

Abstract
Background
The genetic etiologies of the hyper-IgE syndromes are diverse. Approximately 60-70% of patients with hyper-IgE syndrome have dominant mutations in STAT3, and a single patient was reported to have a homozygous TYK2 mutation. In the remaining hyper-IgE syndrome patients, the genetic etiology has not yet been identified.
Methods
We performed genome-wide single nucleotide polymorphism analysis for nine subjects with autosomal recessive hyper-IgE syndrome to locate copy number variations and homozygous haplotypes. Homozygosity mapping was performed with twelve subjects from seven additional families. The candidate gene was analyzed by genomic and cDNA sequencing to identify causative alleles in a total of 27 patients with autosomal recessive hyper-IgE syndrome.
Findings
Subtelomeric microdeletions were identified in six subjects at the terminus of chromosome 9p. In all patients the deleted interval involved DOCK8, encoding a protein implicated in the regulation of the actin cytoskeleton. Sequencing of subjects without large deletions revealed 16 patients from nine unrelated families with distinct homozygous mutations in DOCK8 causing premature termination, frameshift, splice site disruption, single exon- and micro-deletions. DOCK8 deficiency was associated with impaired activation of CD4+ and CD8+ T cells.
Interpretation
Autosomal recessive mutations in DOCK8 are responsible for many, though not all, cases of autosomal recessive hyper-IgE syndrome. DOCK8 disruption is associated with a phenotype of severe cellular immunodeficiency characterized by susceptibility to viral infections, atopic eczema, defective T cell activation and TH17 cell differentiation; and impaired eosinophil homeostasis and dysregulation of IgE.
Keywords: Autosomal recessive hyper-IgE syndrome, human gene mutation, DOCK8, primary immunodeficiency, molluscum contagiosum, recurrent infection, T cells, TH17 cells, eosinophils, IgE regulation, copy number variations, genomic deletions
INTRODUCTION
The hyper-IgE syndromes (HIES) [also called Job’s syndrome; (OMIM) #147060 and #243700] are rare primary immunodeficiencies (estimated prevalence <1:1 million), characterized by the clinical triad of recurrent staphylococcal skin abscesses, recurrent pneumonia and serum IgE-levels of >10 times the upper norm. HIES usually manifest in childhood and have a highly variable expressivity1,2. Although most cases are sporadic, both autosomal dominant (AD) and autosomal recessive (AR) inheritance have been described. The predominant form is AD-HIES caused by heterozygous, dominant-negative mutations in STAT33. In this form of HIES extra-immune manifestations occur, including skeletal abnormalities such as retained primary teeth and a typical facial appearance. A scoring system for these findings has been developed at the NIH4 and refined to a STAT3 score by Woellner et al.5. In contrast, AR-HIES is characterized by recurrent viral and bacterial infections, extreme eosinophilia and elevated IgE without skeletal or dental abnormalities6. The genetic causes of AR-HIES are largely unknown. Minegishi et al. reported a monogenetic defect in the cytoplasmatic tyrosine kinase Tyk2 in a single patient with clinical features of AR- HIES who had consanguineous parents 7. In a follow-up study, however, additional AR-HIES patients did not have mutations in the TYK2 gene8. However, recently a second patient with a related phenotype has been found to be deficient in Tyk2 (J.-L. Casanova, personal communication).
AR-HIES has been described predominantly in consanguineous families from Turkey. We investigated, by genome-wide homozygosity mapping and copy number analysis, 21 patients from 16 families with AR-HIES, defined as a positive NIH HIES score, wild-type STAT3 sequence, and absence of significant skeletal findings. Six additional patients from four families were analyzed after the candidate gene had been identified.
Homozygosity mapping is a method to localize a disease-associated recessive genotype by searching for homozygous haplotypes in consanguineous families; the underlying assumption is that the recessive mutant allele is identical by descent in affected subjects9. If the founder mutation is relatively recent, the causative mutation is likely to be found within the largest stretches of homozygosity. The genotyping required for this approach may be accomplished using high-density oligonucleotide arrays, which have the additional benefit of providing data on copy number at each SNP locus on the array. Copy number variations (CNVs) occur as a result of deletions and insertions of variable size in the genome10. CNVs are common and may be polymorphic in the population or arise de novo in individuals.
Our analysis has demonstrated an AR-HIES genomic locus and made possible detection of mutations in DOCK8 as major defect in AR-HIES.
MATERIALS AND METHODS
Patients and Controls
The phenotypes of the AR-HIES patients analyzed are shown in Table 1. Whole blood samples were taken from patients, family members and healthy volunteers with informed consent. We analyzed DNA from 20 families suspected of having AR-HIES based on clinical assessment. Criteria for AR-HIES were defined as elevated IgE, eczema, hypereosinophilia, and significant infections (particularly with molluscum contagiosum and herpes family viruses) (Fig.1). In some kindreds there were deceased affected siblings, but parents were unaffected. There were 13 families from Turkey, two from Iran, and one each from Lebanon, Oman, Mexico, Italy and Ireland. All affected individuals had an NIH HIES score4 >20. Clinical data on families ARH011 and ARH015 was previously published in Renner et al.6, family ARH011 has also been reported by Zhang et al.11, family ARH010 is family 18 in Grimbacher et al.4, and patients ARH001-004 as well as ARH006-ARH009 were reported in Al-Khatib et al.12
Table 1.
Characteristics of autosomal-recessive hyper-IgE patients with and without detected DOCK8 mutations.
| ARH001 | ARH002 | ARH003 | ARH004 | ARH005 | ARH006 | ARH007 | ARH008 | ARH009 | ARH0010.2 | ARH0010.4 | ARH0010.8 | ARH0010.9 | ARH011.3 | ARH011.4 | ARH011.5 | ARH012 | ARH013.3 | ARH013.4 | ARH014 | ARH015 | ARH016.6 | ARH016.7 | ARH017 | ARH018 | ARH019 | ARH020 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ethnicity | Turkish | Turkish | Turkish | Turkish | Italian | Turkish | Turkish | Turkish | Turkish | Turkish | Turkish | Turkish | Turkish | Mexican | Mexican | Mexican | Irish | Turkish | Turkish | Turkish | Turkish | Turkish | Turkish | Iranian | Iranian | Omani | Lebanese |
| consanguinity | + | − | + | − | − | − | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| age [yrs] | 3.8 | 6,9† | 8,2† | 8.1 | 8† | 9,8† | 9.3 | 8 | 12.9 | 45.4 | 40.8 | 15.9 | 14.8 | 24 | 18† | 16 | 9.5 | 9.6 | 4.4 | 16.7 | 19 | 6 | 3 | 17 | 18 | 9.8 | 23 |
| sex | F | M | M | M | M | M | F | F | F | M | M | F | F | M | F | M | F | F | F | M | F | M | F | F | M | F | F |
|
DOCK8 mutation |
deletion | deletion | deletion | deletion | deletion | heterozygous deletion |
none detected | point mutation in splice site |
single exon deletion (37) |
point mutation in splice site |
point mutation in splice site |
point mutation in splice site |
point mutation in splice site |
point mutation, cryptic splice site |
point mutation, cryptic splice site |
point mutation, cryptic splice site |
point mutation, premature termination |
single exon deletion (46) |
single exon deletion (46) |
deletion | deletion | deletion | deletion | none detected | none detected | none detected | no exonic mutation detected, possibly mutation in promoter |
| HIES score | 47 | 48 | 42 | 58 | 49 | 61 | 49 | 65 | 61 | 40 | 52 | 42 | 49 | 44 | 54 | 42 | 24 | 41 | 23 | 51 | 40 | 38 | 34 | 49 | 53 | 30 | 36 |
| IgE [IU/ml] | 5,000 | 30,000 | 4,970 | 5,000 | 5,400 | 1,163 | 400 | 10,500 | 19,100 | 4,074 | 90,910 | 74,688 | 25,987 | 5,992 | 39,669 | 31,348 | 10,030 | 11,140 | 2,830 | 8,080 | 21,000 | 16,000 | 3,000 | 2,291 | >1000 | 3,325 | 3,703 |
|
Blood eosinophil count [cells/μl] or [%] |
10,600 | 7,400 | 37,880 | 1,400 | N/A | 2,100 | 8,100 | 5,100 | 1,000 | 544 | 1,330 | 1,527 | 932 | 13,600 | 4,730 | 2,610 | 1,069 | 290 | 1,965 | 18% (2-4%) | 4,500 | 25% | 42% | 1,368 | 5,513 | 1,400 | 1,026 |
| Infections | |||||||||||||||||||||||||||
|
upper respiratory infections |
recurrent upper respiratory infections |
recurrent otitis | recurrent otitis | recurrent otitis | sinusitis and otitis |
recurrent upper respiratory infections |
recurrent otitis | recurrent otitis, sinusitis |
recurrent suppurative otitis |
sinusitis, recurrent upper respiratory tract infections, 20/y |
recurrent otitis media and sinusitis |
mild recurrent upper respiratory infections, 2x/y |
mild recurrent upper respiratory infections, 2-3/y |
recurrent upper respiratory infections |
otitis media, sinusitis |
recurrent upper respiratory infections |
− | chronic otitis media |
recurrent otitis media |
− | recurrent otitis media, sinusitis |
recurrent pulmonary infections |
recurrent pulmonary infections (5- 6/year) |
recurrent otitis media |
recurrent upper respiratory infections |
recurrent upper respiratory infections, sinusitis |
− |
|
lower respiratory infections |
1 pneumonia | 2 pneumonia | 2 pneumonia | >5 pneumonias and recurrent bronchitis |
>3 pneumonias | >5 pneumonias | >5 pneumonias | >5 pneumonias | >5 pneumonias | >20 pneumonias, recurrent bronchitis |
>20 pneumonias | pneumonias | frequent pneumonias |
pneumonias | pneumonias | pneumonias | pneumonia | 2 pneumonias, recurrent bronchitis |
− | >3 pneumonias, bronchitis |
>10 pneumonias, bronchitis |
pneumonias (2- 3/year) |
3-4/year | >3 pneumonias | >3 pneumonias | − | pneumonia |
|
Lung abnormalities |
− | − | − | bronchiectasis | bronchiectasis | bronchiectasis | pneumatoceles | − | − | bronchiectasis | bronchiectasis | N/A | N/A | − | − | − | − | − | − | − | − | − | N/A | bronchiectasis | bronchiectasis | − | bronchiectasis |
| abscesses | + | + | + | + | + | − | + | + | + | + | + | + | + | + | + | + | − | + | − | + | + | − | − | + | + | − | + |
| viral | Molluscum contagiosum, recurrent HSV |
herpetic keratitis | − | − | JC virus- associated progressive multifocal leucoencephalop athy (PML) |
Molluscum contagiosum, chronic Hepatitis B virus |
− | recurrent HSV | Heck’s disease associated with chronic oral HPV infection, chronic vulvar HPV infection |
HSV | Molluscum contagiosum |
recurrent HSV | recurrent HSV, herpes zoster |
Papilloma virus | Molluscum contagiosum, HSV, Herpes zoster, Papilloma virus |
Molluscum contagiosum, recurrent HSV |
severe and refractory Molluscum contagiosum |
recurrent HSV, herpetic keratitis, CMV eningitis, papillitis and retinitis |
− | severe and refractory Molluscum contagiosum, HSV |
severe and refractory Molluscum contagiosum, recurrent Herpes zoster, rotavirus enteritis |
JC virus- associated progressive multifocal leucoencephal opathy (PML) |
− | HSV | Molluscum contagiosum |
viral skin infections, oozing ear infections with ulcer |
Molluscum contagiosum, HSV |
| other | candidiasis | candidiasis | candidiasis | candidiasis | candidiasis | N/A | candidiasis | candidiasis, Listeria meningitis |
candidiasis | candidiasis | candidiasis, onychomycosis |
candidiasis | candidiasis, sepsis |
candidiasis, Tinea cruris |
candidiasis, Haemophilus influenza and cryptococcal meningitis, sepsis |
candidiasis | pneumococcal meningitis |
− | − | candidiasis, Klebsiella sepsis |
candidiasis | candidiasis | − | − | − | severe fungal nail bed infections superinfected with bacteria, persistent ear infections with mastoiditis |
candidiasis |
| atopy | eczema | eczema, newborn rash |
eczema | eczema | eczema | eczema, newborn rash |
eczema | eczema | eczema | severe eczema, asthma, polyvalent food allergy |
eczema, newborn rash, asthma |
severe eczema, asthma, polyvalent food allergy, dust mite and latex allergies |
severe eczema, asthma, polyvalent food allergy, dust mite and latex allergies |
eczema | eczema, polyvalent food allergy, drug and environmental allergies |
eczema, polyvalent food allergy, environmental allergies |
eczema, asthma |
eczema, asthma, polyvalent food, drug and environmental allergies |
eczema | eczema, asthma, polyvalent food and animal hair allergy |
eczema, polyvalent food allergy |
pustular neonatal rash, severe polyvalent food allergy |
eczema, severe polyvalent food allergy |
eczema | eczema | eczema | eczema, polyvalent food allergy |
| CNS | − | CNS vasculitis | + | − | severe neurological disease (PML) |
fatal encephalitis | − | brain infarct, meningitis |
− | − | − | − | CNS vasculitis | − | meningitis, post- cryptococcal lesions in brain |
− | meningitis | meningitis | − | − | − | severe neurological disease (PML) |
− | − | − | Headache,Diple gia, chorioform and dystonic movments, |
− |
|
Skeletal/dental features |
characteristic facial features, high palate |
characteristic facial features |
characteristic facial features, high palate |
characteristic facial features, high palate |
retained primary teeth, increased nose width |
increased nasal width, high palate, retained primary teeth |
retained primary teeth |
characteristic facial features |
increased nasal width, high palate, retained primary teeth |
− | − | − | − | − | − | retained primary teeth |
− | clavicula fracture |
− | characteristic facial features, fractures |
− | coarse facies, deep set eyes |
− | hyperflexibility, increased nasal width |
hyperflexibility, increased nasal width |
− | − |
| malignancies | − | − | − | − | − | − | Burkitt lymphoma | − | squamous cell carcinoma |
− | − | − | − | − | − | − | − | Burkitt lymphoma |
− | − | − | − | − | − | − | − | − |
| autoimmunity | − | − | − | − | − | − | − | autoimmune hemolytic anemia |
− | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| others | − | − | exaggerated eosinophilia |
− | − | − | − | − | − | thrombopenia | Dupytren contraction on hands |
paronychia | − | osteomyelitis of the femur |
malabsorption and diarrhea |
− | − | − | microcytic anemia |
retinitis, osteomyelitis |
− | − | − | − | − | mild hypertension, parenchymal kidney disease |
− |
|
Immunological analysis |
|||||||||||||||||||||||||||
| WBC [cells/μl] | 29,400 | 13,700 | 51,200 | 11,180 | 10,300 | 19,100 | 12,200 | 13,600 | 8,900 | 6800 | 9500 | 10300 | 8400 | 6900 | 19200 | 10300 | 8,400 | 5,800 | 13,100 | 15,900 | 4,100 | 13,200 | 16,100 | 7,200 | 14,900 | 9,400 | 14,700 |
| ANC [cells/μl] | 7,200 | 3,100 | 4,096 | 7,210 | 10,900 | 2,000 | 7,800 | 3,750 | 4692 | 6460 | 4635 | 4956 | 3531 | 6509 | 3955 | 4,180 | N/A | N/A | 10,176 | N/A | 9,500 | 5,600 | 4,104 | 6,556 | 4,700 | 9,408 | |
| ALC [cells/μl] | 6,800 | 2,300 | 4,096 | 2,158 | 2,266 | 5,340 | 5,100 | 3,200 | 1,500 | 952 | 1243 | 3811 | 1596 | 963 | 2054 | 2225 | 2,332 | 4,290 | 6,943 | 1,908 | 1,066 | 1,400 | 3,000 | 1,656 | 1,926 | 1,900 | 3,381 |
| PLT [cells/μl] | 596,000 | 413,000 | 350,000 | 49,000 | 263,000 | 684,000 | 172,000 | 393,000 | 327,000 | 188000 | 282000 | 371000 | 211000 | 263000 | 567000 | 370000 | 601,000 | 250,000 | 420,000 | 407,000 | N/A | 423 | 423,000 | 216,000 | 466,000 | 342,000 | 474,000 |
| IgA [mg/dl] | 103 (44-244) | 174 (57-282) | 215 (70-303) | 484 (70-303) | 387 | 468 (62-390) | 428 (62-390) | 239 (70-303) | 20 (67-433) | 165 (40-238) | 94 | 156 | 162 | 175 | 630 | 362 | 211 (40-2,180) | 29 (70-230) | 90 (40-180) | 151 (67-310) | 220 | 8.74 (70-303) | 8 (26-296) | 153 (70-312) | 115 (70-312) | 20.67 (8.2- 45.3) |
351 |
| IgM [mg/dl] | 72 (52-297) | 46,1 (78-261) | 28 (69-387) | 29,2 (69-387) | 35 | 31 (54-392) | 93 (54-392) | 62 (69-387) | 26 (47-484) | 31 (48-228) | 42 | 59 | 30 | 24 | 53 | 44 | <22 (43-190) | 5 (40-150) | 49 (40-180) | 17 (50-190) | 127 | 70.8 (69-387) | 70 (71-235) | 37 (56-352) | <20 (56-352) | 2.6 (4.6-30.4) | 49 |
| IgG [mg/dl] | 1930 (640-2010) | 1740 (745-1804) | 1400 (764-2314) | 2460 (764-2134) | 1,153 | 1190 (842-1943) | 778 (842-1943) | 1500 (764-2134) | 1568 (835-2094) | 1050 (672-1536) | 1050 | 2150 | 1440 | 1710 | 2530 | 3310 | 2,500 (360- 1,520) |
1,090 (700- 1,400) |
1,250 (500- 1,300) |
1,300 (720- 1,560) |
1,450 | 1,580 (764- 2,134) |
1,600 (604- 1,941) |
3,129 (639- 1,349) |
3,150 (639- 1,349) |
184 (75-156) | 1,928 |
| CD3 [%] | 69 (43-76) | 51 (55-78) | 32,5 (55-78) | 52,7 (55-78) | 29 | 25 (55-78) | 70 (55-78) | 46 (55-78) | 54 (52-78) | 60 | 71 (55-83) | 79 | 70 | 58 (57-86) | 65 (57-86) | 59 (57-86) | 47 | 64 (55-78) | 63 (43-76) | 36.2 (52-78) | 53 | 25 | 42 | 50.4 (52-78) | 63 (52-78) | 33 | N/A |
| CD4 [%] | 18 (23-48) | 15 (27-53) | 14,9 (27-53) | 19 (27-53) | 12 | 13 (27-53) | 16 (27-53) | 32 (27-53) | 24 (25-48) | 64 (% of CD3+) | 35 (28-57) | 55 (% of CD3+) | 62 (% of CD3+) | 28 (29-57) | 39 (29-57) | 32 (29-57) | 21 | 28 (27-53) | 31 (23-48) | 24 (25-48) | 16 | 11.7 | 15 | 20.9 (25-48) | 56 (25-48) | 11 | N/A |
| CD8 [%] | 33 (14-33) | 36 (19-34) | 8,4 (19-34) | 31 (19-34) | 15 | 12 (19-34) | 49 (19-34) | N/A | 27 (9-35) | 35 (% of CD3+) | 29 (10-39) | 43 (% of CD3+) | 35 (% of CD3+) | 39 (25-51) | 23 (25-51) | 25 (25-51) | 24 | 35 (19-34) | 29 (14-33) | 5 (9-35) | 26 | 13.6 | 27 | 53.9 (9-35) | 18 (9-35) | 18 | N/A |
| CD19 [%] | 18 (10-31) | 41,6 (10-31) | 20 (10-31) | 29 (10-31) | 55 | 8 (10-31) | 20 (10-31) | 58 (10-31) | 20 (8-24) | 30 | 21 (6-19) | 16 | 25 | 22 (4-16) | 30 (4-16) | 36 (4-16) | 38 | 27 (10-31) | 22 (14-44) | 41 (8-24) | 22 | 52 | 41.4 | 28 (8-24) | 28 (8-24) | 7 | N/A |
| CD16/CD56 [%] | 8 (4-23) | 4 (4-26) | 13 (4-26) | 13 (4-26) | N/A | 54 (4-26) | 6 (4-26) | N/A | 15 (6-27) | 10 | 7 (7-31) | 5 | 5 | 20 (5-30) | 5 (5-30) | 6 (5-30) | 13 | 2 (4-26) | 7 (4-23) | 19.4 (6-27) | 14 | 2 | 5 | N/A | N/A | 59 | N/A |
N/A, not available
denotes deceased individuals.
Figure 1.
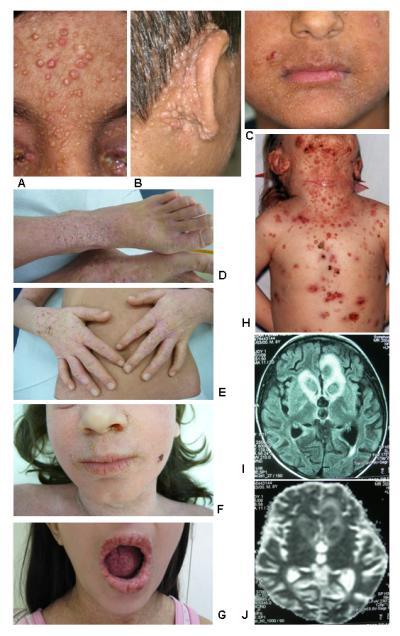
Clinical findings in patients with DOCK8 mutations. Panels A-C show the severe Molluscum contagiosum burden of patient ARH014; panel H illustrates the Molluscum infection of patient ARH012; panels D-F show the severe dermatitis in patients ARH010.8 and ARH010.9; and panel G demonstrates the severe oral papilloma virus infection of patient ARH009. Panels F (MRI) and G (diffusion scan) document the cause of death in patient ARH003 who developed an undefined form of encephalitis.
Methods
A detailed description of the methods can be found in the Online Repository, including homozygosity mapping and copy number analysis, PCR and sequence analysis, immunoblotting, as well as proliferation and CFSE dilution studies.
RESULTS
Search for copy number variations, microdeletions and homozygosity
Representational oligonucleotide microarray analysis (ROMA) for CNVs was performed on eight index patients with AR-HIES previously identified among a cohort of Turkish patients12 and a singleton patient from Italy with HIES (Fig.2a). In the subtelomeric region of chromosome 9p, homozygous deletions (copy number = 0) were identified in four subjects and a large compound heterozygous deletion was identified in the Italian AR-HIES patient. In one additional subject, hemizygosity was identified at this locus. For most subjects, the deletion extended from the most terminal p locus to within the DOCK8 gene (Fig.2b). Homozygous deletions were confirmed by PCR of the affected segments of DOCK8 (see Figure E1 in the Online Repository). The parental origin of the deletion was investigated and is shown in Figure E2 in the Online Repository.
Figure 2.


A. ROMA data demonstrating copy number abnormalities consistent with subtelomeric deletions of 9p in AR-HIES. Individuals ARH001-ARH004 have homozygous deletions, ARH005 has a compound heterozygous deletion, and ARH006 has a heterozygous deletion. The remaining subjects do not have demonstrable deletions. Genome-wide SNP Nsp 250k arrays were used for subjects ARH001-ARH009.
B. HIES patient deletions and homozygous intervals and known and predicted genes at the terminus of chromosome 9p. C9orf66 is an open reading frame, and FAM138C is a noncoding RNA gene. FOXD4 is a transcription factor, and CBWD has a cobalamin binding domain and nuclease function. DOCK8 is described in the text.
To obtain additional evidence that the terminal region of chromosome 9p is associated with AR-HIES, homozygosity mapping was performed on further 14 affected subjects. Twelve subjects were identified to be homozygous for distal chromosome 9p (data not shown).
Positional identification of candidate genes
Taken together, all affected probands had either deletions or extended homozygous haplotypes at 9p24.3, a region encoding at least four genes, the largest of which is DOCK8. The other known genes in this interval include FOXD4, CBWD1, and FAM138C (a non-coding RNA gene). In addition, there are four hypothetical genes and one open reading frame present in this region. The majority of the deletions described in our families (ARH001-006) deleted all of these genes as well as disrupting DOCK8. Because individuals ARH003 and ARH005 had only DOCK8 involved in the deletions, we focused on DOCK8 for further analysis.
Mutation detection in DOCK8
Analysis of DOCK8 genomic and cDNA sequences in individuals with no deletions detectable by CNV analysis revealed a number of mutations and smaller deletions in 16 patients from nine unrelated families out of 14 families analyzed.
The lack of exonic PCR products in families ARH014, ARH015 and ARH016 suggests deletions including parts of the DOCK8 gene (Fig.3). The single affected individual in family ARH014 showed normal PCR amplification products for exons 3-10, a shorter PCR product for exon 11, and no amplification products for exons 12, 13, 15, 16, 26 and 46-48 (Fig.3a,b). We conclude that a large homozygous genomic deletion begins in exon 11 and extends beyond the DOCK8 gene (Fig.5). Likewise, patients from families ARH015 and ARH016 show a deletion up to and including exon 26 and 25, respectively (Fig.3c-f), deleting the first half of the gene (Fig.5).
Figure 3.

Exonic deletions in DOCK8. Pedigrees are shown in panels A, C and E. Squares: males; circles: females. Filled symbols: patients; slash: deceased individuals. The lack of PCR products from patients’ DNA compared to control DNA suggests exonic deletions (B, D, F).
In family ARH010 (Fig.4a), we found a homozygous splice donor site mutation (gta→tta) after exon 25 (Fig.4b) that leads to skipping of exon 25 in the DOCK8 cDNA (Fig.4c). PCR amplification of exons 22-27 from cDNA revealed two products, one normal for wild type individual ARH010.5 and one faster migrating due to lack of exon 25 sequence for the patients, while both products were obtained for heterozygous family members (Fig.4c). Sequence analysis confirmed that exon 25 was missing from mRNA transcribed from the mutated allele (Fig.4c). The mutation perfectly segregated with the phenotype in this family (Fig.4a), with all affected subjects being homozygous, and the mother as well as the healthy sister of the affected girls being heterozygous for the mutation. Family members heterozygous or homozygous for the wild type allele had no signs of HIES. We analyzed this specific splice site mutation in DNA from 148 healthy controls and found no evidence of this sequence variant, indicating it is not a polymorphism. Exon 25 comprises 150 base pairs; hence its excision leads to an in frame deletion of 50 amino acids within the DOCK8 protein that are not part of either of the two predicted functional domains (UniProtKB/Swiss-Prot: DOCK8_HUMAN, Q8NF50) (Fig.5).
Figure 4.

Mutations in DOCK8. Pedigrees are shown in panels A, D, I, L, O, and Q. Squares: males; circles: females. Filled symbols: patients; slash: deceased individuals. Point mutations in the splice site (B, P) or within exons (E, J). Stop codon caused by point mutation (K) or frameshift (H, M). Generation of a cryptic splice site leading to a 4 bp deletion (F, G). Exon skipping shown by cDNA sequencing (C, M, S) and PCR (C, R). Lack of PCR products from genomic DNA suggesting exonic deletions (N, R).
Figure 5.

Cartoon showing the predicted impact of the mutations on DOCK8 protein expression. In three families, the mutation results in a truncated protein affecting both DHR domains, whereas in two families the truncated protein lacks the DHR-1 domain. In family ARH009, 53 amino acids are missing within the DHR-2 domain, and in family ARH0010, 50 amino acids are missing in-between the two DHR domains. In family ARH013, DOCK8 is truncated at the C-terminus.
Sequence analysis of family ARH011 revealed a homozygous point mutation (A→G) at position 133 in exon 12 of DOCK8 that segregated with HIES (Fig.4d), resulting in substitution of arginine for lysine at position 473 (K473R)(Fig.4e). However, in the cDNA of patient ARH011.4, we found a 4 bp deletion directly following the point mutation, which was not seen on the genomic level (Fig.4f). To explain this finding, the point mutation must create an upstream cryptic splice donor site, described as CAG/gc site (http://www.life.umd.edu/labs/mount/RNAinfo/consensus.html), which may be used instead of or in preference over the wild type sequence CAG/gt at the 3′-exon/intron boundary (Fig.4g) and causing the 4 bp deletion in the cDNA. The deletion creates a frameshift followed by a premature stop codon (Fig.4h), predicted to result in the expression of a truncated protein lacking the end of the DHR-1 and the whole DHR-2 domain (Fig.5). The residual full length protein expression reported in this family by Zhang et al.11 suggests that the cryptic splice site is used in preference over, rather than instead of, the wild type splice site. We sequenced exon 12 of DOCK8 in 91 healthy controls and found no evidence that this is a polymorphism.
In the only patient of family ARH012, genomic DNA sequencing revealed a homozygous point mutation (A→T) at position 70 in exon 7 of DOCK8 that segregated with HIES (Fig.4i), resulting in the premature stop codon TAG at amino acid position 271 (Fig.4j,k). Thus, the mutation will cause a truncated form of the DOCK8 protein lacking both DHR domains (Fig.5). We sequenced exon 7 of DOCK8 in 115 healthy controls and found no evidence that this is a polymorphism.
In family ARH013, exon 46 of DOCK8 was absent in the cDNA of both patients (Fig.4m). In contrast to the parents’ DNA, it was impossible to amplify exon 46 with specific primers by PCR from the patients’ genomic DNA (Fig.4n). Sequence analysis of the parents’ PCR products showed no mutation within exon 46 or the flanking splice sites (data not shown). We therefore conclude that the patients harbor a homozygous genomic deletion of exon 46. The lack of exon 46 with its 107 base pairs will lead to a frameshift and cause a truncated protein (Fig.5).
In patient ARH008, a homozygous missense mutation replaced the canonical G in exon 17 splice acceptor site to a C (IVS16-1G→C) (Fig.4p). The mutation segregated with HIES (Fig.4o). The failure to amplify cDNA beyond exon 11 (data not shown) suggested that only proximal shorter messages were expressed. Longer messages would probably be rendered unstable due to exon skipping, leading to out of frame sequences and premature termination, which in turn would trigger nonsense mediated degradation. Consistent with this view, the parental cDNA sequences were normal, suggesting that abnormal messages directed by the mutant allele were lost.
Sequencing of DOCK8 cDNA in the proband ARH009 revealed skipping of exon 37 (Fig.4s). This deletion is predicted to result in an inframe deletion of 53 amino acids. While genomic sequences of exon 36 and 38 and their surrounding intronic junctions were normal in the proband, attempts to amplify exon 37 and its associated splice junctions were unsuccessful (Fig.4r). Normal genomic exon 37 sequences were amplified from both parents, but cDNA analysis revealed two species, one normal and one faster migrating due to lack of exon 37 sequence. These results are consistent with homozygous deletion of exon 37 in the proband, inherited from parents heterozygous for the same deletion (Fig.4q).
Finally, sequencing of genomic DNA of proband ARH006, who suffered from a heterozygous deletion in the DOCK8 locus, failed to reveal exonic mutations, suggesting the presence of a cryptic mutation in the undeleted allele.
DOCK8 deficiency impairs T cell activation
To establish the functional consequences of DOCK8 mutations, we first analyzed the expression of DOCK8 protein in PBMC of probands, their family members and control subjects. Immunoblot analysis of PBMC lysates of control subjects and family members, carried out using an anti-human DOCK8 antibody, revealed a major immunoreactive band at about 180 KDa (Figure 6a, arrowheads) and several less intensely staining smaller bands. In contrast, these bands were missing in two subjects with documented mutations in DOCK8, including one with splice junction mutation leading to early premature termination (ARH008) and another with an inframe exon 37 deletion (ARH009). These results demonstrate a deleterious effect of these DOCK8 mutations on protein expression. Interestingly, another proband (ARH007) with no identified DOCK8 mutations also had absent DOCK8 immunoreactive bands, consistent with absent protein expression. This may be indicative of a hitherto undetected deleterious mutation affecting DOCK8 in the non-exonic regions of the gene or a mutation in another gene that regulates DOCK8 expression and/or stability.
Figure 6.

DOCK8 deficiency impairs T cell activation. A. DOCK8 protein expression in probands, family members and control samples. Lysates from PBMC, normalized for protein content, were analyzed by immunoblotting with an anti-human DOCK8 antibody. A dominant band of about 180 KDa (arrowhead) and several smaller isoforms were detected in control and family members samples but not in those of the probands. B. Proliferative responses of PBMC to anti-CD3 mAb treatment (n=2-5/group; *p=0.02). C. DOCK8 deficiency impairs the activation of both CD4+ and CD8+ T cells. PBMC were loaded with carboxy-fluorescein succinimidyl ester (CFSE) and stimulated with anti-CD3 + anti-CD28 mAb for 3 days. Gated populations of mAb-stimulated CD3+, CD4+ and CD8+ T cells were analyzed for CFSE fluorescence intensity (solid lines), and compared to the respective unstimulated cell population (shaded area).
We next analyzed T cell proliferative responses in probands with documented DOCK8 defects as compared to those of control subjects. Results revealed a profound defect in anti-CD3 mAb induced lymphoproliferation in the probands when compared to control, as measured by 3H-thymidine incorporation into proliferating cells (Figure 6b). Similarly, proband T cells that had been loaded with the chromophore precursor CFSE prior to stimulation with anti-CD3+anti-CD28 mAbs failed to dilute their immunofluorescence signal. Such a dilution is a marker of cell proliferation and was readily detected in similarly treated control T cells (Figure 6c). The proliferative defect affected both the CD4+ and CD8+ subsets, consistent with a prominent role for DOCK8 in T cell activation.
The clinical phenotype of DOCK8 deficiency (see Table 1)
All but two patients with mutations in DOCK8 had upper respiratory tract infections and all but three had recurrent pneumonia on as many as 20 occasions. Four patients developed bronchiectasis. 17/21 patients had skin abscesses and 17/21 patients had a severe susceptibility to recurrent and partially mutilating viral infections mainly caused by Herpes simplex virus or Molluscum contagiosum. Candidiasis was also very prevalent with 17/21 patients affected. A very severe form of atopic dermatitis, often colonized with Staphylococcus aureus, was seen in all 21 patients. 7/21 patients suffered from asthma and 10/21 patients had polyvalent food allergies with six patients having additionally environmental allergies. Cerebral features associated with DOCK8 mutations were CNS vasculitis (2 patients), brain infarction (1 patient), meningitis (4 patients), and JC virus-associated progressive multifocal leucoencephalopathy (2 patients). One patient had a Burkitt lymphoma and one patient had autoimmune hemolytic anemia.
DOCK8 deficiency was associated with elevated serum IgE (range 2,830-90,910 IU/ml; median 20,225 IU/ml; normal <100 IU/ml) and eosinophilia (range 290-37,880 cells/μl; median 5,675 cells/μl). The remainder of the differential blood count was normal in most of the patients. Total T cell numbers were within the normal range in 8/13 patients (62%) and decreased in 5/13 patients (38%). CD4+ T cells were decreased in 7/13 patients (54%), whereas CD8+ T cells were decreased in 3/12 patients (25%) and within the normal range in 7/12 (58%). In contrast to the data reported by Zhang et al.11, B cells were within the normal range in 6/13 patients (46%) and increased in 7/13 patients (54%). NK cells were within the normal range in most (11/12, 92%) patients. IgG serum levels were within the normal range in 11/13 (85%) of patients with mutations in DOCK8. 8/13 patients (62%) had normal IgA. In contrast, 10/13 (77%) of patients had decreased levels of IgM.
DISCUSSION
In 20 families with 27 patients with AR-HIES, a total of 21 patients had biallelic deletions or intragenic small mutations involving DOCK8. Four subjects harbored large homozygous deletions, one had a large compound heterozygous deletion, seven had exonic deletions and nine suffered homozygous point mutations predicted to impair DOCK8 protein expression or function.
The fact that we were not able to demonstrate exonic mutations in the remaining six AR-HIES families may be due to difficulties in detecting slight changes, e.g. compound heterozygous mutations, in a 48 exon gene. Moreover, mutations may lie in intronic regions or in the regulatory regions of the gene. We will pursue this by in depth analysis of patients’ cDNA and protein expression.
By genomic sequence analysis, Griggs et al. had mapped DOCK8 to chromosome 9p2413. They determined that DOCK8 contains 47 exons spanning 190 kb; more recently, however, the Ensembl database (www.ensembl.org) provides evidence for ten different transcripts and 48 exons. Northern blot analysis detected DOCK8 expression in human placenta, lung, kidney, and pancreas and to a lesser degree in brain, heart, and skeletal muscle14.
DOCK8 is one of 11 members of the dedicator of cytokinesis (DOCK) protein family15. DOCK8 interacts with GTP- and GDP-bound forms of Ccd42 and Rac1, and Cdc42 family members TCL and TC10 in a yeast 2-hybrid assay14. The protein contains DOCK Homology Region-1 (DHR-1, or CZH1) and DHR-2 (CZH2) domains, characteristic of DOCK180-related proteins. In some DOCK180-related family members, these domains bind phospholipid and carry out guanine nucleotide exchange factor (GEF) function, respectively15,16. Although the GEF activity of DOCK8 remains to be determined, the high DHR-2 domain homology of the DOCK-C subfamily suggests that like orthologs DOCK6 and DOCK7, DOCK8 may be a GEF specific for Cdc42 and/or Rac117 (Fig.7).
Figure 7.

Hypothetical function of DOCK8. DOCK8 is likely to function as a guanine-nucleotide exchange factor (GEF) for the Rho-GTPases Cdc42 and Rac1, turning them into the active, GTP-bound form upon receptor engagement (e.g. receptor tyrosine kinases, antigen receptors and adhesion receptors). An unknown protein possibly stabilizes the interaction of DOCK8 with Cdc42 and Rac1. GTPase activation induces dynamic filamentous actin rearrangements and lamellipodia formation, leading to cell growth, migration and adhesion. Given the clinical phenotype of the AR-HIES patients with DOCK8 deficiency, we propose an important role of DOCK8 in T-cell actin dynamics, which might be important for the formation of the immunological synapse, leading to T cell activation, proliferation, and differentiation.
DOCK family proteins play roles in regulation of cell migration, morphology, adhesion and growth. Immunofluorescence localized transiently transfected and endogenous DOCK8 to the cytoplasm at cell edges forming lamellipodia, which increased when cells were treated with PDGF or fetal calf serum. Transient transfection of a C-terminal fragment of DOCK8 containing the DHR-2 domain resulted in formation of vesicular structures, suggesting that DOCK8 may play a role in the organization of filamentous actin14 (Fig.7).
Patients with AR-HIES have atopic eczema, severe susceptibility to viral infections, and deficient TH17 cell function12. Our present studies demonstrate that DOCK8 deficiency impairs CD4+ and CD8+ T cell proliferative responses, consistent with a critical role for this protein in T cell activation and effector functions. It can be speculated, based on the protein domain structure, that DOCK8 may fulfill an important cytoskeletal function relevant to T cell activation, such as in the formation of the immunological synapse. Other immune deficiencies, such as the Wiskott-Aldrich syndrome and severe combined immunodeficiency due to lack of Coronin-1A, also involve deficient function of important cytoskeleton-regulating proteins 18,19.
The mechanisms by which abnormal T cell effector functions develop in DOCK8-deficient patients, including impaired TH17 differentiation, remains to be established. However, a role for DOCK8 in the maintenance of memory TH17 cells can be inferred, as it was that component of the TH17 response that appeared most affected in DOCK8-deficient subjects12. It is conceivable that TH17 deficiency in AR-HIES maybe symptomatic of a more widespread derangement of TH cell differentiation, an eventuality that is currently under investigation. The presence of atopic dermatitis in AR-HIES suggests the action of immune dysregulatory mechanisms, although an additional contribution from a defective barrier function may also be involved. Overall, we propose that DOCK8 may play a critical role in cytoskeletal organization, and that its deficiency might result in impaired T cell activation and effector responses.
Two unrelated patients have been previously described with mental retardation and developmental disability associated with heterozygous disruption of DOCK8 by deletion and by a translocation breakpoint, respectively. Immunologic abnormalities were not described in these patients. Mapping of the critical region shared by the two patients showed truncation of the longest isoform of DOCK8. However, these patients had single copy deletions of DOCK8 along with disruptions of other genes13,20, which may explain why their phenotype differed from those of our patients.
After the initial submission of our work, Zhang and colleagues published homozygous and compound heterozygous mutations in DOCK8 in a cohort of twelve patients from eight families11. One of their families (#8 in Zhang et al.) is family ARH011 in our manuscript. They described the phenotype as a “combined T- B- NK-immunodeficiency” and presented evidence of decreased T, B and NK cell numbers. Our assessment of these published cases would suggest that they also suffer from an AR form of HIES, given that the major diagnostic criteria of elevated IgE, eosinophilia, recurrent pneumonia and skin eczema together with a susceptibility to viral infections were fulfilled in all but one patient. However, and in contrast to the findings by Zhang et al.11, studies on the cohort presented herein demonstrate a selective decrease in CD4+ T cell numbers in many but not all patients. The CD8+ T cell population is less affected, while B and NK cells are usually within the normal range. Furthermore, whereas Zhang et al. found a selective defect in CD8+ T cell activation, our findings are consistent with a more comprehensive T cell activation defect involving both the CD4+ and CD8+ T cell subsets. Such a global defect in T cell activation may explain the severe clinical phenotype of AR-HIES.
DOCK8 deficiency is characterized by recurrent infections of the upper and lower respiratory tract, susceptibility to severe, recurrent and mutilating viral infections (especially by Molluscum contagiosum and Herpes viruses), a severe dermatitis that resembles atopic dermatitis and may often be superinfected, elevated IgE levels and eosinophilia. Other clinical features such as asthma, allergies, CNS symptoms, or autoimmune phenomena are variably associated. Whether there is a genotype-phenotype correlation with regard to the size of the deletion or whether or not other deleted genes next to DOCK8 contribute to the phenotype needs to be determined in future studies. It is remarkable that the typical feature of cyst-forming pneumonia (pneumatoceles), as frequently seen in STAT3-mutated HIES, is not typical for DOCK8 deficiency and can be used as a discriminating feature.
Subtelomeric deletions often arise at sites of interspersed repetitive genomic sequences such as Alu, LINE, long terminal repeats, and simple tandem repeats21,22. The region surrounding DOCK8 is particularly rich in these sequences, with 264 such sequences in the region from the p terminus to the end of DOCK8 (Alu database). In particular, there are Alu-Jb sequences near the breakpoints of our patients as mapped by microarray analysis. Abnormal recombination or transposable element activity could contribute to the high frequency of deletions seen. Careful analysis of the breakpoints of these deletions will be helpful in delineating which functions of DOCK8 are affected by the deletions, potenitally shedding light on the role of DOCK8 in immune and integument function.
CLINICAL IMPLICATIONS.
Homozygous mutations in DOCK8 were identified in most patients with autosomal recessive hyper-IgE syndrome. Hence patients with a phenotype of elevated IgE, eosinophilia, and recurrent skin boils, pneumonia, and viral infections (especially molluscum contagiosum and herpes) should be suspected of having mutations in DOCK8.
CAPSULE SUMMARY.
Twenty-one of 27 patients with the autosomal recessive hyper-IgE syndrome had homozygous deletions or point mutations in DOCK8. After STAT3, DOCK8 mutations represent the second most frequent cause of hyper-IgE syndromes.
ACKNOWLEDGEMENTS
We thank Cindy Ng and Anupama Rambhatla for technical support, Jennifer Birmelin for collecting healthy control samples, Erik Glocker for sequencing controls, Alejandro A. Schäffer and Michael Gertz for help with homozygosity mapping, and Othmar Engelhardt for critical reading of the manuscript. This work was supported by National Institutes of Health grants 5R01AI065617 and 1R21AI087627 to T. A. C. and by the EU Marie-Curie grant MEXT-CT-2006-042316 and the European Community’s 7th Framework Programme FP7/2007-2013 grant EURO-PADnet HEALTH-F2-2008-201549 to B.G.
ABBREVIATIONS
- HIES
hyper-IgE syndrome
- AR
autosomal recessive
- AD
autosomal dominant
- DOCK8
dedicator of cytokinesis 8
- SNP
single nucleotide polymorphism
- CNVs
copy number variations
- LINE
long interspersed elements
- ROMA
representational oligonucleotide microarray analysis
- DHR-1
DOCK homology region-1
- DHR-2
DOCK homology region-2
- CNS
central nervous system
- bp
base pairs
- cDNA
complementary DNA
- TH17 cells
T-helper 17 cells
- IgE
immunoglobulin E
- GEF
guanine nucleotide exchange factor
- PBMC
peripheral blood mononuclear cell
- FCS
fetal calf serum
- CFSE
carboxyfluorescein succinimidyl ester
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1).Grimbacher B, Holland SM, Gallin JI, Greenberg F, Hill SC, Malech HL, et al. Hyper-IgE syndrome with recurrent infections--an autosomal dominant multisystem disorder. N Engl J Med. 1999;340:692–702. doi: 10.1056/NEJM199903043400904. [DOI] [PubMed] [Google Scholar]
- 2).Grimbacher B, Holland SM, Puck JM. Hyper-IgE syndromes. Immunol Rev. 2005;203:244–50. doi: 10.1111/j.0105-2896.2005.00228.x. [DOI] [PubMed] [Google Scholar]
- 3).Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med. 2007;357:1608–19. doi: 10.1056/NEJMoa073687. [DOI] [PubMed] [Google Scholar]
- 4).Grimbacher B, Schaffer AA, Holland SM, Davis J, Gallin JI, Malech HL, et al. Genetic linkage of hyper-IgE syndrome to chromosome 4. Am J Hum Genet. 1999;65:735–44. doi: 10.1086/302547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5).Woellner C, Gertz EM, Schäffer AA, Lagos M, Perro M, Glocker EO, et al. Mutations in the signal transducer and activator of transcription 3 (STAT3) and diagnostic guidelines for the Hyper-IgE Syndrome. J Allergy Clin Immunol. 2009 doi: 10.1016/j.jaci.2009.10.059. accepted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).Renner ED, Puck JM, Holland SM, Schmitt M, Weiss M, Frosch M, et al. Autosomal recessive hyperimmunoglobulin E syndrome: a distinct disease entity. J Pediatr. 2004;144:93–9. doi: 10.1016/S0022-3476(03)00449-9. [DOI] [PubMed] [Google Scholar]
- 7).Minegishi Y, Saito M, Morio T, Watanabe K, Agematsu K, Tsuchiya S, et al. Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity. 2006;25:745–55. doi: 10.1016/j.immuni.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 8).Woellner C, Schaffer A, Puck JM, Renner ED, Knebel C, Holland SM, et al. The hyper IgE syndrome and mutations in TYK2. Immunity. 2007;26:535. doi: 10.1016/j.immuni.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 9).Kristiansson K, Naukkarinen J, Peltonen L. Isolated populations and complex disease gene identification. Genome Biol. 2008;9:109. doi: 10.1186/gb-2008-9-8-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10).Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donohoe PK, Qi Y, et al. Detection of large-scale variation in the human genome. Nat Genet. 2004;36:949–51. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 11).Zhang Q, Davis JC, Lamborn IT, Freeman AF, Jing H, Favreau AJ, et al. Combined Immunodeficiency Associated with DOCK8 Mutations. N Engl J Med. 2009 Sep 23; doi: 10.1056/NEJMoa0905506. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12).Al Khatib S, Keles S, Garcia-Lloret M, Karakoc-Aydiner E, Reisli I, Artac H, et al. Defects along the T(H)17 differentiation pathway underlie genetically distinct forms of the hyper IgE syndrome. J Allergy Clin Immunol. 2009;124:342–8. doi: 10.1016/j.jaci.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13).Griggs BL, Ladd S, Saul RA, DuPont BR, Srivastava AK. Dedicator of cytokinesis 8 is disrupted in two patients with mental retardation and developmental disabilities. Genomics. 2008;91:195–202. doi: 10.1016/j.ygeno.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14).Ruusala A, Aspenstrom P. Isolation and characterisation of DOCK8, a member of the DOCK180-related regulators of cell morphology. FEBS Lett. 2004;572:159–66. doi: 10.1016/j.febslet.2004.06.095. [DOI] [PubMed] [Google Scholar]
- 15).Cote JF, Vuori K. Identification of an evolutionarily conserved superfamily of DOCK180-related proteins with guanine nucleotide exchange. J Cell Sci. 2002;115:4901–13. doi: 10.1242/jcs.00219. [DOI] [PubMed] [Google Scholar]
- 16).Yang J, Zhang Z, Roe SM, Marshall CJ, Barford D. Activation of Rho GTPases by DOCK exchange factors is mediated by a nucleotide sensor. Science. 2009;325:1398–402. doi: 10.1126/science.1174468. [DOI] [PubMed] [Google Scholar]
- 17).Miyamoto Y, Yamauchi J, Sanbe A, Tanoue A. Dock6, a Dock-C subfamily guanine nucleotide exchanger, has the dual specificity for Rac1 and Cdc42 and regulates neurite outgrowth. Exp Cell Res. 2007;313:791–804. doi: 10.1016/j.yexcr.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 18).Bouma G, Burns SO, Thrasher AJ. Wiskott-Aldrich Syndrome : Immunodeficiency resulting from defective cell migration and impaired immunostimulatory activation. Immunobiology. 2009;214:778–90. doi: 10.1016/j.imbio.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19).Shiow LR, Roadcap DW, Paris K, Watson SR, Grigrova IL, Lebet T, et al. The actin regulator coronin 1A is mutant in a thymic egress–deficient mouse strain and in a patient with severe combined immunodeficiency. Nature Immunol. 2008;9:1307–15. doi: 10.1038/ni.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20).MacDermot KD, Hulten M. Female with hypohidrotic ectodermal dysplasia and de novo (X;9) translocation: clinical documentation of the AnLy cell line case. Hum Genet. 1990;84:577–9. doi: 10.1007/BF00210814. [DOI] [PubMed] [Google Scholar]
- 21).Stankiewicz P, Lupski JR. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002;18:74–82. doi: 10.1016/s0168-9525(02)02592-1. [DOI] [PubMed] [Google Scholar]
- 22).Yatsenko SA, Brundage EK, Roney EK, Cheung SW, Chinault AC, Lupski JR. Molecular mechanisms for subtelomeric rearrangements associated with the 9q34.3 microdeletion syndrome. Hum Mol Genet. 2009;18:1924–1936. doi: 10.1093/hmg/ddp114. [DOI] [PMC free article] [PubMed] [Google Scholar]