

Abstract
The ideal cryopreservation protocol would combine the benefits of slow freezing with the benefits of vitrification. This report describes a method for the ultra-rapid vitrification of oocytes using slush nitrogen in quartz capillaries. The approach minimizes the thermal mass of the vitrification vessel by using open microcapillaries made of highly conductive quartz and achieves cooling rates of 250,000°C/min. The process of vitrification can be optimized by maximizing the rate at which the sample is cooled, which allows for the use of lower cryoprotectant concentrations. Mouse oocytes can be successfully vitrified using 1.5 mol/l propane-1,2-diol and 0.5 mol/l trehalose and achieve survival rates of 92.5%. Fertilization and blastocyst formation rates of vitrified–warmed and fresh oocytes were not significantly different. A total of 120 blastocysts from each of the vitrified–warmed and fresh oocytes were transferred to surrogate mothers and 23 and 27 offspring were born respectively. All offspring in both groups were healthy, grew and bred normally and gave rise to a second generation of pups. Thus, an ultra-rapid vitrification technique has been developed for mouse oocytes that uses low concentrations of cryoprotectants and slush nitrogen in quartz capillaries, which combines the benefits of slow freezing and vitrification.
Keywords: cryopreservation, oocyte, quartz capillary, slush nitrogen, ultra-rapid, vitrification
Introduction
Assisted reproduction technology has surpassed three decades of successful cryopreservation of mouse embryos (Whittingham et al., 1972) and over two decades of success with human embryos (summarized by Ludwig et al., 1999). The ability to successfully store and bank human embryos and male gametes has revolutionized human assisted reproduction. While the cryopreservation of human embryos has become a routine part of the treatment of infertility and is essential for safe and efficient treatment, advances in the cryopreservation of oocytes have been more limited. Oocyte cryopreservation is one of the most sought after advances in assisted reproductive technologies and has been described as the ‘holy grail’ of reproductive medicine. There have been over 900 verified live births that have resulted from worldwide efforts in oocyte cryopreservation over the last 20 years (Noyes et al., 2009). Most of those births have occurred over the last few years due to a tremendous increase in interest to develop oocyte banks. Success rates with current protocols are not yet high enough to be considered ‘standard of care’ in a clinical setting; thus, clinical oocyte cryopreservation protocols are still judged to be ‘experimental’ (American Society of Reproductive Medicine/Society for Assisted Reproductive Technology, 2008).
Traditional applications of oocyte cryopreservation include: (i) the preservation of future fertility in women at risk of losing their reproductive functions due to cancer treatment (Meirow, 2000) and (ii) the avoidance of the ethical and legal dilemmas surrounding the cryopreservation and long-term storage of human embryos (Ragni et al., 2005). More recent time has witnessed the emergence of a new group of women interested in preserving their oocytes. There has been an increasing social trend among women to delay initiating pregnancy. This delay in initiating pregnancy not only results in higher rates of infertility, but also exposes women and infants to the greater risks of genetic abnormalities associated with advancing maternal age. Thus, oocyte cryopreservation has emerged as a clinical approach to ensure future healthy pregnancies and offspring.
There are currently two approaches to achieving cryopreservation of oocytes: the conventional slow-freezing approach, which uses a low cooling rate of less than 2°C/min, and vitrification, which is a rapid cooling approach that uses cooling rates of over 1000°C/min. With either technique, cryoprotective agents are used. With slow freezing, the cryoprotective agents are used in low concentrations (1–2 mol/l) and are generally non-toxic to the cells. However, the challenge is that slow freezing is associated with injury due to ice formation (Mazur, 1984; Mazur et al., 1984). Oocytes are also sensitive to chilling injury and are damaged when cooled slowly with standard slow-freeze methods (Bernard and Fuller, 1996; Martino et al., 1996; Eroglu et al., 1998). On the other hand, vitrification involves the solidification of a supercooled liquid while maintaining the absence of ice, such that a glass is formed without any deleterious effects of ice formation (Fahy et al., 1984). However, to achieve vitrification, rapid cooling rates and very high concentrations of cocktails of cryoprotective agents (4–8 mol/l) are often required. These high concentrations are toxic to most cells (Fahy et al., 1984; Elmoazzen et al., 2007) and, as a result, multiple steps are required to load and unload the cryoprotective agents and extremely short exposure times to these high concentrations are needed, all of which make the process complicated and difficult to control. The ideal cryopreservation protocol, then, would be one that combines the benefits of conventional slow freezing (i.e., reduced toxicity secondary to low cryoprotective agent concentrations) with the benefits of vitrification (i.e., absence of intracellular ice crystal formation).
There is a relationship between the sample volume, cooling rate and the critical cryoprotectant concentration needed to vitrify. The smaller the sample volume and the higher the cooling rate, the lower the concentration needed to vitrify (Kuwayama et al., 2005; Berejnov et al., 2006; Risco et al., 2007). A number of innovations have been used to increase the cooling rate for vitrification. A minimal volume of cryoprotectant containing the oocyte is exposed directly to liquid nitrogen. Several devices have been developed such as a thin open straw in which the oocyte carrier is a narrow plastic tube and the cryoprotectant is loaded into the open end (Vajta et al., 1998). This method has since been modified to the Cryotop (Kuwayama et al., 2005). Similarly, electron microscopy grids have been used (Martino et al., 1996) which have since been modified to the Cryoloop (Lane et al., 1999). The cooling rates for these devices are in the range of 2000–30,000°C/min and warming rates are around 40,000°C/min (Kuwayama et al., 2005; Kuwayama, 2007). A method and device for oocyte cryopreservation using quartz capillary (QC) vitrification is under development by the authors. The process of vitrification was optimized by maximizing the rate at which the sample cools, which allows for the use of lower cryoprotectant concentrations. The cooling rate needed to achieve ‘ice-free’ conditions directly determines the required concentration of cryoprotectant.
It is of great interest to develop an approach to achieve vitrification of mammalian cells using a low non-toxic concentration of cryoprotectants, which combines the advantages of the existing slow-freezing and vitrification approaches while avoiding their shortcomings. Theoretically, this can be done by ultra-fast cooling (>100,000°C/min) of mammalian cells to a vitrified/glassy state at cryogenic temperatures. This glassy state can be induced in most liquids if cooling occurs rapidly enough and even pure water can be vitrified at cooling rates in the order of 108°C/min (Johari et al., 1987). A technique for mammalian cell cryopreservation that takes advantage of the unique properties of quartz crystal capillaries has been pioneered (Risco et al., 2007; He et al., 2008). The small dimensions of the QC compared with other cooling devices and the extremely high thermal conductivity of quartz (8 W/m/K) compared with other materials including traditionally used plastics (0.2 W/m/K), allows for significantly higher cooling rates. As such, this technique has been shown to be significantly more efficient than conventional vitrification techniques. The thin-walled QC is transparent and has an inner diameter comparable to that of a human oocyte (about 150 μm). These studies have also described a convenient way for determining non-vitrification as the appearance of opacity (or visible ice formation) when solutions are cooled below their freezing point. If there is no observable opacity, it is called ‘apparent vitrification’. Opacity is very difficult to determine for the quartz capillaries due to the small dimension. Therefore, a previously described experimental setup (He et al., 2008) was designed for visualization of opacity in the quartz microcapillary during cooling by directing two focused lights generated from two fibre optic lamps on the capillary held against a dark background. This setup was used to determine the threshold (minimum) cryoprotectant concentration required to completely avoid opacity during cooling of various cryorpotectant solutions. Previous work has also examined the thermal histories of the quartz capillaries when filled with a 1.5 mol/l propane-1,2-diol (PrOH) and 0.3 mol/l sucrose cryoprotectant solution and plunged into slush nitrogen. The thermal history revealed no indication of a freezing or melting process. When the sample was inspected visually after quenching, it appeared transparent, indicating that ‘apparent vitrification’ was achieved. Previous results revealed that the combination of slush nitrogen and quartz capillaries raised the cooling rate to 250,000 ± 30,000°C/min, between 20°C and −150°C. The QC technique seeks to bridge the gap between slow freezing and vitrification.
This study was designed to develop an ultra-rapid vitrification technique for oocytes using quartz capillaries. Two different cryoprotectant solutions were compared and tested as well as the use of liquid compared with slush nitrogen. The optimal protocol was then tested with mouse oocytes.
Materials and methods
Animals
Six-week-old B6CBF1 female mice, 10-week-old male B6CBF1 mice and 8-week-old CD1 female mice were purchased from Charles River Laboratories (Boston, MA, USA). All animal experiments were carried out with the approval of the animal care and use committee at Massachusetts General Hospital.
Retrieval of oocytes
The 6-week-old B6CBF1 female mice were superovulated with 7.5 IU of pregnant mare serum gonadotrophin (Sigma–Aldrich, St Louis, MO, USA) and 7.5 IU of human chorionic gonadotrophin (Sigma–Aldrich) given by intraperitoneal injections 48 h apart. Fourteen hours after human chorionic gonadotrophin injection, females were anaesthetized with avertin (Sigma–Aldrich) then killed by cervical dislocation and their oviducts were removed. The cumulus–oocyte–complex was released from the ampullary region of each oviduct by puncturing the oviduct with a 27-gauge needle. Cumulus cells were removed by exposure to hyaluronidase (80 IU/ml) (Irvine Scientific, Santa Ana, CA, USA) for 3 min and washed three times with human tubal fluid (HTF) medium (Irvine Scientific) with 10% fetal bovine serum (FBS; Gibco, Carlsbad, CA, USA). Oocytes were transferred and cultured in HTF medium (Quinn et al., 1995) containing 10% FBS at 37°C and 5% CO2 in air until they were vitrified.
Cryopreservation and warming
All cryoprotectant solutions were filter-sterilized and stored at 4°C prior to use. The base cryoprotectant solutions used were PrOH (Sigma–Aldrich) and trehalose (Ferro Pfanstiehl Laboratory, Waukegan, IL, USA). The concentrations and types of cryoprotectants used were similar to those used in traditional slow-freezing protocols for human oocytes (Gook et al., 2007) and are not typically vitrifiable. For vitrification, isolated mature oocytes were placed into HTF medium supplemented with 10% FBS. Two vitrification protocols using a HEPES-buffered physiological salt solution (FHM; Specialty Media, Lavallette, NJ, USA) (Lawitts and Biggers, 1993) as a base medium were tested. In the first protocol, the oocytes were equilibrated in a 30 μl droplet of 0.75 mol/l PrOH in FHM20 (FHM medium containing 20% FBS) for 5 min, then transferred to a 30 μl droplet of 0.5 mol/l trehalose + 1.5 mol/l PrOH in FHM20 for 5 min. In the second protocol, oocytes were equilibrated in a 30 μl droplet of 0.75 mol/l PrOH in FHM20 for 5 min, then equilibrated in a 30 μl droplet of 1.5 mol/l PrOH in FHM20 for 5 min before being transferred to a 0.5 mol/l trehalose 2.0 mol/l PrOH in FHM20, for 5 min. The QC straws (The Charles Supper Company, USA) used for vitrification have an outer and inner diameter of approximately 0.2 mm and 0.18 mm, respectively, and a wall thickness of 0.01 mm (He et al., 2008). Figure 1 shows an image of the QC vitrification device. After exposure to the vitrification solution, the oocytes were then loaded into the 200 μm QC and plunged into liquid or slush nitrogen for comparison. For warming, the vitrified oocytes were immediately immersed in a holding medium containing 0.2 mol/l trehalose in 1x phosphate-buffered saline (PBS; Gibco) at 37°C for 1 minute. The oocytes were then expelled in a 30 μl droplet of 0.5 mol/l trehalose in FHM20 for 5 min, then transferred to 0.25 mol/l trehalose containing 20% FBS for 5 min, followed by transfer to 0.125 mol/l trehalose containing 20% FBS for 5 min. The oocytes were washed three times in FHM20 and transferred into HTF with 0.4% bovine serum albumin (BSA; Sigma–Aldrich). The oocytes were then cultured in an incubator at 37°C and with 5% CO2 in air.
Figure 1.
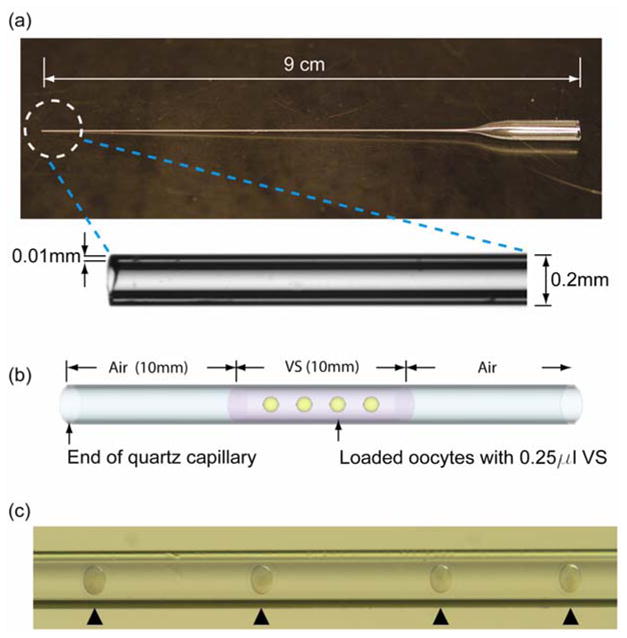
Quartz capillary (QC) vitrification device and oocyte loading method. (a) The QC device; (b) schematic illustration of the QC loaded with oocytes; and (c) photomicrograph of oocytes loaded into QC and loaded oocytes (arrowheads). VS = vitrification solution.
IVF and embryonic development
For IVF, spermatozoa were released from the caudal epididymis of 3-month-old males (B6D2F1) into HTF. The spermatozoa were then dispersed by incubating them in the protein-free medium at 37°C for 15 min and the sperm concentration was determined using a haemocytometer. To obtain a final concentration of 106 spermatozoa/ml, an appropriate volume of sperm suspension was transferred to each insemination drop and allowed to capacitate at 37°C for 1 h before insemination. Oocytes were introduced into the insemination drops containing capacitated spermatozoa. After incubation with spermatozoa for 6 h, oocytes were removed from the insemination drops, washed once in HTF medium containing 0.4% BSA and then transferred to potassium simplex optimized medium with amino acids (KSOM; Specialty Media) (Biggers et al., 2000) containing 0.4% BSA for further culture. Both IVF and subsequent culture of embryos were performed in the modified KSOM medium at 37°C under a humidified atmosphere of 5% CO2 in air. Fertilization was assessed by the presence of male and female pronuclei as well as cleavage to the two-cell stage at 24 h after sperm insemination. Development was then followed through to the blastocyst stage for 72 h.
Blastocyst assessment
Embryos from non-treated and vitrified–warmed groups were stained using the Hoechst nuclear staining for cell numbers (Handyside and Hunter, 1984). Briefly, blastocysts were incubated for 1 min in 1% sodium citrate until the cells swelled visibly. They were moved to a clean slide, and the nuclei of blastocysts were fixed and spread with a drop of 3:1 ethanol:acetic acid. After air-drying, the nuclei were labelled with Hoechst stain and mounted in a medium for the detection of fluorescence. The number of cells in the blastocysts were counted with a fluorescence microscope using ultraviolet excitation (TMD, Nikon, Japan).
Embryo transfer
Blastocysts from non-treated control oocytes and cryopreserved oocytes were transferred into uterine horns of day-3.5 pseudopregnant CD1 females that were obtained by mating them to vasectomized CD1 males. The pseudopregnant CD1 females were anaesthetized by an intraperitoneal injection of avertin (0.4 mg/g body weight), and 10 embryos were transferred to each female. Embryo transfer into oviducts and uterine horns was performed using the method described by Nacy et al. (1993). First-generation pups were allowed to grow to sexual maturity and second-generation pups were produced by cross-mating of the first-generation pups. The litter sizes were compared in untreated and cryopreserved groups. Pups behaviours of both the first and second generations were observed and their fertility was assessed up to the second generation.
Slush nitrogen
Slush nitrogen was made using a vacuum chamber and pump or with the VitMaster (IMT International Ltd, UK). For the vacuum chamber and pump system, a styrofoam container holding 750 ml of liquid nitrogen was placed inside the vacuum chamber. The vacuum pressure in the chamber was then reduced to 6500 Pa. The nitrogen was left at this pressure for 15 min and then removed. As a result, two-phased slush nitrogen was obtained. Liquid nitrogen has a temperature of −196°C. The slush nitrogen has a temperature of roughly −205°C to −210°C. To maximize the effect of slush nitrogen, the slush nitrogen was used within 5 min of removal from the vacuum chamber.
Statistical analysis
The 2×2 and 4×2 contingency tables were analysed using Fisher’s exact test. In those experiments consisting of more than one replicate, the homogeneity of the replicates was assessed with the Cochran–Mantel–Haenszel test. In all cases, the replicates could be assumed to be homogeneous and the data of all replicates pooled and analysed with Fisher’s exact test. The data on the number of cells in the blastocysts were analysed with a t-test. P <0.05 was assumed to be statistically significant. All analyses were performed using StatXact 8 (Cytel, Cambridge, MA, USA).
Results
The morphology of the oocytes before and after exposure to the vitrification solutions was examined. The morphology of the oocytes remained intact after equilibration in 0.75 mol/l PrOH in FHM20, as well as after exposure to both 0.5 mol/l trehalose + 1.5 mol/l PrOH in FHM20 and to 0.5 mol/l trehalose + 2.0 mol/l PrOH in FHM20 (data not shown).
The effects of vitrifying oocytes in liquid and slush nitrogen were examined in simultaneous tests with two different concentrations of PrOH (1.5 mol/l and 2.0 mol/l) + 0.5 mol/l trehalose in the final vitrification solution (Table 1). All of the oocytes that were loaded into the QC were recovered after vitrification. Oocyte survival was significantly higher when plunged into slush nitrogen compared with liquid nitrogen (P = 0.048). Further analyses showed no significant differences in the survival rates observed when either concentration of PrOH were used with liquid nitrogen and the oocyte survival rates in the 0.5 mol/l trehalose + 1.5 mol/l PrOH in FHM20 vitrification solution and in the 0.5 mol/l trehalose + 2.0 mol/l PrOH in FHM20 were 45.0% and 47.5%, respectively. The survival rates observed when slush nitrogen was used with 0.5 mol/l trehalose + 1.5 mol/l PrOH in FHM20 vitrification solution compared with slush nitrogen with 0.5 mol/l trehalose + 2.0 mol/l PrOH in FHM20 were 90.0% and 70.0%, respectively. The difference between the survival rates approached, but did not quite reach, statistical significance.
Table 1.
The effect of liquid and slush nitrogen on survival rates of mouse oocytes.
| Type of nitrogen | Concentration of PrOH (mol/l) | Surviving oocytes |
|---|---|---|
| Liquid | 1.5 | 18/40 (45.0) |
| Liquid | 2.0 | 19/40 (47.5) |
| Slush | 1.5 | 36/40 (90.0) |
| Slush | 2.0 | 28/40 (70.0) |
Values are number/total (%) unless otherwise stated.
PrOH = propane-1,2-diol.
P-values (Fisher’s exact test): overall table <0.001; liquid nitrogen = not statistically significant; slush nitrogen = 0.048.
The oocyte survival rate after vitrification in slush nitrogen in the two different vitrification solutions immediately after warming and 2 h after warming were assessed. Figure 2 shows the survival rates for the two vitrification solutions. Immediately after warming, the survival rates in the 0.5 mol/l trehalose + 1.5 mol/l PrOH in FHM20 vitrification solution and in the 0.5 mol/l trehalose + 2.0 mol/l PrOH in FHM20 were 92.5% and 82.5%, respectively. Two hours after warming, the survival rates in the 0.5 mol/l trehalose + 1.5 mol/l PrOH in FHM20 vitrification solution and in the 0.5 mol/l trehalose + 2.0 mol/l PrOH in FHM20 were 90.0% and 72.5%, respectively.
Figure 2.

Oocyte survival rate after vitrification in slush nitrogen in the two different vitrification solutions, 0.5 mol/l trehalose + 1.5 mol/l propane-1,2-diol (PrOH) in HEPES-buffered physiological salt solution containing 20% fetal bovine serum (FHM20) or 0.5 mol/l trehalose + 2.0 mol/l PrOH in FHM20, immediately and 2 h after warming.
This study was also interested in the effects of the concentration of PrOH in the vitrification medium on the rates of fertilization of mouse oocytes. The results are summarized in Table 2. Fertilization rates observed using the two concentrations of PrOH (1.5 mol/l or 2.0 mol/l) were not significantly different (75.0% versus 61.5%, respectively). However, the efficiency of the method was significantly greater when 1.5 mol/l PrOH was used compared with 2.0 mol/l PrOH (P = 0.024; 67.5% versus 40.0%, respectively).
Table 2.
The effect of propane-1,2-diol concentration on the rate and efficiency of fertilization of vitrified–warmed mouse oocytes.
| Concentration of PrOH (mol/l) | Fertilizationa | Efficiencyb |
|---|---|---|
| 1.5 | 27/36 (75.0) | 27/40 (67.5) |
| 2.0 | 16/26 (61.5) | 16/40 (40.0) |
| P-value | NS | 0.024 |
Fertilization = 100 × no. of pronucleate 2 cell/no. of recovered vitrified–warmed oocytes.
Efficiency = 100 × no. of pronucleate 2 cell/no. of vitrified–warmed oocytes.
NS = not statistically significant.
Based on the above results, the vitrification solution chosen for the subsequent experiments was 0.5 mol/l trehalose + 1.5 mol/l PrOH in FHM20 and plunging was performed in slush nitrogen. The survival rates, fertilization rates and blastocyst development rates of the oocytes after vitrification in 0.5 mol/l trehalose + 1.5 mol/l PrOH in FHM20 were compared with fresh controls. The survival rate of the vitrified–warmed oocytes was 90% compared with the survival rate of 100% of the fresh controls. Table 3 summarizes the fertilization rates and blastocyst formation rates of vitrified–warmed and fresh oocytes. The fertilization rate was 62.9% for the vitrified–warmed oocytes compared with 65.3% for the fresh controls while the blastocyst development rate was 59.1% for the vitrified–warmed oocytes compared with 66.7% for the fresh control. Overall, there was no statistically significant difference in survival rates, fertilization rates or development rates between the vitrified–warmed oocytes and the fresh controls.
Table 3.
A comparison of the rates of fertilization and blastocyst formation of vitrified–warmed and fresh mouse oocytes.
| Oocyte group | Fertilization | Blastocyst formation |
|---|---|---|
| Vitrified–warmed | 22/35 (62.9) | 13/22 (59.1) |
| Fresh | 27/40 (67.5) | 18/27 (66.7) |
Values are number/total (%).
There were no statistically significant differences.
Table 4 summarizes the number of cells in each blastocyst that were derived in vitro after oocyte vitrification compared with the fresh controls. There was no significant difference between the two groups when comparing the number of blastocysts derived and the number of cells of the blastocysts when stained with Hoechst stain. Table 5 summarizes the in-vitro development of the 2-cell embryos and the pups that were derived from the transferred embryos. Of the vitrified–warmed oocytes, 46% developed to the blastocyst stage as compared with 47% of fresh oocytes. A total of 120 blastocysts from each of the vitrified–warmed and fresh oocytes were transferred to surrogate mothers and 23 live offspring (19%) were born from vitrified–warmed oocytes and 27 pups (23%) born from fresh oocytes. All offspring in both oocyte groups were healthy and grew and bred normally and females from first generation gave rise to a second generation of healthy pups (Table 6).
Table 4.
Number of cells in blastocysts derived in vitro.
| Oocyte group | Blastocysts (n) | Cells (mean ± SE) |
|---|---|---|
| Vitrified–warmed | 20 | 78.70 ± 4.80 |
| Fresh | 20 | 81.15 ± 6.12 |
Table 5.
In-vitro development of embryos and offspring derived from blastocysts that were developed using vitrified–warmed or fresh oocytes and then transferred to surrogate mothers.
| Oocyte group | Oocytes inseminated | 2-Cell embryos | Blastocysts | Recipients | Blastocysts transferred | Recipients delivered | Live offspring |
|---|---|---|---|---|---|---|---|
| Vitrified–warmed | 300 | 171 (57) | 137 (46) | 12 | 120 | 6 (50) | 23 (19) |
| Fresh | 300 | 191 (64) | 142 (47) | 12 | 120 | 7 (58) | 27 (23) |
Values are number or number (%).
Table 6.
Second-generation offspring born from vitrified–warmed or fresh oocytes.
| Oocyte group | First-generation females | Pregnancies | Deliveries | Offspring |
|---|---|---|---|---|
| Vitrified–warmed | 10 | 10 (100) | 10 (100) | 72 (7.2 ± 0.36) |
| Fresh | 10 | 10 (100) | 10 (100) | 75 (7.5 ± 0.36) |
Values are number, number (%) or number (mean ± SE).
Discussion
It is well known that one of the main obstacles to vitrification is the deleterious effects of high cryoprotectant concentrations, which are often necessary to induce the glassy vitrified state (Fahy et al., 1984). Consequently, one of the most promising approaches for oocyte vitrification in order to obtain improved survival is to minimize osmotic injury and cryoprotectant toxicity by reducing the cryoprotectant concentration (Kuwayama et al., 2005). An ultra-rapid vitrification technique for the preservation of mammalian cells has been developed using quartz capillaries and slush nitrogen. The concentrations of cryoprotectants used in this vitrification study are similar to those used in standard slow-freeze preservation protocols (Porcu et al., 1997; Tucker et al., 1998; Chen et al., 2003; Fosas et al., 2003; Gook et al., 2007).
The critical cooling rate to achieve vitrification is a function of the cryoprotectant concentration. Previous work has demonstrated that in order for a 1.8 mol/l solution of cryoprotectant to vitrify, cooling rates in the order of 105–106 °C/min are required. In a study by Risco et al. (2007), the cooling rate for a 1.8 mol/l cryoprotectant solution in the quartz capillaries and slush nitrogen was measured to be approximately 250,000 °C/min and was in the range necessary to vitrify the cryopreservation solution. The ultra-rapid vitrification technique has overcome some of the main issues encountered using traditional vitrification protocols, including high cryoptrotectant concentrations, multiple loading steps and the need for short exposure times (seconds) to these high concentrations, all of which make traditional vitrification processes complicated and difficult to control.
Recently, the feasibility of the ultra-rapid vitrification technique using quartz capillaries and low concentrations of cryoprotectants was demonstrated with mouse embryonic stem cells. In the study by He et al. (2008), they showed that using 2 mol/l PrOH and 0.5 mol/l extracellular trehalose was sufficient for vitrification in liquid nitrogen. The proliferation or growth of the attached embryonic stem cells post vitrification was very similar to the proliferation of the control non-vitrified samples.
Alterations in the cryogenic liquid can also increase the desired cooling rates (Martino et al., 1996; Yoon et al., 2007). A previous study demonstrated the capability of QC, plunged into a two-phase slush nitrogen and compared it with the thermal performance of the conventional open pulled straw (OPS) (Risco et al., 2007). Significantly, higher cooling and warming rates were achieved with the QC. The use of QC in slush nitrogen increases the cooling rate one order of magnitude over other approaches developed to reach high cooling rates for cell vitrification (Risco et al., 2007). The QC approach minimizes the thermal mass of the vitrification vessel by using microcapillaries made of highly conductive quartz and achieves cooling rates of 250,000°C/min (Risco et al., 2007). Similarly, there were advantages of warming QC from slush nitrogen that took into account the importance of achieving high warming rates to avoid devitrification and recrystallization (Risco et al., 2007). In the current study, significantly higher oocyte survival was observed when cells were plunged into slush nitrogen compared with liquid nitrogen. Slush nitrogen has been used in the past to reduce the insulating vapour layer that forms on the surface of the sample when plunged from room temperature into liquid nitrogen (Leiden frost effect) (Steponkus et al., 1990). Liquid nitrogen has a temperature of −196°C while slush nitrogen has a temperature of roughly −205°C to −210°C. However, the main benefit of quenching with the slush over liquid nitrogen does not come from this temperature difference, but from a reduction of vapourization when submitted to relatively high temperatures (Luyet, 1960; Sjostrand and Elfvin, 1964; Mazur et al., 1993). This reduction of vapourization exposes the sample to a more direct contact with the cryogenic media and, in this way, increases the cooling rate. Slush nitrogen has been used in the past for the cryopreservation of embryos (Yavin et al., 2009) and oocytes (Martino et al., 1996; Isachenko et al., 2001; Yoon et al., 2007). In a recent study by Yoon et al. (2007), they reported high survival of human oocytes when using slush nitrogen and electron microscope grids. In these studies, the vitrification solution used was 5.5 mol/l ethylene glycol and 1.0 mol/l sucrose.
In summary, an ultra-rapid vitrification technique has have been developed for mouse oocytes using low cryoprotectant concentrations and slush nitrogen. The concentrations of cryoprotectants used in the protocols are similar to those used in traditional oocyte slow-freeze protocols. The fertilization rates and blastocyst formation rates of vitrified–warmed oocytes using a vitrification media of 1.5 mol/l PrOH in FHM20 and 0.5 mol/l trehalose were not significantly different compared with fresh controls. It was found that 46% of the vitrified–warmed oocytes developed to the blastocyst stage as compared with 47% of fresh oocytes. A total of 120 oocytes from each of the vitrified–warmed and fresh oocytes were transferred to surrogate mothers and 23 live offspring were born from vitrified–warmed oocytes and 27 pups born from fresh oocytes. All offspring were healthy, grew and bred normally and gave rise to a second generation of healthy pups. This study has demonstrated the feasibility of using the quartz capillary technique for the ultra-rapid vitrification of oocytes and, as far as is known, this is the first report of oocyte vitrification using slow-freeze cryoprotectant concentrations. The quartz capillary system appears to be a novel ultra-rapid vitrification technique for mouse oocytes that combines the benefits of slow freezing and vitrification.
Acknowledgments
The authors would like to thank Gloria Lee and Dr Kaisa Selesniemi for technical assistance, Dr Jonathan Tilly for guidance of this project and critical reading and Dr Isaac Schiff for his vision and personal support. This study was partially funded by National Institutes of Health and a special thanks are due to Susan and George Domolky for providing additional financial support.
Biography

Ho-Joon Lee has been working in the area of reproductive biology and medicine since 1985, beginning with a human IVF program and as a director of the IVF lab in Cheil General Hospital, Seoul, Korea. He was awarded a PhD in Hanyang University, Seoul, Korea, 1993. He developed co-culture and embryo cryopreservation techniques to improve pregnancy rates. He joined the Vincent Center for Reproductive Biology in Massachusetts General Hospital in 2003, where he is currently working as an investigator. His current interests are oocyte and embryo preservation and he focuses on ovarian stem cells in mammal as well as fertility preservation.
Footnotes
Declaration: Dr J Biggers is a consultant to IVFonline LLC, Guelph, Canada. The other authors report no financial or commercial conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- American Society for Reproductive Medicine, Practice Committee of Society for Assisted Reproductive Technology. Ovarian tissue and oocyte cryopreservation. Fertility and Sterility. 2008;90 (5 Suppl):S241–246. doi: 10.1016/j.fertnstert.2008.08.039. [DOI] [PubMed] [Google Scholar]
- Berejnov V, Husseini NS, Alsaied OA, Thorne RE. Effects of cryoprotectant concentration and cooling rate on vitrification of aqueous solutions. Journal of Applied Crystallography. 2006;39:244–251. [Google Scholar]
- Bernard A, Fuller BJ. Cryopreservation of human oocytes: A review of current problems and perspectives. Human Reproduction Update. 1996;2:193–207. doi: 10.1093/humupd/2.3.193. [DOI] [PubMed] [Google Scholar]
- Biggers JD, McGinnis LK, Raffin M. Amino acids and preimplantation development of the mouse in protein-free potassium simplex optimized medium. Biology of Reproduction. 2000;63:281–293. doi: 10.1095/biolreprod63.1.281. [DOI] [PubMed] [Google Scholar]
- Chen SU, Lien YR, Chao KHHo, et al. Effects of cryopreservation on meiotic spindles of oocytes and its dynamics after thawing: clinical implications in oocyte freezing–a review article. Molecular and Cellular Endocrinology. 2003;202:101–107. doi: 10.1016/s0303-7207(03)00070-4. [DOI] [PubMed] [Google Scholar]
- Elmoazzen HY, Poovadan A, Law GK, et al. Dimethyl sulfoxide toxicity kinetics in intact porcine articular cartilage. Cell and Tissue Banking. 2007;6:125–133. doi: 10.1007/s10561-006-9023-y. [DOI] [PubMed] [Google Scholar]
- Eroglu A, Toth TL, Toner M. Alterations of the cytoskeleton and polyploidy induced by cryopreservation of metaphase II mouse oocytes. Fertility and Sterility. 1998;69:944–957. doi: 10.1016/s0015-0282(98)00030-2. [DOI] [PubMed] [Google Scholar]
- Fahy GM, Macfarlane DR, Angell CA, Meryman HT. Vitrification as an approach to cryopreservation. Cryobiology. 1984;21:407–426. doi: 10.1016/0011-2240(84)90079-8. [DOI] [PubMed] [Google Scholar]
- Fosas N, Marina F, Torres PJ, et al. The births of five Spanish babies from cryopreserved donated oocytes. Human Reproduction. 2003;18:1417–1421. doi: 10.1093/humrep/deg297. [DOI] [PubMed] [Google Scholar]
- Gook DA, Edgar DH. Human oocyte cryopreservation. Human Reproduction Update. 2007;13:591–605. doi: 10.1093/humupd/dmm028. [DOI] [PubMed] [Google Scholar]
- Handyside AH, Hunter S. A rapid procedure for visualizing the inner cell mass and trophectoderm nuclei of mouse blastocysts in situ using polynucleotide-specific fluorochromes. Journal of Experimental Zoology. 1984;231:429–434. doi: 10.1002/jez.1402310317. [DOI] [PubMed] [Google Scholar]
- He XM, Park EY, Fowler A, et al. Vitrification by ultra-fast cooling at a low concentration of cryoprotectants in a quartz micro-capillary: a study using murine embryonic stem cells. Cryobiology. 2008;56:223–232. doi: 10.1016/j.cryobiol.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isachenko V, Alabart JL, Nawroth F, et al. The open pulled straw vitrification of ovine GV-oocytes: positive effect of rapid cooling or rapid thawing or both? Cryo-Letters. 2001;22:157–162. [PubMed] [Google Scholar]
- Johari GP, Hallbrucker A, Mayer E. The glass liquid transition of hyperquenched water. Nature. 1987;330:552–553. [Google Scholar]
- Kuwayama M. Highly efficient vitrification for cryopreservation of human oocytes and embryos: the Cryotop method. Theriogenology. 2007;67:73–80. doi: 10.1016/j.theriogenology.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Kuwayama M, Vajta G, Kato O, Leibo SP. Highly efficient vitrification method for cryopreservation of human oocytes. Reproductive BioMedicine Online. 2005;11:300–308. doi: 10.1016/s1472-6483(10)60837-1. [DOI] [PubMed] [Google Scholar]
- Lane M, Bavister BD, Lyons EA, Forest KT. Containerless vitrification of mammalian oocytes and embryos–Adapting a proven method for flash-cooling protein crystals to the cryopreservation of live cells. Nature Biotechnology. 1999;17:1234–1236. doi: 10.1038/70795. [DOI] [PubMed] [Google Scholar]
- Lawitts JA, Biggers JD. Culture of preimplantation embryos. Methods in Enzymology. 1993;225:153–164. doi: 10.1016/0076-6879(93)25012-q. [DOI] [PubMed] [Google Scholar]
- Ludwig M, Al-Hasani S, Felberbaum R, Diedrich K. New aspects of cryopreservation of oocytes and embryos in assisted reproduction and future perspectives. Human Reproduction. 1999;14:162–185. doi: 10.1093/humrep/14.suppl_1.162. [DOI] [PubMed] [Google Scholar]
- Luyet B. On Various Phase transitions occurring in aqueous solutions at low temperatures. Annals of the New York Academy of Sciences. 1960;85:549–569. doi: 10.1111/j.1749-6632.1960.tb49982.x. [DOI] [PubMed] [Google Scholar]
- Martino A, Songsasen N, Leibo SP. Development into blastocysts of bovine oocytes cryopreserved by ultra-rapid cooling. Biology of Reproduction. 1996;54:1059–1069. doi: 10.1095/biolreprod54.5.1059. [DOI] [PubMed] [Google Scholar]
- Mazur P. Freezing of living cells – mechanisms and implications. American Journal of Physiology. 1984;247:C125–C142. doi: 10.1152/ajpcell.1984.247.3.C125. [DOI] [PubMed] [Google Scholar]
- Mazur P, Cole KW, Schreuders PD, Mahowald AP. Contributions of cooling and warming rate and developmental stage to the survival of Drosophila embryos cooled to −205 degrees. Cryobiology. 1993;30:45–73. doi: 10.1006/cryo.1993.1006. [DOI] [PubMed] [Google Scholar]
- Mazur P, Rall WF, Leibo SP. Kinetics of water-loss and the likelihood of intracellular freezing in mouse ova. Influence of the method of calculating the temperature-dependence of water permeability. Cell Biophysics. 1984;6:197–213. doi: 10.1007/BF02788619. [DOI] [PubMed] [Google Scholar]
- Meirow D. Reproduction post-chemotherapy in young cancer patients. Molecular and Cellular Endocrinology. 2000;169:123–131. doi: 10.1016/s0303-7207(00)00365-8. [DOI] [PubMed] [Google Scholar]
- Nagy A, Rossant J, Nagy R, et al. Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:8424–8428. doi: 10.1073/pnas.90.18.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noyes N, Porcu E, Borini A. Over 900 oocyte cryopreservation babies born with no apparent increase in congenital anomalies. Reproductive BioMedicine Online. 2009;18:769–776. doi: 10.1016/s1472-6483(10)60025-9. [DOI] [PubMed] [Google Scholar]
- Porcu E, Fabbri R, Seracchioli R, et al. Birth of a healthy female after intracytoplasmic sperm injection of cryopreserved human oocytes. Fertility and Sterility. 1997;68:724–726. doi: 10.1016/s0015-0282(97)00268-9. [DOI] [PubMed] [Google Scholar]
- Quinn P, Moinipanah R, Steinberg JM, Weathersbee PS. Successful human in-vitro fertilization using a modified human tubal fluid medium lacking glucose and phosphate ions. Fertility and Sterility. 1995;63:922–924. doi: 10.1016/s0015-0282(16)57504-9. [DOI] [PubMed] [Google Scholar]
- Ragni G, Allegra A, Anserini P, et al. The 2004 Italian legislation regulating assisted reproduction technology: a multicentre survey on the results of IVF cycles. Human Reproduction. 2005;20:2224–2228. doi: 10.1093/humrep/dei011. [DOI] [PubMed] [Google Scholar]
- Risco R, Elmoazzen H, Doughty M, et al. Thermal performance of quartz capillaries for vitrification. Cryobiology. 2007;55:222–229. doi: 10.1016/j.cryobiol.2007.08.006. [DOI] [PubMed] [Google Scholar]
- Sjostrand FS, Elfvin LG. Granular structure of mitochondrial membranes and of cytomembranes as demonstrated in frozen-dried tissue. Journal of Ultrastructure Research. 1964;10:263. doi: 10.1016/s0022-5320(64)80009-5. [DOI] [PubMed] [Google Scholar]
- Steponkus PL, Myers SP, Lynch DV, et al. Cryopreservation of Drosophila melanogaster embryos. Nature. 1990;345:170–172. doi: 10.1038/345170a0. [DOI] [PubMed] [Google Scholar]
- Tucker MJ, Wright G, Morton PC, Massey JB. Birth after cryopreservation of immature oocytes with subsequent in-vitro maturation. Fertility and Sterility. 1998;70:578–579. doi: 10.1016/s0015-0282(98)00205-2. [DOI] [PubMed] [Google Scholar]
- Vajta G, Holm P, Kuwayama M, et al. Open pulled straw (OPS) vitrification: a new way to reduce cryoinjuries of bovine ova and embryos. Molecular Reproduction and Development. 1998;51:53–58. doi: 10.1002/(SICI)1098-2795(199809)51:1<53::AID-MRD6>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Whittingham DG, Leibo SP, Mazur P. Survival of mouse embryos frozen to −196 degrees and −269 degrees C. Science. 1972;178:411. [PubMed] [Google Scholar]
- Yavin S, Aroyo A, Roth Z, Arav A. Embryo cryopreservation in the presence of low concentration of vitrification solution with sealed pulled straws in liquid nitrogen slush. Human Reproduction. 2009;24:797–804. doi: 10.1093/humrep/den397. [DOI] [PubMed] [Google Scholar]
- Yoon TK, Lee DR, Cha SK, et al. Survival rate of human oocytes and pregnancy outcome after vitrification using slush nitrogen in assisted reproductive technologies. Fertility and Sterility. 2007;88:952–956. doi: 10.1016/j.fertnstert.2006.12.071. [DOI] [PubMed] [Google Scholar]