

Abstract
A rapid method to determine fexofenadine concentrations in human plasma using protein precipitation in 96-well plates and liquid chromatography-tandem mass spectrometry was validated. Plasma proteins were precipitated with acetonitrile containing the internal standard fexofenadine-d6, mixed briefly, and then filtered into a collection plate. The resulting filtrate was diluted and injected onto a Phenomenex Gemini C18 (50 × 2.0 mm, 5 micron) analytical column. The mobile phase consisted of 0.1% formic acid, 5 mM ammonium acetate in deionized water and methanol (35:65, v/v). The flow rate was 0.2 ml/min and the total run time was 2 min. Detection of the analytes was achieved using positive ion electrospray ionization and high resolution multiple reaction monitoring mode (H-SRM). The linear standard curve ranged from 1 to 500 ng/ml and the precision and accuracy (intra- and inter-run) were within 4.3% and 8.0%, respectively. The method has been applied successfully to determine fexofenadine concentrations in human plasma samples obtained from subjects administered a single oral dose of fexofenadine. The method is rapid, sensitive, selective and directly applicable to human pharmacokinetic studies involving fexofenadine.
Keywords: Fexofenadine, fexofenadine-d6, protein precipitation, LC-MS, human plasma
1. Introduction
Fexofenadine is a histamine H1-receptor antagonist used therapeutically for the treatment of allergic rhinitis and chronic idiopathic urticaria [1]; it is given orally in doses ranging from 30 to 180 mg/day. Fexofenadine is predominantly eliminated unchanged in bile (80%) and urine (11%)[1]; approximately 5% is metabolized to methyl ester (3.6%) and azacyclonol (1.5%) metabolites [2]. The major determinants of fexofenadine absorption and elimination are the activity of drug transporters located in the intestine and liver [1]. Specifically, fexofenadine is a substrate of the transporters P-glycoprotein (P-gp) and the organic anion transporting polypeptides (OATP) OATP1A2 and OATP1B3 [1, 3–5]. The absorption of fexofenadine in the intestine is limited by the efflux transporter P-gp but is enhanced by the uptake transporter OATP1A2 [3], while the elimination of fexofenadine in the liver is dependent on the hepatic uptake transporter OATP1B3 [5]. Thus, fexofenadine is used as a pharmacologic probe of human drug transporters to characterize transporter activity in interaction studies with concomitant drugs, foods, or herbal products [6–14] and to evaluate the effects of genetic variation or disease state on transporter activity and function [15–17]. As drug transport is increasingly recognized as a critical pathway in the disposition of many drugs, there is a need for simple analytical methods for probes such as fexofenadine to facilitate evaluations of drug transporter activity.
Several HPLC methods for determination of fexofenadine in biologic fluids have been reported [18–31]. A few methods use ultraviolet [18, 19] or fluorescence detection [20–22], but most methods for determination of fexofenadine in human plasma are based on HPLC with mass spectrometric detection [23, 24] or tandem mass spectrometric detection [25–30] because of better sensitivity and selectivity. Sample processing for almost all of the methods reported use costly solid phase extraction (SPE) with C18 cartridges [23] or Oasis™ HLB cartridges [25–30]. The typical range of quantification for the MS-based methods is 1–500 ng/ml and the run times range from 2 to 10 min. Most of the methods reported use structurally related compounds for the internal standard (e.g., diphenhydramine, loratadine, terfenadine) [19, 23–27, 29, 30]; only the method by Fu et al., [28], uses a deuterated internal standard (fexofenadine-d6), but that method requires 0.5 ml of plasma for processing by SPE. Protein precipitation in microcentrifuge tubes was used in a method reported recently by Guo et al., [31], but the oral hypoglycemic drug glipizide was used as the internal standard.
Herein, we present the validation of a sensitive method for fexofenadine determination in human plasma by liquid chromatography-tandem mass spectrometry. The method has several advantages including rapid sample processing based on protein precipitation and filtration in a 96-well plate format, a deuterated internal standard (fexofenadine-d6), a small sample volume requirement (100 μl human plasma) and a total run time of 2 min with isocratic elution. The method is suitable for determination of fexofenadine concentrations in clinical pharmacokinetic studies.
2. Experimental
2.1. Chemicals and reagents
Fexofenadine (>98% chemical purity) was purchased from Sigma-Aldrich Co. (St. Louis, MO, U.S.A) and the deuterated internal standard fexofenadine-d6 (>98% chemical and >99% isotopic purity) was purchased from Toronto Research Chemicals Inc. (North York, ON, Canada). Chemical structures are shown in Figure 1. Acetonitrile and methanol, HPLC grade, and analytical grade formic acid were purchased from VWR International, LLC (West Chester, PA, USA). Human EDTA plasma was obtained from the UF & Shands Hospital blood bank (Gainesville, FL, USA); plasma was screened for the presence of fexofenadine prior to use. HPLC grade deionized water was obtained from a Barnstead Nanopure Diamond UV Ultrapure Water System (Dubuque, IA, USA).
Fig. 1.
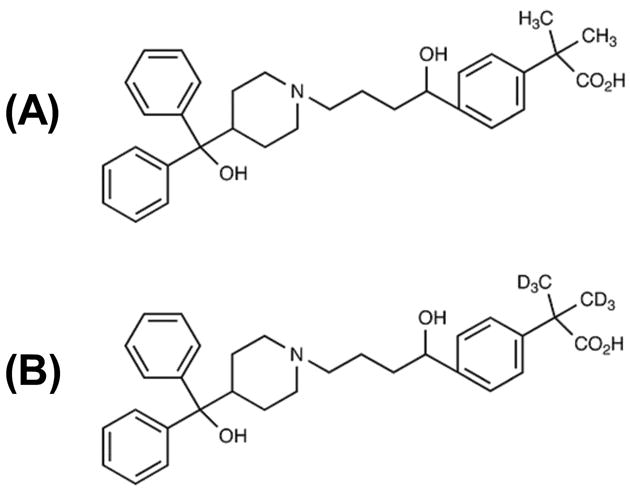
Chemical structures of (A) fexofenadine and (B) fexofenadine-d6 (internal standard).
2.2. Instrumentation and chromatographic conditions
The LC-MS/MS system included a Surveyor HPLC autosampler, Surveyor MS quaternary pump and a TSQ Quantum Discovery triple quadrupole mass spectrometer (Thermo Scientific, San Jose, CA, USA). The TSQ Quantum mass spectrometer was equipped with an electrospray ion source (ESI) with the ESI source spray set orthogonal to the ion transfer capillary tube. The autosampler temperature was maintained at 10°C. The analytical column was a Gemini C18, 50×2.0 mm, 5 μm (Phenomenex, Torrance, CA, USA). The mobile phase consisted of deionized water and methanol (35:65, v/v) that contained 0.1% formic acid and 5mM ammonium acetate and was pumped at a flow rate of 0.2 ml/min. The mobile phase was degassed and filtered through a 0.22μm Nylon 66 membrane prior to use. The MS/MS conditions were optimized by using an infusion system with a mixing tee in which fexofenadine (1 μg/ml at 5 μl/min) was mixed with mobile phase delivered at 0.2 ml/min. For quantification, the TSQ Quantum was operated in high resolution single reaction monitoring mode (H-SRM). The ESI was operated in the positive mode at a spray voltage of 4.6kV and source CID-10V with a heated capillary temperature of 375°C. Nitrogen was used as the sheath and auxiliary gas and the flow rates were set to 35 and 10 units (arbitrary), respectively. The argon collision gas pressure was set to 1.5 mTorr. The collision energy was −41 eV for fexofenadine and fexofenadine-d6 (internal standard). Fexofenadine was monitored at m/z 502.3 → 171.0 and fexofenadine-d6 at m/z 508.3 → 177.0. The instrument was operated in enhanced (high) resolution with peak width (FWHM) set to m/z 0.2 at Q1 and to m/z 0.7 at Q3. The scan time was 300 ms for each transition. SRM data were acquired and processed using ThermoFinnigan XCalibur® software version 1.4, service release 1 (Thermo Scientific, San Jose, CA, USA).
2.3. Standard preparation
A stock solution of fexofenadine was prepared in methanol at a concentration of 1000 μg/ml. Separate dilutions of this stock, made at concentrations of 1, 10 and 100 μg/ml after appropriate dilution with methanol, were used to prepare calibration standards and quality control (QC) samples. The stock solution for the internal standard was prepared by dissolving fexofenadine-d6 in methanol at a concentration of 1 μg/ml and then further diluted with acetonitrile to a concentration of 35 ng/ml. These stock solutions were stored at −20°C. Calibration standards were prepared at concentrations of 1, 2, 5, 25, 50, 100, 250 and 500 ng/ml by spiking blank human plasma with varying quantities of the standard solutions (1, 10 or 100 μg/ml). The lowest calibration standard concentration was considered to be the lower limit of quantitation (LLOQ). QC samples were also prepared in blank human plasma at final concentrations of 1 (LLOQ), 10, 150 and 400 ng/ml. Standards and quality control samples were stored at −20°C until analysis.
2.4. Sample preparation
Acetonitrile (300 μl) containing the internal standard (10.5 ng) and then plasma (100 μl) were pipetted into wells of a 96-well Captiva™ filter plate (Varian Inc., Palo Alto, CA, USA). The filter plate was mixed briefly and then inverted for 5 minutes at room temperature. Next, the filter plate was fitted with a vacuum collar and 1 ml collection plate before filtration by vacuum pressure. The resulting filtrate was diluted with 400 μl water and injected into the HPLC system (10 μl).
2.5. Calibration and linearity
Calibration standards over the concentration range of 1–500 ng/ml were analyzed in duplicate for three runs; the lowest standard was analyzed in triplicate. Back-calculated concentration values for each standard were considered acceptable if both the precision, expressed as the percent relative standard deviation (R.S.D.%), and the accuracy, expressed as the percent relative error (R.E.%), were within 15%. The lower limit of quantification (LLOQ) was acceptable if the R.S.D.% and R.E.% were within ±20%.
2.6. Precision, accuracy and incurred sample analysis
The intra- and inter-run precision (R.S.D.%) and accuracy (R.E.%) of the assay were determined by analyzing QC samples prepared at concentrations of 1 (LLOQ), 10, 150 and 400 ng/ml. Replicate QC samples (n = 12) were analyzed on Day 1 to determine intra-run precision and accuracy. Inter-run precision and accuracy was determined from replicate QC samples analyzed on Day 1 (n = 12), Day 2 (n = 6), and Day 3 (n = 6) for a total of 24 QC samples at each concentration. Means, standard deviations, R.S.D.% and R.E.% were calculated from the QC values by standard methods. Dilution integrity was determined by processing six replicates of a dilution QC (1,000 ng/ml) after a 10-fold dilution.
An incurred sample reanalysis was completed with samples (n=20) randomly selected from fexofenadine pharmacokinetic studies conducted in patients with normal renal function and end stage renal disease. The reproducibility of the assay results is expressed as the mean ratio (MR, accuracy) and the limits of agreement (LsA, precision) as described previously [32]. The assay was considered reproducible if (1) the 95% confidence interval of the mean ratio included 1 and was within the bounds of 0.83 and 1.2 and (2) the 67% limits of agreement of the ratio of sample results, i.e., the range within which the ratio of sample results is expected to fall two thirds of the time, was within 0.83 to 1.2 [32].
2.7. Selectivity and stability
Selectivity was evaluated by processing and analyzing blank plasma samples obtained from six different human plasma lots. Carry-over was evaluated by injections of mobile phase placed in several wells of the analysis set. The autosampler stability of processed samples was evaluated by analyzing QC samples immediately and 24 hours after processing. After the first analysis, the QC samples were stored in the autosampler at 10°C for at least 24 hours and then re-analyzed. The measured concentrations from both analyses were then compared to determine any differences due to the storage conditions. The stability after freeze and thaw was evaluated with low and high-concentration QC samples, which were subjected to three freeze-thaw cycles prior to processing. The effects were measured by the concentrations of each QC relative to a newly processed reference sample.
2.8. Matrix effects and extraction efficiency
The potential for matrix effects (suppression or enhancement of ionization) was evaluated qualitatively by standard post-column infusion experiments [33]. Processed blank plasma samples from six different human plasma lots were injected during a constant post-column infusion of fexofenadine. In addition, to determine the presence of matrix effects quantitatively, the response obtained from plasma samples (n=6) spiked after processing and mobile phase spiked with an equivalent amount of fexofenadine were compared. Responses obtained from the spiked fexofenadine solution were defined as 100%. Extraction efficiency at low and high QC concentrations (10 and 400 ng/ml) was determined by comparing fexofenadine response in plasma samples (n=6) spiked before and after extraction, which was defined as 100% recovery.
2.9. Application to plasma sampling
Fexofenadine pharmacokinetics were evaluated in a research volunteer with renal manifestations of systemic lupus erythematosus. The subject granted written informed consent and the study was approved by the Committee on the Protection of Human Subjects at the University of North Carolina. The study sought to evaluate fexofenadine as a P-glycoprotein probe substrate because glucocorticoids are substrates of this protein and are utilized in combination therapies for lupus nephritis. The subject had been receiving prednisone 60 mg daily for 90 days prior to the pharmacokinetics study. The subject received intravenous cyclophosphamide at the time of fexofenadine administration. The subject was receiving aspirin which is a known inducer of P-glycoprotein; no known inhibitors were prescribed. Fexofenadine 60 mg (Allegra®, Aventis Pharmaceuticals Inc., Bridgewater, NJ, USA) was administered orally with 8 ounces of water. Blood samples were collected at multiple time points over 72 hours; plasma obtained by centrifugation was stored at −70°C until analyzed.
3. Results and discussion
3.1. Chromatography
Representative extracted ion chromatograms of plasma samples are depicted in Fig. 2; Fig. 2A shows a blank plasma sample and Fig. 2B is a plasma sample spiked with fexofenadine at the LLOQ (1 ng/ml). Fig. 2C depicts a plasma sample obtained from a study subject after a single dose of fexofenadine 60 mg. A plasma sample spiked with the internal standard fexofenadine-d6 is shown in Fig. 2D. The retention time for fexofenadine and fexofenadine-d6 was 1.2 minutes.
Fig. 2.

Representative extracted ion chromatograms of: (A) blank plasma (B) fexofenadine lower limit of quantitation (LLOQ; 1.00 ng/ml); (C) plasma sample from a subject obtained 48 hours after oral administration of fexofenadine 60 mg (concentration = 1.62 ng/ml); and (D) fexofenadine-d6 (ISTD).
3.2. Calibration and linearity
The calibration curve was linear over a concentration range of 1–500 ng/ml using weighted (1/y2) linear regression. Duplicate calibration curves were analyzed for three runs and the mean calibration curve equation was y = 0.004069x ± 0.000071 − 0.000867 ± 0.000141; the correlation coefficient (r2) was greater than 0.99.
3.3. Precision, accuracy and incurred sample analysis
The intra- and inter-run accuracy (within ± 8%) and precision (within ± 4.3%) of the back calculated concentrations for the low, medium, and high QC samples demonstrated an accurate and reproducible method (Table 1). The intra- and inter-run accuracy and precision of the LLOQ QC was also acceptable with a mean R.E.% < ± 2.5% and R.S.D.% of 12.6% or less. Dilution integrity was determined by processing six replicates of the dilution QC (1,000 ng/ml) after a 10-fold dilution. The accuracy and reproducibility was found to be acceptable with a mean accuracy of 0.4% (R.E.%) and precision of 3.9% (R.S.D.%).
Table 1.
Intra- and inter-run precision (R.S.D.%) and accuracy (R.E.%) for fexofenadine quality control samples in human plasma.
| Concentration (ng/ml) | Precision (R.S.D.%) | Accuracy (R.E.%) | |
|---|---|---|---|
| Nominal | Found (mean ± SD) | ||
| Intra-run (N=12)a | |||
| 1 (LLOQ) | 1.00 ± 0.11 | 11.1 | −0.3 |
| 10 | 10.45 ± 0.45 | 4.3 | 4.5 |
| 150 | 158.3 ± 2.0 | 1.2 | 5.5 |
| 400 | 429.4 ± 7.3 | 1.7 | 7.3 |
| Inter-run (N=24) b | |||
| 1 (LLOQ) | 1.03 ± 0.13 | 12.6 | 2.5 |
| 10 | 10.41 ± 0.41 | 3.9 | 4.1 |
| 150 | 156.6 ± 2.8 | 1.8 | 4.4 |
| 400 | 431.9 ± 7.8 | 1.8 | 8.0 |
Twelve replicates of each QC were analyzed in one run.
A total of 24 replicates from three separate runs (one run of twelve and two runs of six replicates) were analyzed.
Assay reproducibility was confirmed through an incurred sample reanalysis. The samples tested ranged in concentration from 2.7 to 301.4 ng/ml; the average percent difference in quantitative results was −3.4%. The average fold change in the sample results between the 2 runs (mean-ratio, MR), mean ratio limits (RLs), and the limits of agreement (LsA) were calculated [32]. The results showed a MR of 1.01, RLs of 0.99–1.03 (acceptance range, 0.83–1.20), and LsA of 0.93–1.11 (acceptance range, 0.83–1.20). The acceptance ranges were met for both the RLs and LsA, demonstrating assay reproducibility.
3.4. Selectivity and stability
No interfering peaks were observed for fexofenadine or fexofenadine-d6 in the processed plasma samples from six different plasma lots and there was no evidence of carry-over. Autosampler stability was determined by comparing results for samples that were analyzed immediately and 24 hours after processing. The samples analyzed 24 hours after processing were stored in the autosampler at 10°C and the concentrations found did not deviate by more than 10%. The effect of freeze and thaw was evaluated in QC samples subjected to three freeze-thaw cycles and no degradation of fexofenadine was observed (i.e., less than a 10% difference in measured concentration).
3.5. Matrix effects and extraction efficiency
Recovery of fexofenadine at low and high QC concentrations was measured by comparing the response ratios of plasma samples that were spiked before and after processing. The response in samples spiked after processing were considered to be 100%. We determined the recovery for fexofenadine to be 93.6 ± 6.5% at low and 95.3 ± 10.3% at high concentrations. In addition, there was no evidence of a matrix effect as there was < 10% difference in the fexofenadine response. The post-column infusion experiment supported a lack of matrix effect.
3.6. Application to plasma sampling
This method was used to support a fexofenadine pharmacokinetic study in which fexofenadine was used a probe for transporter activity in patients with glomerulonephritis. The concentration-time profile for a study subject after administration of fexofenadine 60 mg as a single oral dose is shown in Fig. 3. The concentration values were consistent with values reported in the literature [1]. The method was shown to be suitable for pharmacokinetic studies of fexofenadine in human subjects.
Fig. 3.

Concentration-time profile for a study subject administered single oral dose of fexofenadine (60 mg).
4. Conclusion
We have validated a rapid, sensitive, selective, and reproducible LC-MS/MS method for determination of fexofenadine in human plasma. The method uses protein precipitation and filtration in a 96-well format, which from start to finish takes less than an hour to process manually. The rapid sample processing combined with the total chromatographic run time of 2 min facilitates multiple runs per day for high throughput applications. The method is currently being used to support clinical pharmacokinetic studies with the drug transport probe fexofenadine.
Acknowledgments
This work was supported in part by National Institutes of Health Grants K23 DK64888 (MSJ), General Clinical Research Centers program of the Division of Research Resources, National Institutes of Health RR00046, and Clinical and Translational Science Award U54RR024383.
Abbreviations
- H-SRM
high resolution single reaction monitoring
- MR
mean ratio
- RLs
ratio limits
- LsA
limits of agreement
- OATP
organic anion transporting polypeptides
- P-gp
P-glycoprotein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Devillier P, Roche N, Faisy C. Clin Pharmacokinet. 2008;47:217. doi: 10.2165/00003088-200847040-00001. [DOI] [PubMed] [Google Scholar]
- 2.Lippert C, Ling J, Brown P, Burmaster S, Eller M, Cheng L. Pharm Res. 1995;12:S390. [Google Scholar]
- 3.Cvetkovic M, Leake B, Fromm MF, Wilkinson GR, Kim RB. Drug Metab Dispos. 1999;27:866. [PubMed] [Google Scholar]
- 4.Matsushima S, Maeda K, Hayashi H, Debori Y, Schinkel AH, Schuetz JD, Kusuhara H, Sugiyama Y. Mol Pharmacol. 2008;73:1474. doi: 10.1124/mol.107.041459. [DOI] [PubMed] [Google Scholar]
- 5.Shimizu M, Fuse K, Okudaira K, Nishigaki R, Maeda K, Kusuhara H, Sugiyama Y. Drug Metab Dispos. 2005;33:1477. doi: 10.1124/dmd.105.004622. [DOI] [PubMed] [Google Scholar]
- 6.Robertson SM, Davey RT, Voell J, Formentini E, Alfaro RM, Penzak SR. Curr Med Res Opin. 2008;24:591. doi: 10.1185/030079908x260871. [DOI] [PubMed] [Google Scholar]
- 7.Kharasch ED, Walker A, Hoffer C, Sheffels P. J Clin Pharmacol. 2005;45:79. doi: 10.1177/0091270004269705. [DOI] [PubMed] [Google Scholar]
- 8.Glaeser H, Bailey DG, Dresser GK, Gregor JC, Schwarz UI, McGrath JS, Jolicoeur E, Lee W, Leake BF, Tirona RG, Kim RB. Clin Pharmacol Ther. 2007;81:362. doi: 10.1038/sj.clpt.6100056. [DOI] [PubMed] [Google Scholar]
- 9.Dresser GK, Kim RB, Bailey DG. Clin Pharmacol Ther. 2005;77:170. doi: 10.1016/j.clpt.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 10.Bailey DG, Dresser GK, Leake BF, Kim RB. Clin Pharmacol Ther. 2007;81:495. doi: 10.1038/sj.clpt.6100104. [DOI] [PubMed] [Google Scholar]
- 11.Shimizu M, Uno T, Sugawara K, Tateishi T. Br J Clin Pharmacol. 2006;61:538. doi: 10.1111/j.1365-2125.2006.02613.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yasui-Furukori N, Uno T, Sugawara K, Tateishi T. Clin Pharmacol Ther. 2005;77:17. doi: 10.1016/j.clpt.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 13.Wang Z, Hamman MA, Huang SM, Lesko LJ, Hall SD. Clin Pharmacol Ther. 2002;71:414. doi: 10.1067/mcp.2002.124080. [DOI] [PubMed] [Google Scholar]
- 14.van Heeswijk RP, Bourbeau M, Campbell P, Seguin I, Chauhan BM, Foster BC, Cameron DW. J Clin Pharmacol. 2006;46:758. doi: 10.1177/0091270006288733. [DOI] [PubMed] [Google Scholar]
- 15.Niemi M, Kivisto KT, Hofmann U, Schwab M, Eichelbaum M, Fromm MF. Br J Clin Pharmacol. 2005;59:602. doi: 10.1111/j.1365-2125.2005.02354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shon JH, Yoon YR, Hong WS, Nguyen PM, Lee SS, Choi YG, Cha IJ, Shin JG. Clin Pharmacol Ther. 2005;78:191. doi: 10.1016/j.clpt.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 17.Yi SY, Hong KS, Lim HS, Chung JY, Oh DS, Kim JR, Jung HR, Cho JY, Yu KS, Jang IJ, Shin SG. Clin Pharmacol Ther. 2004;76:418. doi: 10.1016/j.clpt.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 18.Emara S, El-Gindy A, Mesbah MK, Hadad GM. J AOAC Int. 2007;90:384. [PubMed] [Google Scholar]
- 19.Miura M, Uno T, Tateishi T, Suzuki T. J Pharm Biomed Anal. 2007;43:741. doi: 10.1016/j.jpba.2006.07.033. [DOI] [PubMed] [Google Scholar]
- 20.Coutant JE, Westmark PA, Nardella PA, Walter SM, Okerholm RA. J Chromatogr. 1991;570:139. doi: 10.1016/0378-4347(91)80208-t. [DOI] [PubMed] [Google Scholar]
- 21.Uno T, Yasui-Furukori N, Takahata T, Sugawara K, Tateishi T. J Pharm Biomed Anal. 2004;35:937. doi: 10.1016/j.jpba.2004.02.036. [DOI] [PubMed] [Google Scholar]
- 22.Pathak SM, Kumar AR, Musmade P, Udupa N. Talanta. 2008;76:338. doi: 10.1016/j.talanta.2008.02.047. [DOI] [PubMed] [Google Scholar]
- 23.Hofmann U, Seiler M, Drescher S, Fromm MF. J Chromatogr B Analyt Technol Biomed Life Sci. 2002;766:227. doi: 10.1016/s0378-4347(01)00468-6. [DOI] [PubMed] [Google Scholar]
- 24.Isleyen EAO, Ozden T, Ozilhan S, Toptan S. Chromatographia. 2007;66:S109. [Google Scholar]
- 25.Nirogi RV, Kandikere VN, Shukla M, Mudigonda K, Maurya S, Komarneni P. Rapid Commun Mass Spectrom. 2006;20:3030. doi: 10.1002/rcm.2701. [DOI] [PubMed] [Google Scholar]
- 26.Nirogi RV, Kandikere VN, Shukla M, Mudigonda K, Maurya S, Komarneni P. Biomed Chromatogr. 2007;21:209. doi: 10.1002/bmc.740. [DOI] [PubMed] [Google Scholar]
- 27.Bharathi VD, Radharani K, Jagadeesh B, Ramulu G, Bhushan I, Naidu A, Mullangi R. Chromatographia. 2008;67:461. [Google Scholar]
- 28.Fu I, Woolf EJ, Matuszewski BK. J Pharm Biomed Anal. 2004;35:837. doi: 10.1016/j.jpba.2004.02.016. [DOI] [PubMed] [Google Scholar]
- 29.Naidong W, Shou WZ, Addison T, Maleki S, Jiang X. Rapid Commun Mass Spectrom. 2002;16:1965. doi: 10.1002/rcm.817. [DOI] [PubMed] [Google Scholar]
- 30.Yamane N, Tozuka Z, Sugiyama Y, Tanimoto T, Yamazaki A, Kumagai Y. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;858:118. doi: 10.1016/j.jchromb.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 31.Guo D, Zou J, Zhu Y, Lou S, Fan H, Qin Q. Biomed Chromatogr. 2009 doi: 10.1002/bmc.1296. [DOI] [PubMed] [Google Scholar]
- 32.Rocci ML, Jr, Devanarayan V, Haughey DB, Jardieu P. AAPS J. 2007;9:E336. doi: 10.1208/aapsj0903040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.King R, Bonfiglio R, Fernandez-Metzler C, Miller-Stein C, Olah T. J Am Soc Mass Spectrom. 2000;11:942. doi: 10.1016/S1044-0305(00)00163-X. [DOI] [PubMed] [Google Scholar]