

Abstract
Enzymatic catalysis of oxygenation reactions in the absence of metal or organic cofactors is a considerable biochemical challenge. The CO-forming 1-H-3-hydroxy-4-oxoquinaldine 2,4-dioxygenase (HOD) from Arthrobacter nitroguajacolicus Rü61a and 1-H-3-hydroxy-4-oxoquinoline 2,4-dioxygenase (QDO) from Pseudomonas putida 33/1 are homologous cofactor-independent dioxygenases involved in the breakdown of N-heteroaromatic compounds. To date, they are the only dioxygenases suggested to belong to the α/β-hydrolase fold superfamily. Members of this family typically catalyze hydrolytic processes rather than oxygenation reactions. We present here the crystal structures of both HOD and QDO in their native state as well as the structure of HOD in complex with its natural 1-H-3-hydroxy-4-oxoquinaldine substrate, its N-acetylanthranilate reaction product, and chloride as dioxygen mimic. HOD and QDO are structurally very similar. They possess a classical α/β-hydrolase fold core domain additionally equipped with a cap domain. Organic substrates bind in a preorganized active site with an orientation ideally suited for selective deprotonation of their hydroxyl group by a His/Asp charge-relay system affording the generation of electron-donating species. The “oxyanion hole” of the α/β-hydrolase fold, typically employed to stabilize the tetrahedral intermediate in ester hydrolysis reactions, is utilized here to host and control oxygen chemistry, which is proposed to involve a peroxide anion intermediate. Product release by proton back transfer from the catalytic histidine is driven by minimization of intramolecular charge repulsion. Structural and kinetic data suggest a nonnucleophilic general-base mechanism. Our analysis provides a framework to explain cofactor-independent dioxygenation within a protein architecture generally employed to catalyze hydrolytic reactions.
Keywords: oxygenase, oxygen chemistry, structural enzymology
Oxygenases operating on diverse substrates have been identified that require neither a metal nor an organic cofactor for catalysis (1, 2). These enzymes are mechanistically intriguing as they are able to activate the spin-forbidden reaction between dioxygen and their organic substrates with limited chemical tools at their disposal. Bacterial cofactor-devoid oxygenases play a crucial role in polyketides tailoring (3, 4) in the biogenesis of nonproteinogenic amino acids found in the vancomycin and teicoplanin families of antibiotics (5, 6), as well as in the degradation of N-heteroaromatic compounds (7).
Arthrobacter nitroguajacolicus Rü61a 1-H-3-hydroxy-4-oxoquinaldine 2,4-dioxygenase (HOD) and Pseudomonas putida 33/1 1-H-3-hydroxy-4-oxoquinoline 2,4-dioxygenase (QDO) were among the first cofactor-independent oxygenases discovered (7). They are approximately 31-kDa monomeric enzymes that catalyze an O2-dependent N-heteroaromatic ring cleavage reaction (Fig. 1A), chemically identical to that catalyzed by the metal-dependent quercetin dioxygenase, of which the copper-dependent form is the best characterized (8). Neither HOD nor QDO are related in sequence to either quercetin dioxygenase or other known oxygenases. However, they are 37% identical to each other, suggesting that they constitute a separate dioxygenase family of similar 3D structure.
Fig. 1.

(A) Scheme of the reaction catalyzed by the bacterial CO-forming cofactor-devoid dioxygenases. HOD catalyzes the conversion of QND (R = CH3) to NAA, whereas QDO catalyzes the conversion of 1-H-3-hydroxy-4-oxoquinoline (R = H, QNL) to N-formylanthranilate. In the dioxygenolytic reaction, the ring A of the N-heteroatomic substrate is disrupted with formation of carbon monoxide as by-product. (B) Cartoon representation of HOD with the α/β-hydrolase fold core domain and cap domain shown in light gray and dark gray, respectively. Residues of the catalytic triad are shown in ball-and-stick representation. Secondary structure elements are labeled. N and C indicate the N and C termini, respectively. (All figures except Figs. 1A and 4 were prepared with Pymol and Adobe Illustrator.)
Comparative sequence analysis and secondary structure predictions (9) have suggested that HOD and QDO belong to the α/β-hydrolase fold superfamily. Enzymes of this large group rely for catalysis on a highly conserved nucleophile/histidine/acidic residue triad similar to that of serine proteases. Albeit functionally heterogeneous, α/β-hydrolase fold enzymes do not typically catalyze oxygenation processes. The latter have a rather distinct chemistry, largely dictated by the diradical nature of dioxygen, unrelated to hydrolytic reactions. The only α/β-hydrolase fold enzyme known to be involved in an oxygen incorporation reaction is the widely employed molecular biology tool Renilla reniformis luciferase monooxygenase whose reaction mechanism is poorly understood (10).
To clarify the mechanism of dioxygenation in the unusual context of the α/β-hydrolase fold, we have solved the crystal structures of both HOD and QDO in their native state as well as the crystal structures of HOD complexed with its natural 1-H-3-hydroxy-4-oxoquinaldine (QND) substrate, its N-acetylanthranilate (NAA) reaction product, and chloride as O2 mimic. Our structural data show that these cofactor-independent dioxygenases employ a simpler His/Asp dyad, rather than the classical triad, to activate their substrates for reaction with dioxygen. They take advantage of the characteristic α/β-hydrolase fold oxyanion hole to host and control dioxygen chemistry, which likely involves a peroxide anion intermediate. Consistent with structural results, kinetic data support a nonnucleophilic general-base mechanism.
Results
Overall Structure and Residues of the Triad.
The crystal structures of HOD and QDO were solved independently using anomalous methods at the resolution of 2.1 and 2.6 Å, respectively. A summary of data collection and model statistics is presented in Table S1. The bacterial cofactor-devoid dioxygenases involved in the degradation of N-heteroaromatic compounds display a typical α/β-hydrolase fold core domain constituted by an extended, mostly parallel, β-sheet flanked by α-helices on both sides, additionally equipped with a cap domain overhanging it (Fig. 1B and Fig. S1). The cap domain is a four-helices insertion located between strand β6 and helix αD of the core domain. HOD and QDO are structurally very similar. The crystal structure of QDO can be superimposed on that of HOD with an rms difference of 1.2 Å for 260 equivalent Cα atoms (Fig. S1). A search in the Protein Data Bank (PDB, August 2009) using the secondary structure matching (SSM) algorithm (11) revealed bacterial haloperoxidases as the closest structural neighbors. Streptomyces aureofaciens bromoperoxidase A2 (12) (PDB Code 1bro) and chloroperoxidase T (13) (1a7u) as well as Streptomyces lividans chloroperoxidase L (13) (1a88) align structurally to HOD with the highest Q-score and rms deviations around 2.75 Å for approximately 230 equivalent Cα atoms.
Residues Ser101/His251/Asp126 and Ser95/His244/Asp120 in HOD and QDO, respectively (Fig. 1B and Fig. S1), located at the interface between the core domain and the cap domain, correspond to the nucleophile/histidine/acidic residue triad required for activity by members of the α/β-hydrolase fold superfamily. Similar to what has been observed in a small set of α/β-hydrolases, HOD and QDO present a relocation of the acidic residue to the end of strand β6 from its typical location at the end of strand β7. An asymmetric hydrogen bond network links the members of the triad (Fig. S2). The histidine residue is connected to the acidic residue with an AspOδ1-HisNδ2 distance of 2.61 and 2.81 Å in HOD and QDO, respectively, whereas the hydroxyl group of the serine residue interacts only weakly with the histidine’s imidazole ring (SerOγ-HisNε2 distance is 3.39 and 3.60 Å in HOD and QDO, respectively). The long SerOγ-HisNε2 distance suggests that an activation of the nucleophile by the His/Asp charge-relay system is not favored in the configuration seen in the native state. A similar weak interaction between the serine and histidine of the triad is also present in the structure of E. coli MhpC hydrolase (14), for which a nonnucleophilic role for the serine residue has been proposed (15).
Active Site Cavity and Its Access.
As seen in Fig. 2A and B for HOD, which we will consider in the remainder of the paper as our reference structure for analysis, a well-defined tunnel approximately 14-Å long connects the bulk solvent to the active site pocket via a narrow passage. The latter is lined by the side chains of Ser101, His102, Phe136, Leu156, Trp160, and His251 (HOD numbering, Fig. S3) and displays a flattened circular section measuring about 3.8 Å at its widest point. The active site pocket is made of a largely flat region, lying across the core domain and the cap domain, from which a roughly hemispherical basin develops toward the core domain (Fig. 2B). Owing to the conformation of the bulky Trp160 side chain, a degree of inward curvature is observed for the top surface of the pocket. The basin corresponds to the α/β-hydrolase fold oxyanion hole which is typically used to stabilize the negatively charged transition state during hydrolysis (16). The side chain of Ile192 is responsible for defining the basin’s shape laterally. Electrostatic potential calculations indicate that the basin has an overall positive charge, whereas the rest of the cavity is predominantly neutral (Fig. 2A and B). Several solvent molecules fill the active site. In particular, three out of the four crystallographic HOD chains present in the crystallographic asymmetric unit are found binding a solvent molecule in the basin. It is stabilized by interactions with other solvent molecules and also by hydrogen bonds with the main chain amide nitrogen atoms of Trp36 and His102, which line the bottom of the basin.
Fig. 2.

(A) Sliced-surface top-view (from the cap domain) highlighting access to the active site cavity. The centroid of the channel leading to the catalytic center is shown as a yellow trace. Residues below the slicing planes are shown as ribbons with core domain and cap domain structural elements in light and dark gray, respectively. Residues of the catalytic triad are shown in ball-and-stick mode. The molecular surface is colored according to the electrostatic potential. Positive and negative potential are shown in blue and red, respectively. (B) Sliced-surface back-view (opposite to the channel entrance) showing a vertical section of the active site cavity corresponding to the middle of the basin. The dotted line indicates the approximate boundary between the core domain and the cap domain. Residues Trp160 and Ile192 of the cap domain, which play an important role in shaping the catalytic pocket, are shown in stick representation. (C– E) Magnified view of the active site section framed in (B) by the gray rectangle with the substrate (C), product (D), and chloride ion as O2 mimic (E) bound to HOD. For clarity, bound molecules are shown unclipped. Tunnel and electrostatic potential calculations were performed with the programs MOLE (34) and APBS (35), respectively.
N-Heteroaromatic Substrate Binding.
Kinetic studies have indicated that HOD follows a compulsory-order ternary-complex mechanism in which the N-heteroaromatic organic substrate binds to the enzyme prior to dioxygen attack (17). The crystal structure of HOD anaerobically complexed with its natural QND substrate (HOD·QND) determined at the 2.15-Å resolution reveals the molecular arrangement in the active site in the first step of catalysis. The QND molecule binds in the flat portion of the active site (Fig. 2C). A combination of hydrogen bonds and hydrophobic interactions holds the substrate in the cavity lined by a total of 20 residues equally contributed by the core domain (Gly35, Trp36, Cys37, His38, His100, Ser101, His102, Gly103, His251, Phe252) and the cap domain (Phe136, Leu140, Leu143, Leu156, Trp160, Met177, Trp185, Ser188, Gly189, Ile192) (Fig. 3A and Fig. S4). In particular, the substrate’s O3 atom is hydrogen bonded to the Nε2 atom of the triad’s histidine at a distance of 2.61 Å, whereas QND’s carbonyl oxygen is stabilized by an H bond (2.68 Å) with the hydroxyl group of the triad’s nucleophile Ser101. HOD’s main chain is also involved in substrate binding. The substrate’s N1 nitrogen atom is hydrogen bonded to Trp36 carbonyl oxygen at a distance of 2.72 Å. The latter interaction suggests that QND binds in the active site with N1 in the protonated state. Overall, the H-bond network involving residues of the triad is unchanged compared to the native state.
Fig. 3.

(A) Active site of the anaerobic HOD·QND (enzyme·substrate) complex. (B) Active site of the HOD·NAA (enzyme·complex) complex. (C) Active site of the HOD·Cl- (enzyme·O2 mimic) complex with superimposed a QND substrate molecule. The simulated-annealing OMIT Fo-Fc electron density for the bound HQD, NAA, and Cl- molecules contoured at the 3.0σ level is shown in green, orange, and cyan, respectively. In (C), an NCS-averaged anomalous difference map (shown in yellow) contoured at the 5.5σ level highlights the location of the chloride ion and nearby sulfur atoms. An approximately 12-Å3 cavity available below the QND molecule is shown in blue as a transparent surface. Side chains of residues lining the active site cavity are shown in stick representation, with residues within 4.0 Å of the substrate and product molecules labeled in green and orange, respectively. Residues of the triad are represented in ball-and-stick representation. Hydrogen bonds are shown as gray dashed lines. The red sphere represents a water molecule. Distances are in angstroms.
To evaluate the role of selected amino acids in catalysis, we measured steady-state kinetic parameters of protein variants generated by site-directed mutagenesis (Table 1; a list of primers used can be found in Table S2). Confirming earlier data (17), we found that the kcat value of the HOD His251 → Ala variant is decreased by several orders of magnitude, supporting the notion that the histidine member of the triad is essential for catalysis. Replacement of the acidic residue of the triad (HOD Asp126 → Ala) yielded a variant strongly impared in its catalytic efficiency. The kcat/Km ratio for this mutant is 0.25% of that of wild-type HOD, suggesting a key role for the His-Asp dyad in catalysis. Because previous data indicated that the equivalent substitution in QDO had only little effect on its catalytic efficiency (9), we tested the QDO Asp120 → Ala variant again. Its catalytic efficiency is 0.70% of that of wild-type QDO, i.e., only marginally higher than that observed for the HOD D126A protein. This confirms the similar requirement of both HOD and QDO for the acidic residue of the triad. HOD Ser101 → Ala replacement impacted negatively only on Km (60-fold increase compared to wild-type HOD), suggesting that this member of the triad has a role in the stabilization of the organic reactant but it is not strictly required for catalysis. We also probed the effect of amino acid substitution for the amino acid facing the methyl group of QND. The imidazole plane of HOD His38 packs roughly perpendicular to QND’s methyl group (Fig. 3A) linking the cap domain to the core domain through hydrogen bonds. The side chain of Gln32 fulfills a similar role in QDO. We found that disrupting this link by HOD His38 → Ala substitution affects both Km and kcat (Table 1). This suggests that the correct relative positioning of the cap domain with respect to the core domain is required for optimal enzyme activity.
Table 1.
Effect of amino acid replacements on the catalytic activity of HOD and QDO
| KM, QND |
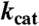 , QND , QND |
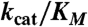 * *
|
|
| HOD | 2.7 | 38.4 | 100 |
| HOD H251A | 59.0 | 0.0034 | 0.0004 |
| HOD D126A | 27.2 | 1.05 | 0.27 |
| HOD S101A | 162.0 | 46.4 | 2.01 |
| HOD H38A | 159.5 | 3.0 | 0.13 |
| HOD H102L | 23.3 | 0.027 | 0.008 |
| KM, QNL |
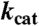 , QNL , QNL |
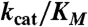 * *
|
|
| QDO | 10.4 | 20.6 | 100 |
| QDO D120A | 180.9 | 2.55 | 0.71 |
The units for KM are μM, and 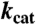 are in s-1.
are in s-1.
*Apparent kcat/Km relative to the wild-type enzyme expressed as percentage.
Product Binding.
Kinetic studies have indicated that release of the organic product is the last catalytic step in the dioxygenolytic process (17). The crystal structure of HOD complexed with its natural NAA product (HOD·NAA) refined at the 2.0-Å resolution allows us to visualize this stage of the reaction cycle. Following CO release from QND, the NAA reaction product features a newly formed amide function and a carboxylate group, each of which contains an oxygen atom derived from the reacted O2 molecule. As seen in Fig. 2D, the bound NAA product adapts to the curvature of the active site top surface by decoupling its B-ring plane from that of the amide portion of the molecule. As a result, the outer portion of NAA’s B ring is positioned more toward the cap domain portion of the active site compared to what is seen in the E·S complex. The E·P complex also reveals a rotation of about 50° of the NAA carboxylate group with respect to the plane of the B ring, resulting in its O2(2) oxygen atom being embedded in the positively charged basin (Fig. 3B). These rearrangements, coupled with a minor shift (0.2 Å) of the triad’s histidine toward the NAA molecule, position the NAAO4 atom at hydrogen bond distance from the catalytic histidine (distance NAAO4-His251Nε2 = 2.83 Å). Interestingly, a deviation from the ideal angle of 120° is observed in the bound product for the QNDC9-N1-C2 angle. The NAA molecule is in a strained conformation with the QNDC9-N1-C2 angle measuring 127.9° (Fig. S5). The most likely explanation for this deviation is a repulsive interaction between the carboxylate and carbonyl groups in the bound NAA molecule.
Chloride Binding.
The shape of the active site pocket together with structural data on the HOD·QND and HOD·NAA complexes indicate that dioxygen must react with the bound substrate on the side of the organic molecule facing the basin. Calculations using the program VOIDOO (18) reveal that, in the HOD·QND complex, a cavity measuring about 12 Å3 is available in the basin for the dioxygen molecule, approximately 2.9 Å below the A ring of the substrate. Considering a volume for molecular oxygen of 7 Å3 (bond length of 1.21 Å, atomic radius of 0.60 Å, and isotropic approximation), the size of the O2 cavity suggests a snug fit in close proximity of the organic molecule.
To obtain a positive assignment for the position of dioxygen during catalysis, we soaked native HOD crystals in a reservoir enriched with NaCl at high concentration. Halides have been effective in identifying dioxygen sites in urate oxidase and in the formylglycine-generating enzyme (19–21). In particular, chloride has polarizability and hydrophobicity similar to those of O2 (21). In urate oxidase, it has been shown binding at the same site as molecular oxygen, whereas xenon, which has also been often employed as dioxygen mimic, did not (19). The structure of HOD complexed with chloride solved at 2.7-Å resolution shows that the ion binds in the active site basin (Fig. 2E) where a water molecule is generally seen in the native state. It is stabilized by interactions with the main-chain nitrogen atoms of Trp36 and His102 at 3.40 and 3.64 Å, respectively, which form the oxyanion hole, and with the hydroxyl group of Ser101 (3.59 Å) (Fig. 3C). The ion is positioned approximately 2.3 Å below the plane occupied by the bound substrate in the HOD·QND structure at roughly the same distance from the substrate’s C2 and C4 atoms (Cl-QNDC2 and Cl-QNDC4 distances are 2.51 and 2.63 Å, respectively). The proximity of H102 imidazole side chain to the chloride ion (Cl-H102Cε2 distance is 3.95 Å) suggested that this residue could play a role in catalysis. We created the HOD H102L variant to increase hydrophobicity at this location without widening the access to the active site. The more than 103-fold decreased apparent kcatof this variant for the organic substrate (Table 1) indicates that a hydrophobic residue at this position significantly impairs the catalytic reaction. Kinetic parameters of HOD-H102L for O2 could not be determined due to a very low slope of the O2 saturation curve.
Discussion
The α/β-hydrolase fold superfamily represents a remarkable example of divergent evolution. Numerous lipases, esterases, dehalogenases, epoxidehydrolases, and haloperoxidases adopt this versatile architecture to accomplish specific chemical tasks. Although the range of reactions catalyzed by members of the superfamily is definitely rich, α/β-hydrolase fold enzymes tend, nevertheless, to be involved in ester or peptide hydrolysis reactions. Oxygenolytic reactions do not share many commonalities with such hydrolytic reactions. HOD and QDO dioxygenases are therefore intriguing for two reasons: First, they belong to a protein family that is not typically involved in oxygen chemistry; second, they are not equipped with metal or organic cofactors generally employed by oxygenases to circumvent the spin restriction of a direct reaction of triplet dioxygen with singlet organic substrates.
Enzymes of the α/β-hydrolase fold superfamily typically employ a strictly conserved nucleophile/histidine/acidic residue triad for catalysis. A nucleophilic mechanism, first experimentally proven with the crystallographic trapping of a covalent alkyl-enzyme ester intermediate in Xanthobacter autotrophicus GJ10 haloalkane dehalogenase soaked with the substrate 1,2-dichloroethane (22), has long represented the paradigm for catalysis at the α/β-hydrolase fold. More recently, however, some α/β-hydrolase fold C-C bond breaking enzymes such as E. coli MhpC hydrolase (15), Burkholderia xenovorans BphD hydrolase (23) and Hevea brasiliensis hydroxynitrile lyase (24) have been proposed to follow a nonnucleophilic general-base mechanism entailing activation by the catalytic histidine of the small molecule component (H2O and HCN) of the bimolecular reaction (25, 26). In the case of BphD, however, this alternative mechanistic proposal is not fully accepted and a nucleophilic mechanism proceeding via an acyl-enzyme intermediate has also been suggested (27, 28). Utilizing the same catalytic triad as the above enzymes are the cofactor-free haloperoxidases which halogenate organic compounds in the presence of halide ions and peroxides such as H2O2. Peroxoacids are probably involved as reaction intermediates (12). The structural similarity of the dioxygenases and the haloperoxidases intriguingly suggests that they share common evolutionary roots.
A possible reaction mechanism for the cofactor-independent 2,4-dioxygenolytic decomposition of N-heteroaromatic substrates at the α/β-hydrolase fold is shown in Fig. 4. Steady-state kinetic data suggest that the initial step of the catalytic cycle requires binding of the organic substrate in the active site. HOD and QDO dioxygenases possess a recognizable triad (Fig. 4A) accessible to the organic substrates via a passage too narrow to allow transit of the organic reactants without some structural rearrangement. Because all complexes presented here were obtained by the soaking technique, such protein movements are not prohibited by the crystalline state. In contrast with earlier suggestions based on model studies (17), the crystal structure of the anaerobic HOD·QND complex shows that the substrate does not bind to the enzyme in the dianionic form. The substrate’s N1 atom forms a well-defined hydrogen bond with the carbonyl oxygen atom of Trp36 (Fig. 3A and Fig. S4), indicative of protonation of the former. Rather, we propose the substrate binds in a formally monoanionic state as a result of selective deprotonation of the 3OH group. The H-bond network connecting the substrate QND to Asp126 via His251 supports the view that the His-Asp charge-relay dyad mediates proton abstraction (Fig. 4B). The residual activity measured for both HOD and QDO acidic residue → Ala variants at pH 8.0 (Table 1) likely reflects the effect of His-dependent solvent-assisted substrate deprotonation. Activity measurements as a function of pH are in agreement with this hypothesis as a further increase in basicity restores activity in HOD Asp126 → Ala to nearly wild-type levels (Fig. S6). The key role of the His/Asp dyad in catalysis at neutral pH is further strengthened by the observation that HOD His251 → Ala variant is catalytically virtually dead in spite of the fact that HOD possesses an alternative histidine residue (His100, not conserved in QDO) at H-bond distance from the substrate’s 3OH group (His100Nε2-QNDO3 distance is 2.81 Å). His100 cannot take over the role of His251 because no negatively charged residue similar to Asp126 is available in its vicinity to form a charge-relay tandem. Whereas the His-Asp dyad is of critical importance for substrate turnover, kinetic data show that the serine residue of the triad is largely dispensable. Consistent with the large effect of the Ser101 → Ala substitution on the apparent Km for QND (Table 1), structural data show that Ser101, corresponding to the triad’s nucleophile, instead plays an important role in the stabilization of the organic substrate.
Fig. 4.

Proposed reaction mechanism for the cofactor-independent dioxygenation of N-heteroaromatic substrates at the α/β-hydrolase fold.
Deprotonation of the substrate likely represents the activation required to bring molecular oxygen into catalysis. Binding of the substrate in the active site defines a “made-to-measure” cavity below the substrate’s A ring available for all subsequent catalytic steps involving dioxygen (Fig. 4C). As suggested by the position of the chloride ion in the HOD·Cl- complex (Fig. 3C), dioxygen can bind in the oxyanion hole. The overall positive electrostatic potential of the basin makes a scenario involving a single electron transfer from the monoanionic substrate to O2 to form a superoxide anion (Fig. 4D) which, following radical recombination, leads to the peroxide shown in Fig. 4E, a possibility. A single electron transfer to dioxygen has also been proposed for the mechanism of the cofactor-independent dioxygenase DpgC(6) and for the reaction of reduced flavin cofactors with O2 (29). Attack of the peroxide on the carbonyl function of the substrate affords the formation of the endoperoxide shown in Fig. 4F. The positive dipole of the αC-helix, the latter beginning just behind the position occupied by carbonyl oxygen of the bound substrate, can act as an electrostatic guide to direct the attack. The conserved histidine residue (His102 and His96 in HOD and QDO, respectively) located at the bottom of the tight entrance leading to the active site (Fig. S3) is also likely to be involved in the catalytic steps involving dioxygen, possibly by assisting the formation/conversion of the E·S·O2 adduct.
The endoperoxide intermediate is the most convenient structure to rationalize the release of carbon monoxide and anthranilate derivative products. Modeling of the rigid endoperoxide structure using the crystal structures of the E·S and E·P complexes as references suggests that a close interaction between the bulky side chain of the conserved Trp160 residue and the leaving carbon monoxide, which upon cyclization raises above the B plane of the substrate in the direction of the cap domain, might destabilize the intermediate favoring CO release. In the final step of the catalytic cycle, the organic product whose carboxylate group is now hydrogen bonded to the triad’s protonated histidine (Fig. 4F) can presumably abstract a proton from the latter, regenerating the enzyme for a new catalytic cycle. The driving force for substrate protonation is likely to be the minimization of the charge repulsion between its carboxylate and carbonyl moieties, which, as seen in the HOD·NAA complex, induce a strained conformation in the bound product.
Our mechanistic proposal for the cofactor-independent α/β-hydrolase fold dioxygenases follows the principles of general-base catalysis. It does not involve a nucleophilic role for the “nucleophile” of the triad. This residue is mainly involved in the stabilization of carbonyl and hydroxylate groups bore by the different species throughout the reaction cycle. We propose that HOD and QDO activate the organic substrate instead of dioxygen. Consequently, they are substrate-assisted oxygenases. As in the case of the chemically identical reaction catalyzed by the metal-dependent dioxygenase on O-heteroaromatic flavonol substrates, the key element for activation is deprotonation of the 3OH hydroxyl group (8) which leads to the formation of species with electron-donating properties able to form resonance-stabilized radicals.
Materials and Methods
Methods used for protein expression, purification, and crystallization, biochemical assays, and site-directed mutagenesis are described in SI Materials and Methods.
Data on HOD and its complexes were collected at the European Synchrotron Radiation Facility (Grenoble, France) and at Diamond Light Source (Didcot, United Kingdom). Data on QDO was collected at the Advanced Photon Source (Argonne, IL). His6-HOD C69S crystals exhibited pseudomerohedral twinning in space group P212121. In an attempt to produce untwinned crystals, we screened a panel of additives (Additive Screening Kit, Hampton Research). Crystals of the catalytically inactive doubly substituted His6-HOD C69S/H251A protein obtained in the presence of CuCl2 showed a different morphology (30). These crystals do not present twinning and belong to the space group P43212. We reasoned that copper ions might be bound to the hexahistidine tag and attempted structure determination using anomalous methods. HOD structure was solved by single anomalous dispersion using data collected at the Cu K edge to 3.5-Å resolution. A noncrystallographic symmetry averaged 3.5-Å map was used to build an initial HOD model, which was subsequently refined against 2.1-Å resolution pseudomerohedrally twinned data using the program REFMAC5 (31, 32). HOD·QND, HOD·NAA, and HOD·Cl- complexes were solved using difference Fourier methods. The structure QDO was solved by the multiple anomalous dispersion method using data from SeMet-derivatized QDO (33). Further details on the methods used for structure solution as well as data collection, processing, and refinement statistics are given in SI Text and Table S1.
Supplementary Material
Acknowledgments.
R.A.S. gratefully acknowledges Prof. R.W. Pickersgill and his research group (Queen Mary University of London) for the use of the anaerobic chamber. We thank Almut Kappius (Muenster) for expert technical assistance. The scientists of the European Synchrotron Radiation Facility BM30A, Diamond I04, Advanced Photon Source 19-ID beamlines are gratefully acknowledged for their support during data collection. This work was supported by a King’s College London incentive grant (R.A.S) and by a grant from the German Research Foundation (FE 383/15-1 to S.F.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0909033107/DCSupplemental.
References
- 1.Fetzner S. Oxygenases without requirement for cofactors or metal ions. Appl Microbiol Biotechnol. 2002;60:243–257. doi: 10.1007/s00253-002-1123-4. [DOI] [PubMed] [Google Scholar]
- 2.Fetzner S. Cofactor-independent oxygenases go it alone. Nat Chem Biol. 2007;3:374–375. doi: 10.1038/nchembio0707-374. [DOI] [PubMed] [Google Scholar]
- 3.Kendrew SG, Hopwood DA, Marsh ENG. Identification of a monooxygenase from Streptomyces coelicolor A3(2) involved in the biosynthesis of actinorhodin: Purification and characterization of the recombinant enzyme. J Bacteriol. 1997;179:4305–4310. doi: 10.1128/jb.179.13.4305-4310.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sciara G, et al. The structure of ActVA-Orf6, a novel type of monooxygenase involved in actinorhodin biosynthesis. EMBO J. 2003;22:205–215. doi: 10.1093/emboj/cdg031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tseng CC, Vaillancourt FH, Bruner SD, Walsh CT. DpgC is a metal- and cofactor-free 3,5-dihydroxyphenylacetyl-CoA 1,2-dioxygenase in the vancomycin biosynthetic pathway. Chem Biol. 2004;11:1195–1203. doi: 10.1016/j.chembiol.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 6.Widboom PF, Fielding EN, Liu Y, Bruner SD. Structural basis for cofactor-independent dioxygenation in vancomycin biosynthesis. Nature. 2007;447:342–345. doi: 10.1038/nature05702. [DOI] [PubMed] [Google Scholar]
- 7.Bauer I, Max N, Fetzner S, Lingens F. 2,4-dioxygenases catalyzing N-heterocyclic-ring cleavage and formation of carbon monoxide. Purification and some properties of 1H-3-hydroxy-4-oxoquinaldine 2,4-dioxygenase from Arthrobacter sp. Rü61a and comparison with 1H-3-hydroxy-4-oxoquinoline 2,4-dioxygenase from Pseudomonas putida 33/1. Eur J Biochem. 1996;240:576–583. doi: 10.1111/j.1432-1033.1996.0576h.x. [DOI] [PubMed] [Google Scholar]
- 8.Steiner RA, Kalk KH, Dijkstra BW. Anaerobic enzyme substrate structures provide insight into the reaction mechanism of the copper-dependent quercetin 2,3-dioxygenase. Proc Natl Acad Sci USA. 2002;99:16625–16630. doi: 10.1073/pnas.262506299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fischer F, Kunne S, Fetzner S. Bacterial 2,4-dioxygenases: New members of the alpha/beta hydrolase-fold superfamily of enzymes functionally related to serine hydrolases. J Bacteriol. 1999;181:5725–5733. doi: 10.1128/jb.181.18.5725-5733.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Loening AM, Fenn TD, Gambhir SS. Crystal structures of the luciferase and green fluorescent protein from Renilla reniformis. J Mol Biol. 2007;374:1017–1028. doi: 10.1016/j.jmb.2007.09.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr Sect. D Biol Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 12.Hecht HJ, Sobek H, Haag T, Pfeifer O, van Pée KH. The metal-ion-free oxidoreductase from Streptomyces aureofaciens has an alpha/beta hydrolase fold. Nat Struct Biol. 1994;1:532–537. doi: 10.1038/nsb0894-532. [DOI] [PubMed] [Google Scholar]
- 13.Hofmann B, et al. Structural investigation of the cofactor-free chloroperoxidases. J Mol Biol. 1998;279:889–900. doi: 10.1006/jmbi.1998.1802. [DOI] [PubMed] [Google Scholar]
- 14.Dunn G, et al. The structure of the C-C bond hydrolase MhpC provides insights into its catalytic mechanism. J Mol Biol. 2005;346:253–265. doi: 10.1016/j.jmb.2004.11.033. [DOI] [PubMed] [Google Scholar]
- 15.Li C, et al. Catalytic mechanism of C-C hydrolase MhpC from Escherichia coli: Kinetic analysis of His263 and Ser110 site-directed mutants. J Mol Biol. 2005;346:241–251. doi: 10.1016/j.jmb.2004.11.032. [DOI] [PubMed] [Google Scholar]
- 16.Nardini M, Dijkstra BW. Alpha/beta hydrolase fold enzymes: The family keeps growing. Curr Opin Struct Biol. 1999;9:732–737. doi: 10.1016/s0959-440x(99)00037-8. [DOI] [PubMed] [Google Scholar]
- 17.Frerichs-Deeken U, Ranguelova K, Kappl R, Huttermann J, Fetzner S. Dioxygenases without requirement for cofactors and their chemical model reaction: Compulsory order ternary complex mechanism of 1H-3-hydroxy-4-oxoquinaldine 2,4-dioxygenase involving general base catalysis by histidine 251 and single-electron oxidation of the substrate dianion. Biochemistry. 2004;43:14485–14499. doi: 10.1021/bi048735u. [DOI] [PubMed] [Google Scholar]
- 18.Kleywegt GJ, Jones TA. Detection, delineation, measurement and display of cavities in macromolecular structures. Acta Crystallogr Sect. D Biol Crystallogr. 1994;50:178–185. doi: 10.1107/S0907444993011333. [DOI] [PubMed] [Google Scholar]
- 19.Colloc’h N, et al. Oxygen pressurized x-ray crystallography: Probing the dioxygen binding site in cofactorless urate oxidase and implications for its catalytic mechanism. Biophys J. 2008;95:2415–2422. doi: 10.1529/biophysj.107.122184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roeser D, Schmidt B, Preusser-Kunze A, Rudolph MG. Probing the oxygen-binding site of the human formylglycine-generating enzyme using halide ions. Acta Crystallogr Sect. D Biol Crystallogr. 2007;63:621–627. doi: 10.1107/S0907444907009961. [DOI] [PubMed] [Google Scholar]
- 21.Roeser D, et al. A general binding mechanism for all human sulfatases by the formylglycine-generating enzyme. Proc Natl Acad Sci USA. 2006;103:81–86. doi: 10.1073/pnas.0507592102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verschueren KH, Seljee F, Rozeboom HJ, Kalk KH, Dijkstra BW. Crystallographic analysis of the catalytic mechanism of haloalkane dehalogenase. Nature. 1993;363:693–698. doi: 10.1038/363693a0. [DOI] [PubMed] [Google Scholar]
- 23.Speare DM, Fleming SM, Beckett MN, Li JJ, Bugg TD. Synthetic 6-aryl-2-hydroxy-6-ketohexa-2,4-dienoic acid substrates for C-C hydrolase BphD: Investigation of a general base catalytic mechanism. Org Biomol Chem. 2004;2:2942–2950. doi: 10.1039/B410322J. [DOI] [PubMed] [Google Scholar]
- 24.Gruber K, Gartler G, Krammer B, Schwab H, Kratky C. Reaction mechanism of hydroxynitrile lyases of the alpha/beta-hydrolase superfamily: The three-dimensional structure of the transient enzyme-substrate complex certifies the crucial role of Lys236. J Biol Chem. 2004;279:20501–20510. doi: 10.1074/jbc.M401575200. [DOI] [PubMed] [Google Scholar]
- 25.Bugg TD. Diverse catalytic activities in the alphabeta-hydrolase family of enzymes: Activation of H2O, HCN, H2O2, and O2. Bioorg Chem. 2004;32:367–375. doi: 10.1016/j.bioorg.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 26.Li JJ, Bugg TD. Investigation of a general base mechanism for ester hydrolysis in C-C hydrolase enzymes of the alpha/beta-hydrolase superfamily: A novel mechanism for the serine catalytic triad. Org Biomol Chem. 2007;5:507–513. doi: 10.1039/b615605c. [DOI] [PubMed] [Google Scholar]
- 27.Horsman GP, et al. The tautomeric half-reaction of BphD, a C-C bond hydrolase. Kinetic and structural evidence supporting a key role for histidine 265 of the catalytic triad. J Biol Chem. 2007;282:19894–19904. doi: 10.1074/jbc.M702237200. [DOI] [PubMed] [Google Scholar]
- 28.Horsman GP, et al. Kinetic and structural insight into the mechanism of BphD, a C-C bond hydrolase from the biphenyl degradation pathway. Biochemistry. 2006;45:11071–11086. doi: 10.1021/bi0611098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Massey V. Activation of molecular oxygen by flavins and flavoproteins. J Biol Chem. 1994;269:22459–22462. [PubMed] [Google Scholar]
- 30.Steiner RA, Frerichs-Deeken U, Fetzner S. Crystallization and preliminary x-ray analysis of 1H-3-hydroxy-4-oxoquinaldine 2,4-dioxygenase from Arthrobacter nitroguajacolicus Rü61a: A cofactor-devoid dioxygenase of the alpha/beta-hydrolase-fold superfamily. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2007;63:382–385. doi: 10.1107/S174430910701353X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 32.Steiner RA, Lebedev AA, Murshudov GN. Fisher’s information in maximum-likelihood macromolecular crystallographic refinement. Acta Crystallogr D Biol Crystallogr. 2003;59:2114–2124. doi: 10.1107/s0907444903018675. [DOI] [PubMed] [Google Scholar]
- 33.Qi R, Fetzner S, Oakley AJ. Crystallization and diffraction data of 1H-3-hydroxy-4-oxoquinoline 2,4-dioxygenase: a cofactor-free oxygenase of the alpha/beta-hydrolase family. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2007;63:378–381. doi: 10.1107/S1744309107013760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petrek M, Kosinova P, Koca J, Otyepka M. MOLE: A Voronoi diagram-based explorer of molecular channels, pores, and tunnels. Structure. 2007;15(11):1357–1363. doi: 10.1016/j.str.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 35.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.