

Abstract
The neuraminidase inhibitors oseltamivir and zanamivi are used to treat H5N1 influenza. However, oseltamivir-resistant H5N1 viruses have been isolated from oseltamivir-treated patients. Moreover, reassortment between H5N1 viruses and oseltamvir-resistant human H1N1 viruses currently circulating could create oseltamivir-resistant H5N1 viruses, rendering the oseltamivir stockpile obsolete. Therefore, there is a need for unique and effective antivirals to combat H5N1 influenza viruses. The investigational drug T-705 (favipiravir; 6-fluoro-3-hydroxy-2-pyrazinecarboxamide) has antiviral activity against seasonal influenza viruses and a mouse-adapted H5N1 influenza virus derived from a benign duck virus. However, its efficacy against highly pathogenic H5N1 viruses, which are substantially more virulent, remains unclear. Here, we demonstrate that T-705 effectively protects mice from lethal infection with oseltamivir-sensitive or -resistant highly pathogenic H5N1 viruses. Furthermore, our biochemical analysis suggests that T-705 ribofuranosyl triphosphate, an active form of T-705, acts like purines or purine nucleosides in human cells and does not inhibit human DNA synthesis. We conclude that T-705 shows promise as a therapeutic agent for the treatment of highly pathogenic H5N1 influenza patients.
Keywords: antiviral compounds, murine lethal-infection model, neuraminidase
To treat influenza, which causes respiratory disease with significant morbidity and mortality especially among the elderly and young (1), the M2 ion channel blockers (aminoadamantanes: amantadine and rimantadine) have been used for more than two decades (2, 3). However, aminoadamantane-resistant viruses, which readily arise (4, 5), limit the efficacy of these drugs. Most currently-circulating human H1N1 and H3N2 viruses, and some H5N1 viruses, are aminoadamantane-resistant (6). The neuraminidase inhibitors, oseltamivir and zanamivir, are efficacious for the treatment of patients infected with seasonal influenza viruses, including aminoadamantane-resistant variants (7). Oseltamvir is an oral drug, whereas zanamivir is inhaled; therefore, oseltamivir is the more extensively used. Although the prevalence of viruses resistant to these drugs is lower than that for the aminoadamantanes (7, 8), a higher proportion of oseltamivir-resistant viruses has been detected among treated patients than was expected (9), and person-to-person transmission of oseltamivir-resistant influenza B viruses has occurred (10). Furthermore, oseltamivir-resistant H1N1 viruses emerged during the 2007–2008 season (11), and currently most H1N1 viruses are oseltamivir-resistant (12, 13). Oseltamivir has been used to treat H5N1 influenza, with some patients recovering, but others died and shed oseltamivir-resistant virus despite early initiation of the drug (14, 15) (see also www.emro.who.int/csr/media/pdf/ai_press_22_01_07.pdf). Clearly, alternative antiviral agents that are effective against oseltamivir-resistant viruses are urgently needed.
Favipiravir (6-fluoro-3-hydroxy-2-pyrazinecarboxamide), known as T-705, has potent inhibitory activity against influenza A, B, and C viruses in vitro and in vivo (16–21). Favipiravir negatively affects the synthesis of influenza viral RNA, but not cellular DNA or RNA (17). Sidwell et al. (19) demonstrated that T-705 is effective against a mouse-adapted H5N1 virus, A/duck/Minnesota/1525/81, which is a benign duck virus. Authentic “highly pathogenic” H5N1 viruses, however, are substantially more virulent. In fact, the efficacy of oseltamivir against highly pathogenic H5N1 viruses in animal models was limited, with substantial lethality even if the drug was initiated within 1 h of infection (22). Therefore, animal studies with highly pathogenic H5N1 viruses are essential for the evaluation of T-705 for the treatment of H5N1 virus infection.
Here, we assessed the efficacy of T-705 against highly pathogenic H5N1 viruses and their oseltamivir-resistant mutants in a murine lethal-infection model. The mechanism of action of T-705 in human cells was also evaluated.
Results
T-705RTP Production in Human Cells.
T-705 ribofuranosyl triphosphate (T-705RTP) is a T-705 intracellular metabolite that inhibits influenza viral RNA synthesis in Madin-Darby canine kidney (MDCK) cells (17, 21). To clarify the mechanism of action of T-705 in humans, we treated human lung-derived A549 cells with T-705 in the absence (Fig. S1A) or presence (Fig. S1 B–D) of excess nucleoside. Even though the effect of thymidine on intracellular T-705RTP production was limited (Fig. S1B), purine nucleosides (adenosine or guanosine) severely decreased T-705RTP production (Fig. S1 C and D). Because the activity of the cellular nucleotide-kinase should mediate T-705RTP production (17) and should be balanced by the concentration of substrates (e.g., nucleoside) and products (e.g., nucleotide), these results indicate that T-705RTP behaves as a pseudopurine or a pseudopurine nucleoside or nucleotide in human cells, as has been shown in canine cells (17).
Effect of T-705RTP on Human DNA Polymerase Activity.
Furuta et al. (17) demonstrated that T-705 does not inhibit cellular DNA synthesis in MDCK cells. To evaluate the effect of T-705 on DNA synthesis in humans, we compared inhibition rates of T-705RTP and Ribavirin 5′-triphosphate (Ribavirin TP), an active form of the widely used broad-spectrum antiviral ribonucleoside Ribavirin (23), against the activity of human DNA polymerase α, β, and γ in vitro (Table S1). Inhibition of human DNA polymerase α and β by T-705RTP (inhibition rates at 1 mM: −9.12 to 13.5%) was appreciably lower than that by Ribavirin TP (inhibition rates at 1 mM: 50.8 to 65.0%). These results suggest that T-705 administrated to humans would not severely affect cellular DNA synthesis.
Efficacy of T-705 Against Highly Pathogenic H5N1 Influenza Viruses in Mice.
To assess the efficacy of T-705 against highly pathogenic H5N1 influenza viruses in vivo, we used a murine lethal-infection model and the highly pathogenic H5N1 influenza viruses A/Vietnam/UT3040/04 (VN3040) and A/Hanoi/30408/05 clone7 (HN30408cl7), which were isolated from deceased (24) and hospitalized (15) patients, respectively. Fifty-percent mouse lethal doses (MLD50) of VN3040 and HN30408cl7 were 0.25 and 1.3 pfu, respectively, indicating that these two viruses are highly pathogenic in mice. BALB/c mice intranasally infected with 10 MLD50 of virus were orally administered T-705 twice daily for 5 or 8 days, beginning 1 h postinfection. We also treated a group of infected mice with oseltamivir phosphate (GS4104), whose efficacy against the highly pathogenic H5N1 influenza virus A/Vietnam/1203/04 has been reported (25). Mouse survival was monitored daily for 21 days (Fig. 1). The effect of both drugs on survival was similar for the VN3040- and HN30408cl7-infected mice. The number of surviving animals increased with T-705 administration in a dose-dependent manner. Five days of T-705 at 300 mg/kg per day was almost as effective as GS4104 at 50 mg/kg per day. However, with the 8-day regimen, we observed enhanced efficacy, particularly in the T-705-treated groups. All mice that received T-705 at 300 mg/kg per day survived and were asymptomatic throughout the entire monitoring period. T-705 at 100 mg/kg per day also showed substantial efficacy with a high survival rate. Based on these results, the 8-day regimen was chosen for subsequent assessment.
Fig. 1.

Efficacy of T-705 and GS4104 against highly pathogenic H5N1 influenza viruses in mice. Ten mice per group were intranasally infected with 10 MLD50 of VN3040 (A and B) or HN30408cl7 (C and D). Infected mice were orally administrated T-705 or GS4104 at the indicated doses or methylcellulose (control) twice daily for 5 (A and C) or 8 days (B and D), beginning 1 h postinfection. Green bars indicate the period of drug administration. Survival was monitored daily for 21 days.
To assess lung damage upon infection, we measured proinflammatory cytokines/chemokines in the lungs of VN3040-infected mice on days 3 and 6 postinfection and examined the lung pathology. The levels of most of the cytokines/chemokines induced by virus infection were reduced with T-705 administration in a dose-dependent manner (Fig. S2). Furthermore, pathological examination revealed that T-705 administration mitigated lung lesions in H5N1 virus-infected mice (Fig. 2 and Table S2). The control, untreated mice showed apparent tracheitis and bronchopneumonia (Fig. 2A); these lung lesions were detected in most of the lung lobes of all of the control mice. In the lungs of mice inoculated with 30 mg/kg per day of T705, we found tracheitis and bronchopneumonia in some lobes. Moreover, the affected areas were smaller than those in the untreated control mice (Fig. 2B). In the T705 100-mg group, two of the three mice did not develop bronchopneumonia at both 3 and 6 days postinfection. In the mice that did develop tracheitis and bronchopneumonia, the lesions were restricted to a few lobes (Fig. 2C). Furthermore, mice in the T705 300-mg group had no tracheal or lung lesions (Fig. 2D). Mice in the GS4104 group had lung lesions that were restricted to a small number of lung lobes of one of the three mice at 3 days postinfection and two of the three mice at 6 days postinfection (Fig. 2E).
Fig. 2.
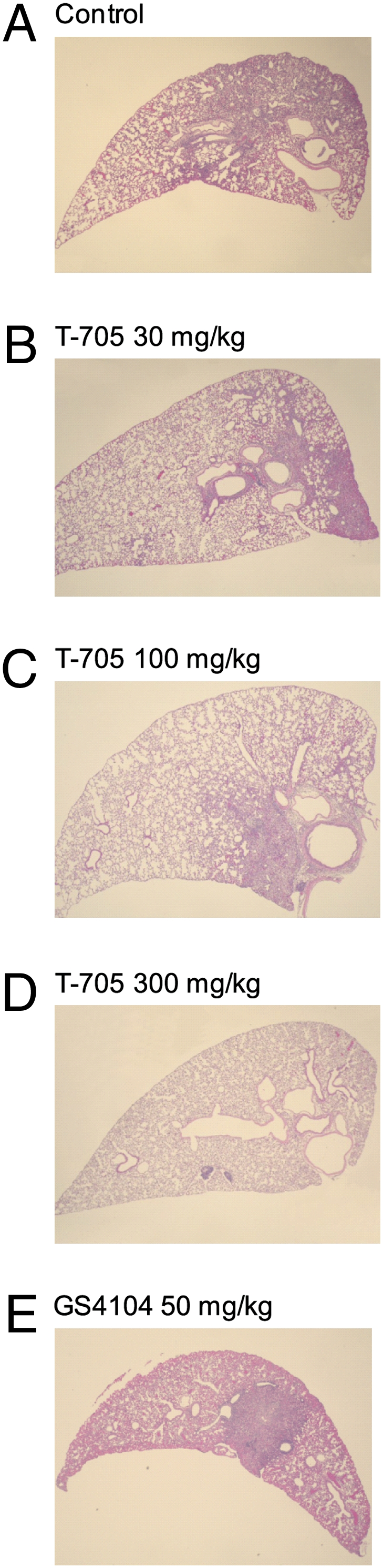
Pathological findings for the lungs of infected mice. Three mice per group were intranasally infected with 10 MLD50 of VN3040, and then orally administered methylcellulose (control) (A), T-705 (B–D), or GS4104 (E) at the indicated doses, twice daily for 6 days, beginning 1 h postinfection. Shown are representative histopathologic findings with H&E stain for the lungs of infected mice for each group. (A) In this section, two thirds of the area is affected with bronchopneumonia under microscopic examination. Similar lesions were observed in most of the lung lobes in this group. (B) In two bronchi (on Right) bronchitis, peribronchitis, and bronchopneumonia can be observed. These kinds of lesions can be seen scattered in several, but not all, of the lung lobes. (C) Some of the lung lobes have bronchopneumonia, which can be observed to the right-hand side of this section. (D) Mice in this group were free from lung lesions and had normal-looking lungs, as exemplified by this section. (E) Lesions of bronchopneumonia can be seen sparsely scattered in the lung lobes. (Magnification: x40.)
Efficacy of T-705 Against Oseltamivir-Resistant Viruses.
To assess the efficacy of T-705 against oseltamivir-resistant highly pathogenic H5N1 influenza viruses, we generated wild-type A/Vietnam/1203/04 (H5N1; VN1203) and its two variants, one possessing the histidine to tyrosine substitution at position 274 and the other the asparagine to serine substitution at position 294 in NA (VN1203-H274Y and -N294S), which were identified in oseltamivir-resistant H5N1 viruses (15). These viruses are highly virulent in mice (26, 27); the MLD50 of wild-type VN1203, VN1203-H274Y, and VN1203-N294S were 4.7, 2.1, and 4.7 pfu, respectively. Mice intranasally infected with 10 MLD50 of viruses were orally administered T-705 or GS4104 twice daily for 8 days, beginning 1 h postinfection. Regardless of the infected virus strain, the number of surviving animals increased with T-705 administration in a dose-dependent manner (Fig. 3). In contrast, GS4104 exhibited limited efficacy against oseltamivir-resistant mutants (Fig. 3 B and C). These results suggest that T-705 could be used to treat patients infected with oseltamivir-resistant highly pathogenic H5N1 influenza viruses.
Fig. 3.

Efficacy of T-705 and GS4104 against oseltamivir-resistant highly pathogenic H5N1 influenza viruses in mice. Ten mice per group were intranasally infected with 10 MLD50 of VN1203 (A), VN1203-H274Y (B), and VN1203-N294S (C). Infected mice were orally administrated T-705 or GS4104 at the indicated doses twice daily for 8 days, beginning 1 h postinfection. Green bars indicate the period of drug administration. Survival was monitored daily for 21 days.
Effect of Delayed Administration on T-705 Efficacy.
To evaluate the effect of delaying initial T-705 administration, VN3040-infected mice were treated with T-705 or GS4104 twice daily for 8 days, beginning 1, 24, 48, or 72 h postinfection. Mice were completely protected from the lethal virus infection by T-705 at 300 mg/kg per day, even when the drug was initiated 72 h postinfection. Under these conditions, GS4104 protected only 50% of infected mice at best (Fig. 4). Furthermore, T-705 administration reduced the level of body-weight loss experienced by the infected mice (Fig. 5). Throughout the course of these experiments, all mice that received T-705 at 300 mg/kg per day experienced transient body-weight loss.
Fig. 4.

Effect of delayed administration on the efficacy of T-705 and GS4104 against highly pathogenic H5N1 influenza viruses in mice. Eight mice per group were intranasally infected with 10 MLD50 of VN3040. Infected mice were orally administrated T-705 or GS4104 at the indicated doses twice daily for 8 days following virus infection, beginning 1 (A), 24 (B), 48 (C), or 72 (D) h postinfection. Control mice received methylcellulose, beginning 1 h postinfection. Green bars indicate the period of drug administration. Survival was monitored daily for 21 days.
Fig. 5.

Body weight changes in infected mice. Four mice per group were intranasally infected with 10 MLD50 of VN3040. Infected mice were orally administrated with T-705 or GS4104 at the indicated doses twice daily for 8 days following virus infection, beginning 1 (A), 24 (B), 48 (C), or 72 (D) h postinfection. Body weights were monitored daily. Green bars indicate the period of drug administration. The values are mean ± SDs for four mice.
On days 3, 6, and 9 postinfection, virus titers in lungs and brains of infected mice were determined (Table 1). The virus titers in lungs were reduced by T-705, particularly at the highest dose, whereas dose-dependency of GS4104 was not especially obvious. Furthermore, delaying the initial administration of drug led to the detection of viruses in the brains of both T-705- and GS4104-treated mice. These results indicate that T-705 provides reasonable efficacy against highly pathogenic H5N1 influenza viruses under conditions where oseltamivir shows limited efficacy.
Table 1.
Effect of delayed administration of drugs on their efficacy as measured by virus titers in the lungs and brains of infected mice
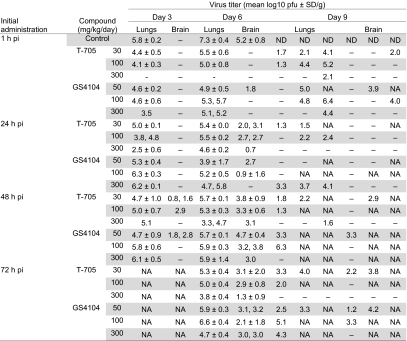 |
Ten mice per group were intranasally infected with 10 MLD50 of VN3040. Infected mice received oral T-705, GS4104, or methylcellulose (control) at the indicated doses beginning 1, 24, 48, or 72 h postinfection for 8 days after virus infection. Animals (three per group) were killed on days 3, 6, and 9 postinfection for virus titration. When virus was not recovered from all three mice on days 3 and 6, individual titers were recorded. The virus titers on day 9 postinfection are from individual animals because the other animals had died by this time point.
–, Virus not detected (detection limit: 1 log10 pfu/g); NA, not applicable (some animals died between days 6 and 9); ND, no data; pi, postinfection.
Discussion
In preparation for a highly pathogenic avian influenza H5N1 outbreak among humans, many countries have stockpiled oseltamivir. However, the isolation of oseltamivir-resistant H5N1 viruses from fatal or severe cases in Vietnam and Egypt (14, 15) (see also www.emro.who.int/csr/media/pdf/ai_press_22_01_07.pdf) necessitates the development of novel antivirals that are effective against oseltamivir-resistant viruses. In this study, we examined the mechanism of action of T-705 in vitro and its efficacy against highly pathogenic H5N1 viruses, including oseltamivir-resistant variants, in a murine lethal-infection model.
Our results suggest that T-705RTP, metabolized from T-705 via a cellular phosphorylation pathway, may act as a pseudopurine or a pseudopurine nucleoside or nucleotide in human cells (Fig. S1). Similar results have been obtained in canine cells (17). To date, the potent inhibitory effects of T-705 have been demonstrated against various RNA viruses, including H1N1, H2N2, and H3N2 influenza A virus, influenza B and C viruses, poliovirus, rhinovirus, and respiratory syncytial virus (16), West Nile virus (28), yellow fever virus (29), and arena virus (30) in cultured cells and mammalian animal models, but not against DNA viruses, such as herpes simplex virus type 1, human cytomegalovirus, and adenovirus (16). Although further mechanistic studies are required with these RNA viruses, it is likely that T-705 inhibits their RNA-dependent RNA polymerase activity (20).
T-705RTP showed only a mild effect on the activity of human DNA polymerase (Table S1), suggesting that any negative effect of T-705 in humans would be limited. In fact, uninfected-mice given T-705 at 300 mg/kg per day twice daily for 8 days gained body weight during the monitoring period and exhibited no signs of toxicity. This dosage provided reasonable protection against lethal H5N1 influenza viruses in mice, even when the drug was initiated 72 h postinfection (Figs. 1–5, Table 1, and Table S2). Therefore, we suggest this dosage should be the benchmark for dose escalation studies in higher mammals, such as ferrets and macaques.
The efficacy of T-705 against a recombinant A/Puerto Rico/8/34 (H1N1) oseltamivir-resistant virus was demonstrated previously (16). Although the MLD50 of A/Puerto Rico/8/34 is over 103 pfu, we confirmed that T-705 is also effective against oseltamivir-resistant highly pathogenic H5N1 influenza viruses whose MLD50 is less than 5 pfu (Fig. 3). Furthermore, T-705 exhibited marked efficacy, even when dosing was delayed for up to 72 h postinfecton (Figs. 4 and 5). By contrast, oseltamivir should be given within 36 to 48 h of onset of symptoms for adequate therapeutic effect (31), although, in our study, the protective efficacy against highly pathogenic H5N1 viruses did not substantially differ whether oseltamivir was administered 1 h or 72 h postinfecton. Although the most effective dosage in this study (300 mg/kg per day) seems to be much greater than the dosage applicable to human use, the viral dose used for challenge (10 MLD50) was also quite high. Furthermore, an optimal dosage in humans does not always reflect that in mice. The important point of our findings is that T-705, whose mechanism of action differs from those of currently-licensed M2 ion channel blockers and neuraminidase inhibitors, demonstrated considerable activity against highly pathogenic H5N1 influenza virus infection in a mouse model.
In conclusion, our results suggest that T-705 is safe and effective in combating H5N1 influenza viruses and is, thus, a promising candidate antiviral for the treatment of highly pathogenic H5N1 patients.
Materials and Methods
Viruses and Cells.
Influenza viruses A/Vietnam/UT3040/04 (H5N1; VN3040), which was isolated from the same specimen from which A/Vietnam/1203/04 (H5N1) (24) was isolated, and A/Hanoi/30408/05 clone 7 (H5N1; HN30408cl7), which was cloned as an oseltamivir-sensitive variant of A/Hanoi/30408/05 (H5N1) (15, 32), were propagated in MDCK cells. A/Vietnam/1203/04 (H5N1; VN1203) was generated by reverse genetics, as described below in human embryonic kidney 293T cells. MDCK cells were maintained in Eagle's MEM (MEM) supplemented with 5% newborn calf serum (Sigma). 293T cells and human lung adenocarcinoma A549 cells were maintained in DMEM (Sigma) supplemented with 10% FCS. All cells were cultured at 37°C in 5% CO2. All experiments with H5N1 viruses were performed in a biosafety level 3 containment laboratory at the University of Tokyo (Japan), which is approved for such use by the Ministry of Agriculture, Forestry, and Fisheries, Japan.
Compounds.
T-705, favipiravir (6-fluoro-3-hydroxy-2-pyrazinecarboxamide), T-705 ribofuranosyl triphosphate (T-705RTP), and oseltamivir phosphate (GS4104) were synthesized at Toyama Chemical Co., Ltd. Ribavirin 5′-triphosphate (Ribavirin TP) was purchased from Jena Bioscience GmbH. For animal experiments, T-705 and GS4104 were suspended in 0.5% methylcellulose and saline, respectively, and stored at 4°C until use.
Evaluation of Intracellular T-705RTP in T-705–Treated A549 Cells.
A549 cells were incubated with DMEM containing 1% BSA, 0.5 μg/mL trypsin and T-705 at 5 μM alone or in combination with 50-μM nucleosides at 37°C for 24 h. The medium was then removed and the cell layers were washed with ice-cold PBS (PBS). T-705RTP was extracted with 70% ice-cold methanol. The extract was warmed at 25°C for 1 h and then loaded onto a SAX column (Mega Bond Elut; GL Sciences Inc.). The fraction from the loaded column was eluted with 0.5 M triethylammonium hydrogen carbonate solution, dried, and stored at -40°C for further use. Samples were separated on an Inertsil ODS-3 column (4.6 × 100 mm; LC-20AD; SHIMADZU Corporation) with the mobile phase of acetonitrile/methanol/0.5 M potassium phosphate buffer (pH 6.0)/water/tetra-n-butylammonium bromide (70:90:100:800:3.22 vol/vol/vol/vol/w). The mobile phase was pumped through the column at 1.0 mL per min. The column eluate was monitored continuously using an FP-2025 fluorescence detector (JASCO Corporation) at an excitation wavelength of 370 nm and an emission wavelength of 445 nm. Under these conditions, the retention time for the pure standard of T-705RTP was 14.6 min.
Effect of T-705RTP and Ribavirin TP on Human DNA Polymerase Activity.
One unit each of human DNA polymerases α and β (CHIMERx) were incubated at 37°C for 1 h in 30 μl of 75 mM Tris-HCl buffer (pH 7.5) containing 6.5 mM MgCl2, 83 μM dATP, 83 μM dCTP, 83 μM dGTP, 1.67 mM 2-mercaptoethanol, 0.011 mg of activated calf thymus DNA/mL, 0.42 mg of BSA/mL, 40 mM KCl, 1 μM [methyl-3H]dTTP (including cold dTTP) and the test compounds. One unit of human DNA polymerase γ (CHIMERx) was incubated at 37°C for 1 h in 30 μL of 75 mM Tris-HCl buffer (pH 7.5) containing 6.5 mM MgCl2, 83 μM dATP, 83 μM dCTP, 83 μM dGTP, 1.67 mM 2-mercaptoethanol, 0.011 mg of activated calf thymus DNA/mL, 0.42 mg of BSA/mL, 50 mM KCl, 1 μM [methyl-3H]dTTP (including cold dTTP) and the test compounds. In the dGTP incorporation assay, dTTP and [methyl-3H]dGTP were used instead of dGTP and [methyl-3H]dTTP, respectively. Reactions were stopped by placing 20 μL of reaction mixture on a DE81 filter (Whatman Japan Ltd.) soaked in 500-mM EDTA. The papers were dried, washed three times for 10 min each with 5% Na2HPO4, and rinsed twice for 5 min each with distilled water and once with methanol. Air-dried filters were mixed with ≈10 mL of PCS Scintillation Mixture (GE Healthcare U.K. Ltd.) and radioactivity was measured using a liquid scintillation counter.
Reverse Genetics.
Plasmid construction for the expression of viral RNAs of VN1203 was described previously (27). Two amino acid substitutions that confer oseltamivir-resistance [histidine to tyrosine at position 274 and asparagine to serine at position 294 in NA (15)] were independently introduced into the plasmid for the VN1203 NA gene. Wild-type and mutant VN1203 were generated by reverse genetics in 293T cells as described previously (33). The transfectant viruses were propagated in MDCK cells twice and stored at −80°C for subsequent use. All constructed plasmids were sequenced to ensure that no unwanted mutations were introduced.
Drug Efficacy in Mice.
Female 6-week-old BALB/c mice (Japan SLC Inc.) (8–10 per group) were intranasally infected with 10 MLD50 of viruses, and then orally administered T-705 (10, 30, 100, or 300 mg/kg per day) or GS4104 (50, 100, or 300 mg/kg per day) twice daily for 5 or 8 days, beginning 1 h postinfection unless otherwise stated. Control mice orally received methylcellulose. Survival of infected mice was monitored daily for 21 days.
On days 3, 6, and 9 postinfection, mice were killed for virologic examinations. To determine virus titers in the lungs and brains of infected mice, each organ was homogenized in PBS containing antibiotics. After cellular debris was removed by centrifugation at 3,000 × g for 10 min, supernatants were subjected to plaque assays in MDCK cells. The research protocol for animal experiments was approved by the Animal Experiment Committee of the Institute of Medical Science, the University of Tokyo (approval number 19–29).
Cytokine and Chemokine Measurements.
Three mice per group were infected with 10 MLD50 of VN3040, and then orally administered T-705 (30, 100, or 300 mg/kg per day) or GS4104 (50 mg/kg per day) twice daily for 3 or 6 days, beginning 1 h postinfection. Control mice received methylcellulose orally. On days 3 and 6 postinfection, the lungs of infected mice were treated with the Bio-Plex Cell Lysis Kit (Bio-Rad Laboratories) according to the manufacturer's instructions. Concentrations of IFN-α and -β in mouse lung homogenates were determined by using the Mouse IFN Alpha or Beta ELISA Kit (Invitrogen). The concentrations of other cytokines/chemokines were determined by using the Bio-Plex Mouse Cytokine 23-Plex and 9-Plex panel (Bio-Rad Laboratories) and by an array analysis with the Bio-Plex Protein Array system (Bio-Rad Laboratories).
Pathological Examination.
Three mice per group were infected with 10 MLD50 of VN3040, and then orally administered T-705 (30, 100, or 300 mg/kg per day) or GS4104 (50 mg/kg per day) twice daily for 3 or 6 days, beginning 1 h postinfection. Control mice received methylcellulose orally. On days 3 and 6 postinfection, mice were killed, and lung tissues were fixed by perfusion of 10% phosphate-buffered formalin through the heart. Collected lung tissues were cut, dehydrated, and embedded in paraffin. Paraffin blocks were cut into sections, and stained with standard H&E.
Body-Weight Monitoring.
Four mice per group were infected with 10 MLD50 of VN3040, and then orally administered T-705 (30, 100, or 300 mg/kg per day) or GS4104 (50 mg/kg per day) twice daily for 8 days, beginning 1, 24, 48, or 72 h postinfection. Control mice received methylcellulose orally. Body weights of infected mice was monitored daily for 14 days.
Supplementary Material
Acknowledgments
We thank Susan Watson for editing this manuscript. This work was supported, in part, by Grants-in-Aid for Specially Promoted Research and for Scientific Research, by a Contract Research Fund for the Program of Founding Research Centers for Emerging and Reemerging Infectious Diseases, by Exploratory Research for Advanced Technology (Japan), by the Special Coordination Funds for Promoting Science and Technology from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, and by National Institute of Allergy and Infectious Diseases Public Health Service research grants. M.O. was partially supported by a Japan Society for the Promotion of Science Postdoctoral Fellowship for Research Abroad.
Footnotes
Conflict of interest statement: Y.K. has received speaker’s honoraria from Chugai Pharmaceuticals, Novartis, Daiichi-Sankyo Co., Ltd., Toyama Chemical, Wyeth, and GlaxoSmithKline; a grant of support from Chugai Pharmaceuticals, Daiichi Sankyo Co., Ltd. and Toyama Chemical; and is a consultant for Theraclone and is a founder of FluGen.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0909603107/DCSupplemental.
References
- 1.Simonsen L. The global impact of influenza on morbidity and mortality. Vaccine. 1999;17(Suppl 1):S3–S10. doi: 10.1016/s0264-410x(99)00099-7. [DOI] [PubMed] [Google Scholar]
- 2.Dolin R, et al. A controlled trial of amantadine and rimantadine in the prophylaxis of influenza A infection. N Engl J Med. 1982;307:580–584. doi: 10.1056/NEJM198209023071002. [DOI] [PubMed] [Google Scholar]
- 3.Hay AJ, Wolstenholme AJ, Skehel JJ, Smith MH. The molecular basis of the specific anti-influenza action of amantadine. EMBO J. 1985;4:3021–3024. doi: 10.1002/j.1460-2075.1985.tb04038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bright RA, Shay DK, Shu B, Cox NJ, Klimov AI. Adamantane resistance among influenza A viruses isolated early during the 2005–2006 influenza season in the United States. JAMA. 2006;295:891–894. doi: 10.1001/jama.295.8.joc60020. [DOI] [PubMed] [Google Scholar]
- 5.Shiraishi K, et al. High frequency of resistant viruses harboring different mutations in amantadine-treated children with influenza. J Infect Dis. 2003;188:57–61. doi: 10.1086/375799. [DOI] [PubMed] [Google Scholar]
- 6.He G, et al. Amantadine-resistance among H5N1 avian influenza viruses isolated in Northern China. Antiviral Res. 2008;77:72–76. doi: 10.1016/j.antiviral.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 7.Zambon M, Hayden FG, Global Neuraminidase Inhibitor Susceptibility Network Position statement: global neuraminidase inhibitor susceptibility network. Antiviral Res. 2001;49:147–156. doi: 10.1016/s0166-3542(01)00124-3. [DOI] [PubMed] [Google Scholar]
- 8.Varghese JN, Epa VC, Colman PM. Three-dimensional structure of the complex of 4-guanidino-Neu5Ac2en and influenza virus neuraminidase. Protein Sci. 1995;4:1081–1087. doi: 10.1002/pro.5560040606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kiso M, et al. Resistant influenza A viruses in children treated with oseltamivir: descriptive study. Lancet. 2004;364:759–765. doi: 10.1016/S0140-6736(04)16934-1. [DOI] [PubMed] [Google Scholar]
- 10.Hatakeyama S, et al. Emergence of influenza B viruses with reduced sensitivity to neuraminidase inhibitors. JAMA. 2007;297:1435–1442. doi: 10.1001/jama.297.13.1435. [DOI] [PubMed] [Google Scholar]
- 11.Hauge SH, Dudman S, Borgen K, Lackenby A, Hungnes O. Oseltamivir-resistant influenza viruses A (H1N1), Norway, 2007-08. Emerg Infect Dis. 2009;15:155–162. doi: 10.3201/eid1502.081031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nicoll A, Ciancio B, Kramarz P, Influenza Project Team Observed oseltamivir resistance in seasonal influenza viruses in Europe interpretation and potential implications. Euro Surveill. 2008;13:8025. doi: 10.2807/ese.13.05.08025-en. [DOI] [PubMed] [Google Scholar]
- 13.Lackenby A, Thompson CI, Democratis J. The potential impact of neuraminidase inhibitor resistant influenza. Curr Opin Infect Dis. 2008;21:626–638. doi: 10.1097/QCO.0b013e3283199797. [DOI] [PubMed] [Google Scholar]
- 14.de Jong MD, et al. Oseltamivir resistance during treatment of influenza A (H5N1) infection. N Engl J Med. 2005;353:2667–2672. doi: 10.1056/NEJMoa054512. [DOI] [PubMed] [Google Scholar]
- 15.Le QM, et al. Avian flu: isolation of drug-resistant H5N1 virus. Nature. 2005;437:1108. doi: 10.1038/4371108a. [DOI] [PubMed] [Google Scholar]
- 16.Furuta Y, et al. In vitro and in vivo activities of anti-influenza virus compound T-705. Antimicrob Agents Chemother. 2002;46:977–981. doi: 10.1128/AAC.46.4.977-981.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Furuta Y, et al. Mechanism of action of T-705 against influenza virus. Antimicrob Agents Chemother. 2005;49:981–986. doi: 10.1128/AAC.49.3.981-986.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takahashi K, et al. In vitro and in vivo activities of T-705 and oseltamivir against influenza virus. Antivir Chem Chemother. 2003;14:235–241. doi: 10.1177/095632020301400502. [DOI] [PubMed] [Google Scholar]
- 19.Sidwell RW, et al. Efficacy of orally administered T-705 on lethal avian influenza A (H5N1) virus infections in mice. Antimicrob Agents Chemother. 2007;51:845–851. doi: 10.1128/AAC.01051-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Furuta Y, et al. T-705 (favipiravir) and related compounds: Novel broad-spectrum inhibitors of RNA viral infections. Antiviral Res. 2009;82:95–102. doi: 10.1016/j.antiviral.2009.02.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smee DF, et al. Intracellular metabolism of favipiravir (T-705) in uninfected and influenza A (H5N1) virus-infected cells. J Antimicrob Chemother. 2009;64:741–746. doi: 10.1093/jac/dkp274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yun NE, et al. Injectable peramivir mitigates disease and promotes survival in ferrets and mice infected with the highly virulent influenza virus, A/Vietnam/1203/04 (H5N1) Virology. 2008;374:198–209. doi: 10.1016/j.virol.2007.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crotty S, et al. The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat Med. 2000;6:1375–1379. doi: 10.1038/82191. [DOI] [PubMed] [Google Scholar]
- 24.Maines TR, et al. Avian influenza (H5N1) viruses isolated from humans in Asia in 2004 exhibit increased virulence in mammals. J Virol. 2005;79:11788–11800. doi: 10.1128/JVI.79.18.11788-11800.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yen HL, Monto AS, Webster RG, Govorkova EA. Virulence may determine the necessary duration and dosage of oseltamivir treatment for highly pathogenic A/Vietnam/1203/04 influenza virus in mice. J Infect Dis. 2005;192:665–672. doi: 10.1086/432008. [DOI] [PubMed] [Google Scholar]
- 26.Yen HL, et al. Neuraminidase inhibitor-resistant recombinant A/Vietnam/1203/04 (H5N1) influenza viruses retain their replication efficiency and pathogenicity in vitro and in vivo. J Virol. 2007;81:12418–12426. doi: 10.1128/JVI.01067-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hatta M, et al. Growth of H5N1 influenza A viruses in the upper respiratory tracts of mice. PLoS Pathog. 2007;3:1374–1379. doi: 10.1371/journal.ppat.0030133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morrey JD, et al. Efficacy of orally administered T-705 pyrazine analog on lethal West Nile virus infection in rodents. Antiviral Res. 2008;80:377–379. doi: 10.1016/j.antiviral.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Julander JG, Shafer K, Smee DF, Morrey JD, Furuta Y. Activity of T-705 in a hamster model of yellow fever virus infection in comparison with that of a chemically related compound, T-1106. Antimicrob Agents Chemother. 2009;53:202–209. doi: 10.1128/AAC.01074-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gowen BB, et al. Treatment of late stage disease in a model of arenaviral hemorrhagic fever: T-705 efficacy and reduced toxicity suggests an alternative to ribavirin. PLoS One. 2008;3:e3725. doi: 10.1371/journal.pone.0003725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moscona A. Neuraminidase inhibitors for influenza. N Engl J Med. 2005;353:1363–1373. doi: 10.1056/NEJMra050740. [DOI] [PubMed] [Google Scholar]
- 32.Le QM, Sakai-Tagawa Y, Ozawa M, Ito M, Kawaoka Y. Selection of H5N1 influenza virus PB2 during replication in humans. J Virol. 2009;83:5278–5281. doi: 10.1128/JVI.00063-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neumann G, et al. Generation of influenza A viruses entirely from cloned cDNAs. Proc Natl Acad Sci USA. 1999;96:9345–9350. doi: 10.1073/pnas.96.16.9345. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.