 43 |  |
3.69 | 101 | |
| 44 |  |
> 10 | 84 | |
| 45 | 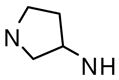 |
> 15 | 82 | |
| 46 | 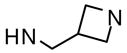 |
3.05 | 108 | |
| 47 | 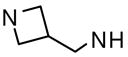 |
4.47 | 93 |
AC50 values were determined utilizing the luminescent pyruvate kinase-luciferase coupled assay (ref. 22) and the data represents the results from three separate experiments. See the supporting information section for comparative values from the fluorescent pyruvate kinase-lactate dehydrogenase coupled secondary assay (ref. 27). Max Res. (Maximum Response) is %activity at 57 μM of compound. See Methods for normalization. See supporting information section for standard deviations.
Several piperazine rings were synthesized and evaluated with a single methyl addition proximal to either the 2,6-difluorophenyl or 6-(2,3-dihydrobenzo[b][1,4]dioxine) heterocycle. Additional consideration was given to the absolute stereochemistry of the methyl group. The results are detailed in Table 3 and show that these analogues were less potent than the unmodified ring systems. Another piperazine ring modification was the incorporation of a carbonyl moiety alpha to the ring nitrogens. Here, the amine to lactam conversion proximal to the 6-(2,3-dihydrobenzo[b][1,4]dioxine) heterocycle resulted in an active derivative (analogue 52, AC50 = 114 nM, maximum response = 105%). The same amine to lactam conversion adjacent to the 2,6-difluorobenzene resulted in a loss of potency (analogue 53, AC50 = 2.42 μM, maximum response = 97%). The activities of these agents again demonstrate the lack of symmetric SAR for this chemotype.
Table 3.
SAR of selected N,N′-diarylsulfonamides with substitutions on the piperazine ring.
| # | A | B | C | D | X | Y | hPK, M2 AC50 (μM)a | hPK, M2 M ax. Res.b | |
|---|---|---|---|---|---|---|---|---|---|
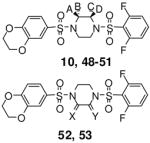 |
10 | H | H | H | H | NA | NA | 0.065 | 94 |
| 48 | Me | H | H | H | NA | NA | 4.34 | 109 | |
| 49 | H | Me | H | H | NA | NA | 3.10 | 99 | |
| 50 | H | H | Me | H | NA | NA | 9.18 | 108 | |
| 51 | H | H | H | Me | NA | NA | 2.93 | 108 | |
| 52 | NA | NA | NA | NA | O | NA | 0.114 | 105 | |
| 53 | NA | NA | NA | NA | NA | O | 2.42 | 97 |
AC50 values were determined utilizing the luminescent pyruvate kinase-luciferase coupled assay (ref. 22) and the data represents the results from three separate experiments. See the supporting information section f or comparative values from the fluorescent pyruvate kinase-lactate dehydrogenase coupled secondary assay (ref. 27). Max Res. (Maximum Response) is % activity at 57 μM of compound. See Methods for normalization. See supporting information section for standard deviations.
An important consideration was the aqueous solubility of the analogues presented in this study. To assess the solubility of selected analogues including 10 and 34 we utilized a commercial provider of aqueous solubility profiles.26 The majority of analogues examined had solubility levels (measured in both μg/mL and μM) below the detectable limit. The solubility of the seven-member 34 registered at the low values of 5.6 μg/mL (Table 4). The lack of aqueous solubility is, therefore, a major liability for these agents as molecular probes of PKM2. To rectify this we designed and synthesized numerous analogues incorporating solubilizing functionalities on one or both aryl groups of selected compounds (including pyridines, anilines and N-acetyl anilines). Table 4 presents selected results. In general, aniline substitutions placing the amine meta relative to the sulfonamide functionality resulted in highly active agents including 55 (AC50 = 43 nM, maximum response = 84%), 56 (AC50 = 99 nM, maximum response = 84%) and 58 (AC50 = 38 nM, maximum response = 82%). Gratifyingly, selected analogues incorporating this moiety also had amplified water solubilities including 57 (26.3 μg/mL) and 58 (51.2 μg/mL) (Table 4). Overall, 58 possesses the best combination of activity and solubility of all the analogues presented in this study.
Table 4.
SAR of selected N,N′-diarylsulfonamides including solubility assessment.
| # | n | Ar1 | Ar2 | hPK, M2 AC50 (μM)a | hPK, M2 M ax. Res.b | Solubilityc(μM) | Solubilityc(μg/mL) | |
|---|---|---|---|---|---|---|---|---|
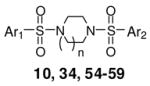 |
10 | 1 | 2,6-difluorobenzene | 6-(2,3-dihydrobenzo[b][1,4]dioxine) | 0.065 | 94 | < 1.1 | < 0.5 |
| 34 | 2 | 2,6-difluorobenzene | 6-(2,3-dihydrobenzo[b][1,4]dioxine) | 0.866 | 120 | 5.6 | 2.7 | |
| 54 | 1 | 3-aniline | 2,6-difluoro-4-methoxybenzene | 0.026 | 90 | < 0.7 | < 0.4 | |
| 55 | 1 | 3-aniline | 6-(2,3-dihydrobenzo[b][1,4]dioxine) | 0.043 | 84 | 7.3 | 4.1 | |
| 56 | 1 | 3-aniline | 2,6-difluorobenzene | 0.099 | 84 | 5.7 | 3.0 | |
| 57 | 2 | 3-aniline | 2,6-difluoro-4-methoxybenzene | 0.223 | 92 | 26.3 | 15.1 | |
| 58 | 2 | 3-aniline | 6-(2,3-dihydrobenzo[b][1,4]dioxine) | 0.038 | 82 | 51.2 | 29.0 |
AC50 values were determined utilizing the luminescent pyruvate kinase-luciferase coupled assay (ref. 22) and the data represents the results from three separate experiments. See the supporting information section for comparative values from the fluorescent pyruvate kinase-lactate dehydrogenase coupled secondary assay (ref. 27).
Max. Res. value represents the % activation at 57 μM of compound.
kinetic solubility analysis was performed by Analiza Inc. and are based upon quantitative nitrogen detection as described (www.analiza.com). The data represents results from three separate experiments with an average intraassay %CV of 4.5%.
LogD analysis was performed by Analiza Inc. and are based upon octanol/buffer partitioning and quantitative nitrogen detection of sample content as described (www.analiza.com). NT = not tested.
Selectivity of chosen N,N′-diarylsulfonamide analogues
With a better understanding of the SAR for this chemotype, we next concerned ourselves with the selective activation of PKM2 versus PKM1, PKR and PKL. An appropriate tool compound aimed at further delineating the role of PKM2 as a critical contributor in the Warburg effect requires a high degree of selective activation of PKM2 relative to other PK targets with particular consideration for PKM1. Members of each chemotype were assayed versus PKM1, PKR and PKL (the assay design was similar as that described for PKM2 and details are located in the Methods and Supplemental Information section). All analogues in the N,N′-diarylsulfonamides class were found to be inactive versus PKM1. This is consistent with the lack of allosteric regulation for the PKM1 isoform. The activity versus PKR and PKL varied from compound to compound but generally showed maximum responses below 30% for both isozymes in both assay formats. The selectivity for 2 is shown in figure 4.
Figure 4.

Selectivity assessment for 2 versus PKM2 (open circles), PKM1 (filled squares), PKL (open squares), and PKR (filled circles).
Conclusion
Warburg’s finding that cancer cells show altered cellular respiration and metabolism ranks as one of the earliest observations in cancer biology. A key realization associated with the Warburg effect is the re-expression of PKM2 in all cancer cells leading to increased availability of glycolyctic intermediates for biosynthesis of amino acids, nucleic acids and lipid building blocks for cellular construction. The expression of PKM2 in tumor cells makes this enzyme an attractive target for therapeutic intervention in cancer. We have described here the discovery and optimization of a series of substituted N,N′-diarylsulfonamide activators of PKM2 which should be useful pharmacological tools to study the antiproliferative effects of PKM2 activation. This chemotype activates the PKM2 isoform by increasing the affinity for phosphoenolpyruvate (PEP) with little effect on kinetics of ADP binding similar to the natural activator FBP. The synthesis of the lead structure and numerous analogues allowed the identification of compounds with low nanomolar potencies and activation responses similar to full activation afforded by the native allosteric activator FBP. In addition, unlike FBP, these agents were found to be selective for PKM2 versus PKM1, PKL and PKR isoforms. SAR explorations included both evaluations of PKM2 activation and aqueous solubility. From these studies analogues 10 (AC50 = 65 nM), 12 (AC50 = 28 nM), 14 (AC50 = 52 nM), 28 (AC50 = 66 nM), 54 (AC50 = 26 nM), 55 (AC50 = 43 nM), 56 (AC50 = 99 nM) and 58 (AC50 = 38 nM) were identified as highly potent activators of PKM2. NCGC00185916-01 (58) was identified as a small molecule PKM2 activator that combined good potency as a PKM2 activator with appropriate aqueous solubility (51.2 μg/mL). The PKM2 activators described here represent first-in-class small molecule activators of PKM2 and studies to examine these compounds’ antiproliferative capacity and their ability to alleviate the Warburg effect are currently underway.
METHODS
Reagents
Kinase-Glo was obtained from Promega (Madison, Wi). ATP, PEP, LDH and NADH were from Sigma. Reagents and solvents were purchased from Sigma, Alfa Aesar, Acros, Enamine, Oakwood Products, Matrix Scientific or Chem-Impex International. Pyruvate kinase (EC 2.7.1.40), human M2 isoform, was expressed as a N-terminal His6-tagged fusion protein in BL21(DE3)pLys cells. Preparation and purification was as described in Christofk et al.18
Luminescent pyruvate kinase-luciferase coupled assay
Production of a luminescent signal based on the generation of ATP by pyruvate kinase was determined by using the ATP-dependent enzyme firefly luciferase.21 Three μL of substrate mix (at r.t.) in assay buffer (50 mM imidazole pH 7.2, 50 mM KCl, 7 mM MgCl2, 0.01% Tween 20, 0.05% BSA) was dispensed into Kalypsys white solid bottom 1,536 well microtiter plates using a bottle-valve solenoid-based dispenser (Kalypsys). The final concentrations of substrates in the assay were 0.1 mM ADP and 0.5 mM PEP. 23 nL of compound were delivered with a 1,536-pin array tool28 and 1 μL of enzyme mix in assay buffer (final concentration, 0.1 nM pyruvate kinase, 50 mM imidazole pH 7.2, 0.05% BSA, 4 °C) was added. Microtiter plates were incubated at r.t. for 1 hour and 2 uL of luciferase detection mix (Kinase-Glo, Promega21 at 4° C protected from light) was added and luminescence was read with a ViewLux (Perkin Elmer) using a 5 second exposure/plate. Data was normalized for AC50 values to control columns containing uninhibited enzyme (n), and AC100 inhibition (i) according the following equation: Activation (%) = [(c−n)/(n−i)]*100 where c = compound, n = DMSO neutral, i = no enzyme control. A % activity of 100% is approximately a 2-fold increase over basal assay signal (% Activation by FBP was variable but averaged 100%). Monitoring of activation was accomplished using enzyme at 3x the final concentration. The primary qHTS data and confirmatory data are available in PubChem (AIDs: 1631, 1634, and 1751). Follow-up of synthesized analogs was determined using the same protocol with the exception that the enzyme concentrations for isoforms PKM1, L and R were 1 nM, 0.1 nM, and 0.1 nM respectively (PubChem AIDs for M1, L and R bioluminescent assays are 1780, 1781, and 1782, respectively).
Fluorescent pyruvate kinase-lactate dehydrogenase coupled secondary assay
All compounds were also tested in a kinetic mode by coupling the generation of pyruvate by pyruvate kinase to the depletion of NADH through lactate dehydrogenase.27 For PKM2, 3 μL of substrate mix (final concentration, 50 mM Tris-Cl pH 8.0, 200 mM KCl, 15 mM MgCl2, 0.1 mM PEP, 4.0 mM ADP, and 0.2 mM NADH) was dispensed into Kalypsys black-solid 1,536 well plates using the Aurora Discovery BioRAPTR Flying Reagent Dispenser (FRD; Beckton-Dickenson, Franklin Lakes, NJ)28 and 23 nL of compounds were delivered using a Kalypsys pin tool29 and then 1 μL of enzyme mix (final concentrations, 10 nM hPK-M2 and 1 μM of LDH) was added. Plates were immediately placed in ViewLux (Perkin Elmer) and NADH fluorescence was determined at 30 second exposure intervals for between 3 and 6 minutes. Data were normalized to the uninhibited and EC100 activation using known activators such as FBP. The data has been deposited in PubChem (AID: 1540). Follow-up of synthesized analogs was determined using the same protocol (PubChem AIDs for L, M1 and R LDH-coupled assays are 1541, 1542, and 1543, respectively). This assay was also used to determine the KM’s for PEP and ADP in the presence and absence of activator. Data was fit in GraphPad prism. Conversion of fluorescent units to pmols of NADH was performed using a standard curve of known NADH concentrations. Data was collected on the Perkin Elmer Viewlux.
Synthesis of 2 and analogues
Complete procedures for the synthesis of 2 and all analogues are presented in the supporting information section. Proton NMR, HRMS and purity (as judged by peak integration on two separate HPLC gradients) for all compounds are listed in the supporting information section. 2: 1H NMR (400 MHz, DMSO-d6)δ: 2.94 (s, 8H), 3.86 (s, 3H), 4.23-4.45 (m, 4H), 7.01-7.08 (m, 1H), 7.08-7.19 (m, 4H), 7.57- 7.67 (m, 2H). HRMS; Calculated for C19H22N2O7S2 (M+): 454.0868, found: 454.08718.
Supplementary Material
Acknowledgments
The authors thank Jeremy Smith, Paul Shinn, and Danielle van Leer for assistance with compound management. We thank Ms. Allison Mandich for critical reading of this manuscript. This research was supported by the Molecular Libraries Program of the National Institutes of Health Roadmap for Medical Research and the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health and R03 (1R03MH085679-01). The Structural Genomics Consortium is a registered charity (number 1097737) that receives funds from the Canadian Institutes for Health Research, the Canadian Foundation for Innovation, Genome Canada through the Ontario Genomics Institute, GlaxoSmithKline, Karolinska Institutet, the Knut and Alice Wallenberg Foundation, the Ontario Innovation Trust, the Ontario Ministry for Research and Innovation, Merck & Co., Inc., the Novartis Research Foundation, the Swedish Agency for Innovation Systems, the Swedish Foundation for Strategic Research and the Wellcome Trust.
Abbreviations
- PK
pyruvate kinase
- ATP
adenosine triphosphate
- ADP
adenosine diphosphate
- PEP
phosphoenolpyruvate
- FBP
fructose-1,6-bis-phosphate
- qHTS
quantitative high-throughput screen
- LDH
lactate dehydrogenase
Footnotes
Supporting Information Available: Assay protocols, experimental procedures and spectroscopic data (1H, LC/MS and HRMS) are listed for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 2.Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–270. [PubMed] [Google Scholar]
- 3.Devita VT Jr, Hellman S, Rosenberg SA, editors. Cancer Principles & Practice of Oncology. 7. Lippincott Williams & Wilkins; Philadelphia (PA): 2005. [Google Scholar]
- 4.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mazurek S, Boschek CB, Hugo F, Eigenbrodt E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin Cancer Biol. 2005;15:300–308. doi: 10.1016/j.semcancer.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 6.Takenaka M, Noguchi T, Sadahiro S, Hirai H, Yamada K, Matsuda T, Imai E, Tanaka T. Isolation and characterization of the human pyruvate kinase M gene. Eur J Biochem. 1991;198:101–106. doi: 10.1111/j.1432-1033.1991.tb15991.x. [DOI] [PubMed] [Google Scholar]
- 7.Takenaka M, Yamada K, Lu T, Kang R, Tanaka T, Noguchi T. Alternative splicing of the pyruvate kinase M gene in a minigene system. Eur J Biochem. 1996;235:366–371. doi: 10.1111/j.1432-1033.1996.00366.x. [DOI] [PubMed] [Google Scholar]
- 8.Noguchi T, Yamada K, Inoue H, Matsuda T, Tanaka T. The L- and R-type isozymes of rat pyruvate kinase are produced from a single gene by the use of different promotors. J Biol Chem. 1987;262:14366–14371. [PubMed] [Google Scholar]
- 9.Noguchi T, Inoue H, Tanaka T. The M1 and M2-type isoenzymes of rat pyruvate kinase are produced from the same gene by alternative RNA splicing. J Biol Chem. 1986;261:13807–13812. [PubMed] [Google Scholar]
- 10.Yamada K, Noguchi T. Regulation of pyruvate kinase M gene expression. Biochem Biophys Res Commun. 1999;256:257–262. doi: 10.1006/bbrc.1999.0228. [DOI] [PubMed] [Google Scholar]
- 11.Dombrauckas JD, Santarsiero BD, Mesecar AD. Structural basis for tumor pyruvate kinase M2 allosteric regulation and catalysis. Biochemistry. 2005;44:9417–9429. doi: 10.1021/bi0474923. [DOI] [PubMed] [Google Scholar]
- 12.Reinacher M, Eigenbrodt E. Immunohistological demonstration of the same type of pyruvate kinase isoenzyme (M2-PK) in tumors of chicken and rat. Virchows Arch B Cell Pathol Incl Mol Pathol. 1981;37:79–88. doi: 10.1007/BF02892557. [DOI] [PubMed] [Google Scholar]
- 13.Staal GEJ, Rijksen G. Pyruvate kinase in selected human tumors. In: Pretlow TG, Pretlow TP, editors. Biochemical and molecular aspects of selected cancers. Academic Press; San Diego: 1991. pp. 313–337. [Google Scholar]
- 14.Steinberg P, Klingelhöffer A, Schäfer A, Wüst G, Weisse G, Oesch F, Eigenbrotd E. Expression of pyruvate kinase M2 in preneoplastic hepatic foci of N-nitrosomorpholine- treated rats. Virchows Arch. 1999;434:313–337. doi: 10.1007/s004280050330. [DOI] [PubMed] [Google Scholar]
- 15.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 16.DeBerardinis RJ, Sayed N, Ditsworth D, Thompson CB. Brick by Brick: metabolism and tumor cell growth. Curr Opin Genet Dev. 2008;18:54–61. doi: 10.1016/j.gde.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumor growth. Nature. 2008;452:230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- 18.Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC. Pyruvate kinase M2 is a phosphotyrosine binding protein. Nature. 2008;452:181–186. doi: 10.1038/nature06667. [DOI] [PubMed] [Google Scholar]
- 19.Fan F, Wood KV. Bioluminescent assays for high-throughput screening. Assay Drug Dev Technol. 2007;5:127–136. doi: 10.1089/adt.2006.053. [DOI] [PubMed] [Google Scholar]
- 20.Singh P, Harden BJ, Lillywhite BJ, Broad PM. Identification of kinase inhibitors by an ATP depletion method. Assay Drug Dev Technol. 2004;2:161–169. doi: 10.1089/154065804323056503. [DOI] [PubMed] [Google Scholar]
- 21.Auld DS, Zhang YQ, Southall NT, Rai G, Landsman M, Maclure J, Langevin D, Thomas CJ, Austin CP, Inglese JA. A basis for reduced chemical library inhibition of firefly luciferase obtained from directed evolution. J Med Chem. 2009;52:1450–1458. doi: 10.1021/jm8014525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Inglese J, Auld DS, Jadhav A, Johnson RL, Simeonov A, Yasgar A, Zheng W, Austin CP. Quantitative high-throughput screening: A titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc Natl Acad Sci USA. 2006;103:11473–11478. doi: 10.1073/pnas.0604348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Austin CP, Brady LS, Insel TR, Collins FS. Science. 2004;306:1138. doi: 10.1126/science.1105511. [DOI] [PubMed] [Google Scholar]
- 24.Shukla SJ, Nguyen DT, MacArthur R, Simeonov A, Frazee WJ, Hallis TM, Marks BD, Singh U, Eliason HC, Printen J, Austin CP, Inglese J, Auld DS. ASSAY Drug Dev Technol. 2009;7:143–169. doi: 10.1089/adt.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Proisy N, Taylor S, Nelson A, Collins I. Rapid Synthesis of 3- Aminoisoquinoline-5-sulfonamides Using the Buchwald-Hartwig Reaction. Synthesis. 2009;4:561–566. [Google Scholar]
- 26.http://www.analiza.com/
- 27.Hannaert V, Yernaux C, Rigden DJ, Fothergill-Gilmore LA, Opperdoes FR, Michels PA. The putative effector-binding site of Leishmania Mexicana pyruvate kinase studied by site-directed mutagenesis. FEBS Lett. 2002;514:255–259. doi: 10.1016/s0014-5793(02)02374-8. [DOI] [PubMed] [Google Scholar]
- 28.Niles WD, Coassin PJ. Piezo-and solenoid valve-based liquid dispensing for miniaturized assays. ASSAY Drug Dev Technol. 2005;3:189–202. doi: 10.1089/adt.2005.3.189. [DOI] [PubMed] [Google Scholar]
- 29.Cleveland PH, Koutz PJ. Nanoliter dispensing for uHTS using pin tools. ASSAY Drug Dev Technol. 2005;3:213–225. doi: 10.1089/adt.2005.3.213. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.