

Abstract
Ferritin is a multimeric nanocage protein that directs the reversible biomineralization of iron. At the catalytic ferroxidase site two iron(II) ions react with dioxygen to form diferric species. In order to study the pathway of iron(III) from the ferroxidase site to the central cavity a new NMR strategy was developed to manage the investigation of a system composed of 24 monomers of 20 kDa each. The strategy is based on 13C-13C solution NOESY experiments combined with solid-state proton-driven 13C-13C spin diffusion and 3D coherence transfer experiments. In this way, 75% of amino acids were recognized and 35% sequence-specific assigned. Paramagnetic broadening, induced by iron(III) species in solution 13C-13C NOESY spectra, localized the iron within each subunit and traced the progression to the central cavity. Eight iron ions fill the 20-Å-long iron channel from the ferrous/dioxygen oxidoreductase site to the exit into the cavity, inside the four-helix bundle of each subunit, contrasting with short paths in models. Magnetic susceptibility data support the formation of ferric multimers in the iron channels. Multiple iron channel exits are near enough to facilitate high concentration of iron that can mineralize in the ferritin cavity, illustrating advantages of the multisubunit cage structure.
Keywords: ¹³C direct detection, high molecular weight NMR, iron channels, solid-state NMR, paramagnetic NMR
Ferritins are a family of protein nanocages that concentrate iron in biominerals for controlled release and use in enzyme iron cofactors. A large cavity that occupies about 30% of the total protein volume at the center of the cage is the mineralization site of ferritin ferrihydrite. The importance of ferritin is illustrated by embryonic lethality of gene deletion in mammals (1) and resistance to oxidants in animals, plants, bacteria, and archea that reflects consumption of iron(II) and dioxygen or hydrogen peroxide (2–5). Some classes of nanomaterials use ferritin nanocages as templates (6). The protein nanocages self-assemble from four-helix-bundle subunits into cages of two sizes: 12-subunit, hydrogen peroxide–consuming miniferritins of bacteria and archea, and 24-subunit maxiferritins of archea, bacteria, and higher organisms (3, 4, 7). Miniferritins may be the more primitive and are alternatively named DNA-binding proteins because some of them coat DNA.
The first step in the conversion of iron(II) and dioxygen to ferric oxide mineral, in the 24-subunit maxiferritins, occurs at catalytic oxidoreductase (ferroxidase) sites in the center of each of the four-helix bundles, which have long axes oriented almost parallel to the protein surface (Fig. 1A). Each active site has residues contributed by each of the four helices (Fig. 1B) that create a diiron(II) site similar to the diiron cofactor sites in ribonucleotide reductases and stearoyl desaturases (8). When dioxygen, the second substrate, binds to the active site after iron(II) binding, diferric oxo products form via a diferric peroxo intermediate. The active site has been studied extensively, e.g., with ferrous ion substrate analogues in cocrystals, subtractive mutagenesis, and protein chimeras (4, 8–12), although little is known about where the ferroxidase products are until they appear in the mineral in the protein cavity. Simple inspection of the protein cage does not show an obvious path from the active site to the mineralization cavity. Therefore, we sought the path by NMR spectroscopy by taking advantage of the paramagnetic effects induced by the iron(III) species released at the catalytic site, on the resonances of nearby residues. 13C-13C NOESY experiments acquired in solution with increasing ratios of iron:protein were combined with sequence-specific assignments of the protein residues distributed throughout the ferritin subunits (Fig. 1C). Partial sequence-specific assignments for the iron-free protein cage used data from our earlier side-chain identification in solution (13) and a unique approach where solid-state and solution NMR data were combined to overcome the spectroscopic problems for a protein of the size of ferritin (480 kDa).
Fig. 1.

Sequential assignment of ferritin by solid-state NMR. (A) 3D structure of recombinant frog (R. catesbeiana) M ferritin (24 subunits, PDB 1MFR; PyMOL 0.99rc6). The four-helix-bundle subunits are displayed as gray helices and red loops. The green subunit illustrates the orientation of the monomeric units along the surface of the hollow sphere. (B) Representation of the four-helix-bundle subunit NMR-assigned residues (blue spheres) and ferroxidase residues (red spheres). (C) Primary and secondary structures of frog M ferritin-ferroxidase site residues (red font), NMR sequence-specific assigned residues (blue font), α-helices (bars), and loops (broken lines).
Results
A homopolymeric, 24-subunit ferritin cage was studied, the recombinant protein composed of catalytically active M subunits (175 amino acids each) from R. catesbeiana. This protein has been extensively used as a model for oxidoreductase activity (4). Given the homooligomeric nature of this ferritin and the symmetry properties of the cage (Fig. 1A), the number of NMR resonances in the spectra of this large protein assembly corresponds to that of a single subunit. However, the unequivocal identification of the signals of amino acids to be used as probes for the paramagnetic effects required the development of a unique NMR approach because of the high molecular weight of the assembled protein (480 kDa).
The large size of ferritin causes slow tumbling of the molecule in solution and prevents 1H NMR signal detection. We applied 13C-13C NOESY in solution for the identification of about 75% of amino acid spin systems (13). On the other hand, slow molecular tumbling induces rapid transverse relaxation and hampers solution NMR experiments for sequence-specific assignment based on coherence transfer. Immobilization of the protein in the solid state overcomes the problems arising from the rapid transverse relaxation, and magic angle spinning (MAS) solid-state NMR (14, 15) provided relatively well-resolved spectra (Fig. 2A). The 13C-13C correlation solid-state experiments, proton-driven spin diffusion (PDSD) (16) or the dipolar assisted rotational resonance (DARR) (17), and the 13C-13C solution NOESY (Fig. 2A), are very similar and provide the same kind of information about spin patterns because the experiments are all based on spin diffusion effects along amino acid side chains. A 3D Ni-Cαi-CXi spectrum (18) confirmed the intraresidue connectivities from solution 13C-13C NOESY and enables the identification of the intraresidue backbone amide nitrogen.
Fig. 2.

Combined use of solid-state and solution NMR. (A) Superimposition of the aliphatic region of the ferritin 13C-13C NOESY spectrum acquired in solution (blue trace) and in the solid-state13C-13C DARR spectrum (red trace) permits the transfer of the solid-state assignments to solution data and vice versa. The two 2D maps were overlaid by looking for the best superimposition of the aliphatic part. (B) Superimposition of the aliphatic region of the ferritin 13C-13C NOESY spectra acquired in solution before (blue) and after (green) the addition of 1 equivalent of iron (II) (2 iron(II)/active site; 48 iron(II)/nanocage); resonances disappearing upon formation of iron (III) products are labeled.
Sequential assignments were first obtained through a 3D 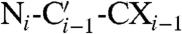 spectrum (18) using a process that requires linking spin systems in the two spectra by matching 15N shifts, an approach that in ferritin suffers from the large degeneracy in 15N chemical shifts due to the four-helix-bundle structure (19). More effective was a 3D
spectrum (18) using a process that requires linking spin systems in the two spectra by matching 15N shifts, an approach that in ferritin suffers from the large degeneracy in 15N chemical shifts due to the four-helix-bundle structure (19). More effective was a 3D 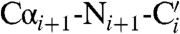 spectrum (18) that provides direct connectivities between these backbone atoms on adjacent residues (Figs. S1–S3). As a result, 59 residues, distributed throughout each subunit, were sequence-specific assigned (Fig. 1B and C). The chemical shift index is consistent with their location on the expected secondary structure elements, while the use of ferritin expressed in [2-13C]-labeled glycerol confirmed the chemical nature of the observed residues (20). The transfer of sequence-specific assignments derived from solid-state data to the solution 13C-13C NOESY is relatively straightforward if one takes into account the deuterium isotope shift (21–23) and overlays the spectra searching for the best superimposition of the aliphatic region (Fig. 2A), which is characterized by a good resolution both in solution and in the solid state. Our strategy is based on the recognition of side-chain spin patterns. The several factors that affect the completeness of the assignment are (i) the intrinsically low chemical shift dispersion related to the four-helix-bundle structure of the monomers (19); (ii) the primary sequence of ferritin (Fig. 1C), which has large numbers of residues of the same type (the most abundant amino acids are 19 Leu, 18 Glu, 13 Lys, and 13 Asp, and account for 36% of the total amino acids); (iii) the extremely narrow distribution in 13C chemical shifts within each of the most abundant amino acids and within the Lys/Leu and Glu/Asp groups; and (iv) the difficulty in discriminating between Glu and Gln and between Asp and Asn. More extensive assignments will require repetition of the experiments using samples prepared with selective “unlabeling” of different amino acids (24) and is beyond the scope of this article. Nevertheless, the present results represent a large step forward in the assignment of big molecular complexes.
spectrum (18) that provides direct connectivities between these backbone atoms on adjacent residues (Figs. S1–S3). As a result, 59 residues, distributed throughout each subunit, were sequence-specific assigned (Fig. 1B and C). The chemical shift index is consistent with their location on the expected secondary structure elements, while the use of ferritin expressed in [2-13C]-labeled glycerol confirmed the chemical nature of the observed residues (20). The transfer of sequence-specific assignments derived from solid-state data to the solution 13C-13C NOESY is relatively straightforward if one takes into account the deuterium isotope shift (21–23) and overlays the spectra searching for the best superimposition of the aliphatic region (Fig. 2A), which is characterized by a good resolution both in solution and in the solid state. Our strategy is based on the recognition of side-chain spin patterns. The several factors that affect the completeness of the assignment are (i) the intrinsically low chemical shift dispersion related to the four-helix-bundle structure of the monomers (19); (ii) the primary sequence of ferritin (Fig. 1C), which has large numbers of residues of the same type (the most abundant amino acids are 19 Leu, 18 Glu, 13 Lys, and 13 Asp, and account for 36% of the total amino acids); (iii) the extremely narrow distribution in 13C chemical shifts within each of the most abundant amino acids and within the Lys/Leu and Glu/Asp groups; and (iv) the difficulty in discriminating between Glu and Gln and between Asp and Asn. More extensive assignments will require repetition of the experiments using samples prepared with selective “unlabeling” of different amino acids (24) and is beyond the scope of this article. Nevertheless, the present results represent a large step forward in the assignment of big molecular complexes.
The location of the assigned residues on different structural elements (Fig. 1B and C) makes the group of 59 identified residues structurally representative. Many of the assigned residues (62%) are on protein loops because of the intrinsically lower chemical shift dispersion characteristic of amino acids located on the α-helices (19). However, residues assigned on α-helices are distributed throughout the subunit: helix 1—residues 10, 11, 14–16, 26, 27, 31, and 32; helix 3—117, 118, and 119; helix 4–141, 144–146, and 148–151; and helix 5—161, 163, and 167–169. The set of signals for the sequence-assigned amino acids was used to monitor the fate of the iron(III) products in solution after iron(II) is added and reacts with dioxygen at the active sites.
When iron(II) and oxygen substrates are converted to ferric species in ferritin, selective changes are observed of the 13C-13C solution NOESY (Fig. 2B). The titration used saturating amounts of ferrous substrate for each catalytic cycle, beginning with the addition of 48 atoms of iron(II) per ferritin nanocage, i.e., 2 iron(II) ions per ferroxidase site. The iron(II) ions and oxygen substrates are immediately converted into iron(III) species, which is accompanied by the disappearance of some resonances in well-resolved spectral regions, well outside the noise level in the spectrum of the apoprotein. This behavior is consistent with the presence of paramagnetic species that cause broadening beyond detection of resonances of nearby residues (25). Except for the disappearance of a few cross peaks, the 13C-13C NOESY spectra are identical with and without iron(III), indicating localized paramagnetic effects. Good indicators of the location of the product are provided by the well-resolved side-chain peaks of isoleucine residues; all four of the isoleucines present in ferritin are assigned. Among the four isoleucine residues, only the resonances of the side chain of I144 disappeared at this stage of the titration, indicating that the ferric products leave the catalytic site and migrate toward a new site close to I144, which has a side chain protruding toward the interior of the helix bundle. I141, by contrast, adjacent to the ferroxidase site and unaffected at this stage of the titration, has a side chain directed toward the exterior of the bundle, toward the internal cavity of the cage. Paramagnetic broadening is also induced in A26, another residue near the active site with a side chain pointing toward the interior of the bundle. These effects suggest that the ferric products leave the catalytic site and move inside the four-helix bundle, from the ferroxidase site toward the short fifth helix at the C-terminus of each subunit (Fig. 3A). Addition of another two iron ions per active site, for a total of 96 per nanocage, affects a larger number of residues. Besides I144 and A26, W89, T92, I148, and L156 are also broadened beyond detection (Fig. 3A). The side chains of these residues, as for A26 and I144, are located inside the helix-bundle but farther from the ferroxidase site. Adding a total of four more iron ions per active site, in two additions, for a total of 192 iron atoms per nanocage, results in the disappearance of additional resonances, e.g., T149 and L154 (Fig. 3A). These residues, however, are located outside the helix bundle at the junction of the channel with the inner surface of the mineralization cavity and probably represent the gate for iron exit from the channel into the internal protein cavity. T149 and L154 are also at interfaces with other subunits, and, at this level of the titration, other residues in close contact to iron(III) complexes are in different subunits. For example, the resonances of V42 in one subunit, which is almost in contact with T149 on a perpendicular subunit (Fig. 3B), disappear. The relative position of the subunits in the protein architecture is such that multiple exits are close enough to nucleate mineralization inside the central compartment through fusion of the emerging products (Fig. 3C).
Fig. 3.

Tracing the iron channel in ferritin by paramagnetic effects. (A) NMR resonances that disappear in 13C-13C NOESY solution spectra, as the iron:protein ratio increases, are mapped onto the ribbon structure of a ferritin subunit as colored spheres (1 equivalent of iron = 2 iron(II)/active site = 48 iron(II)/nanocage). 1 equivalent: light blue, 2 equivalents: blue, 4 equivalents: dark blue. (B) View of the internal surface of the ferritin protein cage showing the relative spatial relationship near channel exits into the cavity; iron(III) products emerging from the channel of the red subunit have paramagnetic effects on residue V42 in the blue subunit and on R72 and G74 in the green subunit, after 4 equivalents of iron are added. Paramagnetically broadened residues are shown as spheres. (C) Ferritin channel exits in four adjacent subunits surround the four fold axes of the ferritin protein cage: V42 (blue spheres); I148, T149, L154, and L156 (red spheres). (D) Effective magnetic moments per subunit (μeff, red bars) and average magnetic moment per iron atom (μeff/iron atom, blue bars) obtained by Evans measurements at increasing concentrations of iron.
The first addition of iron (48/nanocage; 2/active site) causes no hyperfine shift in any of the resolved resonances, consistent with the presence of iron(III) species characterized by low anisotropy, which are incapable of influencing nuclei at distances larger than those broadened beyond detection. With 96–192 iron atoms a few resonances experience small chemical shifts (≤ 0.2 ppm). These signals belong to amino acids that have some signals broadened by paramagnetic effects or that are close in space to amino acids with signals broadened beyond detection.
To explore the nuclearity of the ferric products in ferritin after oxidation of iron(II), bulk magnetic susceptibility in solution was measured at different steps in the iron titration (Fig. 3D, Table S1), by the Evans method (26, 27). The species formed upon addition of the first two irons per active site has an effective magnetic moment μeff of 4.49 ± 0.22 μB, i.e., significantly lower than expected for two noncoupled high-spin iron(III) centers (4.5 μB vs. 8.3 μB) and indicative of the presence of an antiferromagnetically coupled dimer (28, 29), as also observed by Mössbauer spectroscopy (30). The small magnetic susceptibility is consistent with a μ-oxo complex (28). This species accounts for the observed paramagnetic line broadening on nearby residues (estimated to be within 5–6 Å). Addition of the second two irons per active site is associated with a μeff of 5.61 ± 0.28 μB, i.e., an average μeff of 1.40 ± 0.07 μB per iron. The decrease in the average μeff per iron atom from 2.24 ± 0.11 μB to 1.40 ± 0.07 μB suggests the presence of a four-iron(III) cluster with antiferromagnetically coupled metal ions. The presence of four-iron ions induces paramagnetic line broadening of a greater number of amino acids, as observed in Fig. 3A. Further additions of pairs of iron atoms per active site are accompanied by a continual increase in total μeff and decrease in the average μeff per iron atom, as expected for antiferromagnetically coupled clusters of increasing nuclearity. The μeff of 7.85 ± 0.39 μB obtained with 8 iron/subunit is comparable to the value reported for the Fe8(μ4-O)4 cluster (31).
Insights on the kinetics of products formation could be obtained with Evans measurements as a function of time. The magnetic moments in Fig. 3D are equilibrium values (Table S2). Equilibration of the dimeric ferric products produced by the first equivalent of iron is fast (about 1.5 h). As more iron(II) is added, there is an initial increase in magnetic susceptibility attributable to the formation of a new dimer, followed by a slow decrease until a stable value is reached in about 24 h. This behavior suggests that the first product at the catalytic site is always a dimer, which then interacts with other products already present within the four-helix-bundle channel.
Discussion
Protein nanocages are subcellular compartments for chemical reactions; an example is provided by ferritin that concentrates iron in a solid mineral. Formation of ferryhydrite mineral through hydrolysis occurs in the protein cavity and is a well-studied inorganic reaction. In ferritin, water is coordinated to iron in diiron sites or to other metals bound at the active sites (4, 8), but where or when the water participates in hydrolytic coupling among the ferric oxo mineral precursors is unknown. Moreover, little direct structural information is available about ferrous ions moving within the cage after entering at pores near the threefold axes (32) to the active sites buried in the center of four-helix bundles of each subunit or, until this study, how the diferric products leave the active site and move to the cavity. The NMR approach of combining solid-state experiments for partial sequence-specific assignment and solution 13C-13C NOESY spectra for side-chain observation has provided the identification of an iron channel that guides the directional transport of the multimeric iron(III) products from the active site toward the nanocage. The four-helix-bundle structure of the ferritin subunits provides the scaffold that hosts the ferroxidase sites and also guides the catalytic products toward the mineralization site. Based on the observed paramagnetic effects on residues lining the internal face of the four-helix bundle, the functional channel corresponds to the interior of the four-helix bundle. Iron catalytic products proceed along the 20-Å-long path inside the four-helix bundle of each subunit, roughly parallel to the protein surface, until they reach the C-terminal edge, where the short fifth helix, angled about 60° from the axes of the bundle, creates a kink that may guide the iron(III) clusters into the mineralization cavity. The iron channel we observed is much longer than current models, which predict a short hop across the protein cage, between the active site and the cavity.
Formation and migration of ferric products at the active sites of the 24 subunits appear to be synchronized in all the subunits at all titration steps. After the first catalytic turnover (2Fe/catalytic center), the iron(III) complexes have only moved a few angstroms into the channels from the active centers. Two Ile residues are close to the active site, I141 and I144, with I141 closer to the metal ions at the catalytic center. (Distances between the metal ion coordinated to Asp140/Gln137/Glu103 and I141 nuclei are Cα 7.7 Å, Cβ 8.0 Å, Cδ1 7.6 Å, and Cγ1 7.1 Å, whereas distances between the same metal ion and I144 are Cα 10.2 Å, Cβ 9.2 Å, Cδ1 7.1 Å, and Cγ1 8.2 Å; PDB 1MFR.) If the iron(III) ions produced at the catalytic site maintained the same coordination of the ferrous substrate, I141 should be more affected by the paramagnetic effects. However, resonances of I141 are unchanged while those of I144, further from the active site, are broadened beyond detection. A possible driving force for the change in the coordination environment of the iron upon oxidation may be interaction with Y30 and H54; distances between the metal ion coordinated to Asp140/Gln137/Glu103 in the active site and potential donor atoms of these residues are: Y30 Oη 5.9 Å; H54 Nδ1 4.8 Å, and Nε2 6.1 Å (PDB 1MFR). Migration of the iron(III) products from the active site, which as we now know occurs inside the long channel of each subunit, is required to complete turnover at the active site. In the channel, a coordination site with different affinities for substrate ferrous (lower than the active site) and ferric product (higher than the active site) is needed for the migration of the diferric species. This mechanism is consistent with the reported tenfold reduction in iron oxidation rate upon substitution of Tyr with Phe at position 30 (33). When a second equivalent of iron(II) is added (2 irons/active site), NMR showed paramagnetic species penetrating further into the subunit channels. In addition, the resonances of A26 and I144 remained broadened beyond detection, suggesting that the second cycle of diferric products are at the same site of the diferric products from the first catalytic cycle. The ferritin iron channels contain the products of up to four catalytic turnovers before iron reaches the central cavity. Equilibrium among different coordination states, with comparable and possibly increasingly lower affinity, is needed for the migration of the iron(III) species, when new diferric products are formed and released at the active site.
The magnetic susceptibility data suggest interactions between diferric reaction products in the protein channels and indicate iron aggregation in the protein cage. Newly formed iron(III) dimers from the active site, upon addition of more iron(II) equivalents, react with earlier iron(III) products, on a time scale of the order of several hours, possibly by hydrolysis of water coordinated to ferric ions. The time required may reflect the slow kinetics of ligand exchange typical of iron(III), and may be ascribed to the establishment of interactions, although weak, between the ferric products and the several potential ligands present in the channel. In addition to the already discussed Y30 and H54, other potential ligands are M33, T92, M96, E163, and S170.
The exits of the ferritin channels, where iron(III) catalytic products migrate from active sites to the cavity, are close enough, one to the other, to promote fusion/hydrolysis of the emerging multimeric iron(III) complexes into larger biomineral. The multisubunit structure of ferritin and the symmetry properties of the protein nanocage, thus, are essential elements for the concentration of iron products in the nanocage cavity.
Materials and Methods
Protein Expression and Purification.
Frog (R. catesbieana) M ferritin protein was expressed from a pET3a plasmid (33). Uniformly 13C, 15N-labeled protein for solid-state and 2H, 13C, 15N-labeled protein (deuteration > 90%) for solution studies were expressed in minimal medium with label algal hydrolysate as described in (13). Unlabeled protein was used for Evans magnetic susceptibility measurements. A ([2-13C] glycerol, 15N) ferritin sample was prepared according to the published procedure (17).
Protein Crystallization for Solid-State NMR.
A reaction tube was filled with 1 mL of a solution of PEG 3350 at 25% wt/vol in Na2CO3 100 mM pH 6.5. A 0.2-mL drop of 40 mg/mL ferritin was deposited on the upper surface of this solution. The solution was kept at room temperature for 24 h before the precipitate was separated by centrifugation. In this form, about 50 mg of protein was transferred into a 4-mm MAS rotor.
MAS NMR Spectra.
All the NMR experiments were performed at a field of 16.4 T (700 MHz 1H Larmor frequency, 176.0 MHz 13C Larmor frequency) on a Bruker Avance 700 wide-bore spectrometer equipped with a 4-mm CP-MAS probe. All the experiments were acquired at MAS frequency of 9 kHz. Accurate determination of sample temperatures in solid-state NMR with MAS is known to be problematic, mainly due to frictional and RF heating that make the internal sample temperature significantly different from the measured temperature outside the MAS rotor (34). In order to define the best temperature for our measurements we acquired preliminary data over the 273- to 298-K range of measured temperature values outside the MAS rotor. The best resolution, signal-to-noise, and agreement with solution chemical shift values occurred for data acquired at a measured temperature of 298 K. Standard sequences were used. In all experiments a ramped cross polarization (CP) from protons was used (35). The relaxation delays were of 3–4 s. Acquisition times in the direct dimension were in the range of 15–35 ms. For 2D13C-13C correlation experiments, PDSD and DARR were employed for 13C-13C mixing (16, 36). For the 15N-13C correlation experiments (NCO providing  correlations and NCA providing NiCαi correlations) a double CP (37) sequence was utilized, in which the 15N-13C transfer was achieved using the tangent amplitude modulated pulse on 13C channel. The above mentioned building blocks (NCO or NCA and DARR) were combined to obtain the 3D Ni-Cαi-CXi (NCACX) and 3D
correlations and NCA providing NiCαi correlations) a double CP (37) sequence was utilized, in which the 15N-13C transfer was achieved using the tangent amplitude modulated pulse on 13C channel. The above mentioned building blocks (NCO or NCA and DARR) were combined to obtain the 3D Ni-Cαi-CXi (NCACX) and 3D  (NCOCX) experiments (18). Finally, a 3D Cαi-Ni-CXi-1 (CANCO) experiment (18) was acquired. For 2D DARR experiments, mixing times of 20 and 40 ms were used. The 1H radiofrequency field strength during mixing was matched to the MAS speed to satisfy the n = 1 condition. Each 2D 13C-13C spectrum was acquired with 32 scans, 3744 points in the direct dimension, and 1024 experiments in the indirect dimension and spectral widths of 305 × 305 ppm. The 2D NCO and NCA experiments were acquired with 256 scans, 512 points in the direct dimension, and 160–256 experiments in the indirect dimension and spectral widths of 149 × 141 ppm, respectively. The 3D NCACX and 3D NCOCX were recorded with 32 scans, 2710 points in the direct dimension, 80 experiments in the 15N indirect dimension, and 64 in the 13C indirect dimension (spectral widths of 354 × 45 × 20 ppm). Both were acquired with a 22-ms DARR mixing time. The 3D CANCO was recorded with 128 scans for each experiment, 2710 points in the direct dimension, 32 experiments in the 15N dimension, and 54 in the 13C indirect dimension (spectral widths of 354 × 45 × 30 ppm). All pulse sequences were implemented with 100-kHz spinal 64 1H decoupling (36) during indirect evolution and acquisition periods.
(NCOCX) experiments (18). Finally, a 3D Cαi-Ni-CXi-1 (CANCO) experiment (18) was acquired. For 2D DARR experiments, mixing times of 20 and 40 ms were used. The 1H radiofrequency field strength during mixing was matched to the MAS speed to satisfy the n = 1 condition. Each 2D 13C-13C spectrum was acquired with 32 scans, 3744 points in the direct dimension, and 1024 experiments in the indirect dimension and spectral widths of 305 × 305 ppm. The 2D NCO and NCA experiments were acquired with 256 scans, 512 points in the direct dimension, and 160–256 experiments in the indirect dimension and spectral widths of 149 × 141 ppm, respectively. The 3D NCACX and 3D NCOCX were recorded with 32 scans, 2710 points in the direct dimension, 80 experiments in the 15N indirect dimension, and 64 in the 13C indirect dimension (spectral widths of 354 × 45 × 20 ppm). Both were acquired with a 22-ms DARR mixing time. The 3D CANCO was recorded with 128 scans for each experiment, 2710 points in the direct dimension, 32 experiments in the 15N dimension, and 54 in the 13C indirect dimension (spectral widths of 354 × 45 × 30 ppm). All pulse sequences were implemented with 100-kHz spinal 64 1H decoupling (36) during indirect evolution and acquisition periods.
Spectra were processed with the program TopSpin (Bruker). The 2D and 3D maps were analyzed with the CARA program (38).
Solution NMR.
13C-13C NOESY.
Ferritin solutions, 3 mM in subunits (125 μM in nanocages) in 100-mM MOPS, pH 7.0, were used to acquire 13C-13C NOESY maps (13, 39) at increasing iron concentrations. Freshly prepared solutions of ferrous sulfate in 1-mM HCl were added to the protein solution at a ratio of 2 iron atoms/active site (48 iron atoms/protein nanocage). The conditions were comparable to those used previously for solution Mössbauer and EXAFS spectroscopies (40, 41), which showed complete and fast oxidation of iron. In order to facilitate homogenous diffusion of iron and oxygen, protein samples were titrated according to the following procedure: (i) taking out the sample from the NMR tube, (ii) adding the iron(II) solution and gently pipetting up and down the entire solution, and (iii) spinning the solution. EXAFS data (to be published) confirmed that the iron products obtained with this procedure were all in the iron(III) oxidation state. Our samples were incubated at 298 K for 2 d before collecting the 13C-13C NOESY spectra. After measurements were made on the sample with 48 iron atoms/nanocage, a second aliquot of 2 iron atoms/active site was added and the process repeated until 192 iron atoms/nanocage were analyzed through the 13C-13C NOESY. The 13C-13C NOESY maps at each step of the titration were acquired on a 16.4-T Bruker AVANCE 700 spectrometer equipped with a triple-resonance observe cryoprobe optimized for 13C direct detection, at 298 K, using two different mixing times: 500 ms on the full spectral width (200 ppm), for 18 h, and 150 ms on the aliphatic region (90 ppm) in order to increase resolution, for 16 h to achieve good signal-to-noise ratios.
Magnetic Susceptibility Measurements.
The magnetic susceptibility of ferritin with increasing amounts of iron was measured by the modified Evans method (26, 27) as previously reported (42). Coaxial NMR tubes were used with tert-butyl alcohol and 1,4-dioxane as internal references. The paramagnetic and diamagnetic protein samples were prepared from the same stock solution, i.e., 320 μM in subunit monomer (13.3 μM in protein nanocages) in 100 mM of MOPS, pH 7 with 5 mM of tert-butyl alcohol and 5 mM of 1,4-dioxane. The protein solution was split into two aliquots. The inner capillary contained the diamagnetic apoferritin solution. Freshly prepared solutions of ferrous sulfate in 1 mM of HCl were added to the second aliquot of ferritin in steps of 2 iron/active site (48 iron atoms/protein nanocage) in the outer tube. Addition of iron to the protein solution followed the procedure described for the 13C-13C NOESY. Ten additions of two iron(II)/subunit (up to a total of 20 iron atoms/subunit) were analyzed. The shifts of the proton signals of the two reference molecules were measured on the 16.4-T Bruker AVANCE 700 spectrometer (the same used for 13C-13C NOESY) at 298 K. The set of Evans measurements was repeated twice. An additional set was measured on the 18.7-T Bruker AVANCE 800 spectrometer. The inner-outer tube peak separation (Δδ, expressed in ppm) for each standard was measured and assigned to the bulk susceptibility shift. The paramagnetic contribution to the molar susceptibility of the solute (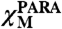 ) was related to the bulk susceptibility shift Δδ as indicated in the following equation:
) was related to the bulk susceptibility shift Δδ as indicated in the following equation:
| [1] |
in which CM is the millimolar concentration of the protein and 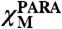 is given in m3 mol-1. The magnetic moment in solution (μeff) was then calculated according to Eq. 2:
is given in m3 mol-1. The magnetic moment in solution (μeff) was then calculated according to Eq. 2:
| [2] |
where k is the Boltzman constant, T is absolute temperature, NA is Avogadro’s number, and μ0 is the vacuum permeability.
Supplementary Material
Acknowledgments.
The authors are grateful to Dr. Manolis Matzapetakis for his contributions to earlier stages of the research. This work has been supported by NIH-DK20251 (E.C.T.), the SPINE2 Contract 031220, the EU-NMR Contract RII3-026145, and the MIUR PRIN-2007 Contract 2007M5MWM9 (I.B., I.C.F., D.L., and P.T.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0908082106/DCSupplemental.
References
- 1.Ferreira C, et al. Early embryonic lethality of H ferritin gene deletion in mice. J Biol Chem. 2000;275:3021–3024. doi: 10.1074/jbc.275.5.3021. [DOI] [PubMed] [Google Scholar]
- 2.Arnaud N, et al. An iron-induced nitric oxide burst precedes ubiquitin-dependent protein degradation for Arabidopsis AtFer1 ferritin gene expression. J Biol Chem. 2006;281:23579–23588. doi: 10.1074/jbc.M602135200. [DOI] [PubMed] [Google Scholar]
- 3.Lewin A, Moore GR, Le Brun NE. Formation of protein-coated iron minerals. Dalton Trans. 2005:3597–3610. doi: 10.1039/b506071k. [DOI] [PubMed] [Google Scholar]
- 4.Liu X, Theil EC. Ferritins: Dynamic management of biological iron and oxygen chemistry. Acc Chem Res. 2005;38:167–175. doi: 10.1021/ar0302336. [DOI] [PubMed] [Google Scholar]
- 5.Hintze KJ, Theil EC. DNA and mRNA elements with complementary responses to hemin, antioxidant inducers, and iron control ferritin-L expression. Proc Natl Acad Sci USA. 2005;102:15048–15052. doi: 10.1073/pnas.0505148102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flenniken ML, et al. A library of protein cage architectures as nanomaterials. Curr Top Microbiol Immunol. 2009;327:71–93. doi: 10.1007/978-3-540-69379-6_4. [DOI] [PubMed] [Google Scholar]
- 7.Moore GM, Gaballa A, Hui M, Ye RW, Helmann JD. Genetic and physiological responses of Bacillus subtilis to metal ion stress. Mol Microbiol. 2005;57:27–40. doi: 10.1111/j.1365-2958.2005.04642.x. [DOI] [PubMed] [Google Scholar]
- 8.Schwartz JK, Liu XS, Tosha T, Theil EC, Solomon EI. Spectroscopic definition of the ferroxidase site in M ferritin: Comparison of binuclear substrate vs. cofactor active sites. J Am Chem Soc. 2008;130:9441–9450. doi: 10.1021/ja801251q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ha Y, Shi D, Small GW, Theil EC, Allewell NM. Crystal structure of bullfrog M ferritin at 2.8 Å resolution: analysis of subunit interactions and the binuclear metal center. J Biol Inorg Chem. 1999;4:243–256. doi: 10.1007/s007750050310. [DOI] [PubMed] [Google Scholar]
- 10.Stillman TJ, et al. Insights into the effects on metal binding of the systematic substitution of five key glutamate ligands in the ferritin of Escherichia coli. J Biol Chem. 2003;278:26275–26286. doi: 10.1074/jbc.M207354200. [DOI] [PubMed] [Google Scholar]
- 11.Bou-Abdallah F, et al. Facilitated diffusion of iron(II) and dioxygen substrates into human H-chain ferritin. A fluorescence and absorbance study employing the ferroxidase center substitution Y34W. J Am Chem Soc. 2008;130:17801–17811. doi: 10.1021/ja8054035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tosha T, Hasan MR, Theil EC. The ferritin Fe2 site at the diiron catalytic center controls the reaction with O2 in the rapid mineralization pathway. Proc Natl Acad Sci USA. 2008;105:18182–18187. doi: 10.1073/pnas.0805083105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matzapetakis M, Turano P, Theil EC, Bertini I. 13C-13C NOESY spectra of a 480 kDa protein: Solution NMR of ferritin. J Biomol NMR. 2007;38:237–242. doi: 10.1007/s10858-007-9163-9. [DOI] [PubMed] [Google Scholar]
- 14.Etzkorn M, et al. Secondary structure, dynamics, and topology of a seven-helix receptor in native membranes, studied by solid-state NMR spectroscopy. Angew Chem Int Ed. 2007;46:459–462. doi: 10.1002/anie.200602139. [DOI] [PubMed] [Google Scholar]
- 15.Pintacuda G, et al. Solid-state NMR of a paramagnetic protein: Assignment and study of the human dimeric oxidized Cu(II), Zn(II) superoxide dismutase. Angew Chem Int Ed. 2007;46:1079–1082. doi: 10.1002/anie.200603093. [DOI] [PubMed] [Google Scholar]
- 16.Szeverenyi NM, Sullivan MJ, Maciel GE. Observation of spin exchange by 2D FT 13C CP/MAS. J Magn Reson. 1982;47:462–475. [Google Scholar]
- 17.Takegoshi K, Nakamura S, Terao T. 13C-1H dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem Phys Lett. 2001;344:631–637. [Google Scholar]
- 18.Li Y, Berthold DA, Frericks HL, Gennis RB, Rienstra CM. Partial 13C and 15N chemical-shift assignments of the disulfide-bond-forming enzyme DsbB by 3D magic-angle spinning NMR spectroscopy. ChemBioChem. 2007;8:434–442. doi: 10.1002/cbic.200600484. [DOI] [PubMed] [Google Scholar]
- 19.Zhang H, Neal S, Wishart DS. RefDB: A database of uniformly referenced protein chemical shifts. J Biomol NMR. 2003;25:173–195. doi: 10.1023/a:1022836027055. [DOI] [PubMed] [Google Scholar]
- 20.Castellani F, et al. Structure of a protein determined by solid-state magic-angle-spinning NMR spectroscopy. Nature. 2002;420:98–102. doi: 10.1038/nature01070. [DOI] [PubMed] [Google Scholar]
- 21.Venters RA, Farmer BT, II, Fierke CA, Spicer LD. Characterizing the use of perdeuteration in NMR studies of large proteins: 13C, 15N and 1H assignments of human carbonic anhydrase II. J Mol Biol. 1996;264:1101–1116. doi: 10.1006/jmbi.1996.0699. [DOI] [PubMed] [Google Scholar]
- 22.Garrett DS, et al. Solution structure of the 30-kDa N-terminal domain of enzyme I of the Escherichia coli phosphoenolpyruvate:sugar phosphotransferase system by multidimensional NMR. Biochemistry. 1997;36:2517–2530. doi: 10.1021/bi962924y. [DOI] [PubMed] [Google Scholar]
- 23.Gardner KH, Rosen MK, Kay LE. Global folds of highly deuterated, methyl-protonated proteins by multidimensional NMR. Biochemistry. 1997;36:1389–1401. doi: 10.1021/bi9624806. [DOI] [PubMed] [Google Scholar]
- 24.Atreya HS, Chary KV. Selective “unlabeling” of amino acids in fractionally 13C labeled proteins: an approach for stereospecific NMR assignments of CH3 groups in Val and Leu residues. J Biomol NMR. 2001;19:267–272. doi: 10.1023/a:1011262916235. [DOI] [PubMed] [Google Scholar]
- 25.Bertini I, Luchinat C, Parigi G. Solution NMR of Paramagnetic Molecules. Amsterdam: Elsevier; 2001. [Google Scholar]
- 26.Evans DF. The determination of the paramagnetic susceptibility of substances in solution by nuclear magnetic resonance. J Chem Soc. 1959:2003–2005. [Google Scholar]
- 27.Phillips WD, Poe M. Contact shifts and magnetic susceptibilities in iron-sulfur proteins as determined from NMR spectra. Methods Enzymol. 1972;24:304–317. doi: 10.1016/0076-6879(72)24077-0. [DOI] [PubMed] [Google Scholar]
- 28.Mukherjee RN, Stack TDP, Holm RH. Angle dependence of the properties of the [Fe2X]4+ bridge unit (X = O,S): structures, antiferromagnetic coupling, and properties in solution. J Am Chem Soc. 1988;110:1850–1861. [Google Scholar]
- 29.Dawson JW, et al. Magnetic susceptibility study of hemerythrin using an ultrasensitive magnetometer. Biochemistry. 1972;11:461–465. doi: 10.1021/bi00753a026. [DOI] [PubMed] [Google Scholar]
- 30.Jameson GN, et al. Stoichiometric production of hydrogen peroxide and parallel formation of ferric multimers through decay of the diferric-peroxo complex, the first detectable intermediate in ferritin mineralization. Biochemistry. 2002;41:13435–13443. doi: 10.1021/bi026478s. [DOI] [PubMed] [Google Scholar]
- 31.Baran P, et al. Synthesis, characterization, and study of octanuclear iron-oxo clusters containing a redox-active Fe4O4-cubane core. Inorg Chem. 2008;47:645–655. doi: 10.1021/ic7020337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Theil EC, Liu XF, Tosha T. Gated pores in the ferritin protein nanocage. Inorg Chim Acta. 2008;361:868–874. doi: 10.1016/j.ica.2007.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fetter J, Cohen J, Danger D, Sanders-Loehr J, Theil EC. The influence of conserved tyrosine 30 and tissue-dependent differences in sequence on ferritin function: Use of blue and purple Fe(III) species as reporters of ferroxidation. JBIC. 1997;2:652–661. [Google Scholar]
- 34.Thurber KR, Tycko R. Measurement of sample temperatures under magic-angle spinning from the chemical shift and spin-lattice relaxation rate of 79Br in KBr powder. J Magn Reson. 2009;196:84–87. doi: 10.1016/j.jmr.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pauli J, Baldus M, van Rossum B, de Groot H, Oschkinat H. Backbone and side-chain 13C and 15N signal assignments of the alpha-spectrin SH3 domain by magic angle spinning solid-state NMR at 17.6 Tesla. ChemBioChem. 2001;2:272–281. doi: 10.1002/1439-7633(20010401)2:4<272::AID-CBIC272>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 36.Fung BM, Khitrin AK, Ermolaev K. An improved broadband decoupling sequence for liquid crystals and solids. J Magn Reson. 2000;142:97–101. doi: 10.1006/jmre.1999.1896. [DOI] [PubMed] [Google Scholar]
- 37.Schaefer J, McKay RA, Stejskal EO. Double-cross-polarization NMR of solids. J Magn Reson. 1979;34:443–447. [Google Scholar]
- 38.Keller R, Wüthrich K. A New Software for the Analysis of Protein NMR Spectra. 2002.
- 39.Bertini I, Felli IC, Kümmerle R, Moskau D, Pierattelli R. 13C-13C NOESY: An attractive alternative for studying large macromolecules. J Am Chem Soc. 2004;126:464–465. doi: 10.1021/ja0357036. [DOI] [PubMed] [Google Scholar]
- 40.Pereira AS, et al. Direct spectroscopic and kinetic evidence for the involvement of a peroxodiferric intermediate during the ferroxidase reaction in fast ferritin mineralization. Biochemistry. 1998;37:9871–9876. doi: 10.1021/bi980847w. [DOI] [PubMed] [Google Scholar]
- 41.Hwang J, et al. A short Fe-Fe distance in peroxodiferric ferritin: Control of Fe substrate versus cofactor decay? Science. 2000;287:122–125. doi: 10.1126/science.287.5450.122. [DOI] [PubMed] [Google Scholar]
- 42.Bertini I, Luchinat C, Turano P, Battaini G, Casella L. The magnetic properties of myoglobin as studied by NMR spectroscopy. Chem-Eur J. 2003;9:2316–2322. doi: 10.1002/chem.200204562. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.