

Abstract
Bacterial single-stranded DNA-binding proteins (SSBs) help to recruit a diverse array of genome maintenance enzymes to their sites of action through direct protein interactions. For all cases examined to date, these interactions are mediated by the evolutionarily conserved C terminus of SSB (SSB-Ct). The essential nature of SSB protein interactions makes inhibitors that block SSB complex formation valuable biochemical tools and attractive potential antibacterial agents. Here, we identify four small molecules that disrupt complexes formed between Escherichia coli SSB and Exonuclease I (ExoI), a well-studied SSB-interacting enzyme. Each compound disrupts ExoI/SSB-Ct peptide complexes and abrogates SSB stimulation of ExoI nuclease activity. Structural and biochemical studies support a model for three of the compounds in which they compete with SSB for binding to ExoI. The fourth appears to rely on an allosteric mechanism to disrupt ExoI/SSB complexes. Subsets of the inhibitors block SSB-Ct complex formation with two other SSB-interaction partners as well, which highlights their utility as reagents for investigating the roles of SSB/protein interactions in diverse DNA replication, recombination, and repair reactions.
Keywords: Exonuclease I, inhibitor, PriA, protein complex, RecQ
To provide cellular genome maintenance machinery access to genomic information, DNA must be unwound to form single-stranded (ss) intermediates (1–3). Although DNA unwinding processes are obligatory, they also present intrinsic risks to cells: ssDNA is sensitive to damage that can be difficult to repair and can self-associate to create structural impediments to genome maintenance. To protect and stabilize unwound ssDNA, cells have evolved specialized ssDNA-binding proteins (SSBs) that bind DNA with high affinity and in a sequence-independent manner. In addition to DNA binding, SSBs have a second activity in which they physically associate with over a dozen different genome maintenance proteins (3). This latter function helps target SSB’s protein partners to their sites of action and, in many cases, stimulates the biochemical activities of the recruited proteins. Both DNA- and protein-binding activities are essential for SSB cellular functions.
Most bacterial SSBs function as homotetramers, with each subunit containing an N-terminal DNA-binding/oligomerization (OB) domain and a C-terminal segment (SSB-Ct) that mediates SSB interactions with other genome maintenance proteins (Fig. 1A) (3, 4). This structural arrangement is distinct from the major eukaryotic SSB (Replication Protein A), which functions as a heterotrimer and lacks the SSB-Ct element found in bacterial SSBs (5). The SSB-Ct sequence includes highly conserved acidic and hydrophobic segments (Asp-Asp-Asp-Ile-Pro-Phe in E. coli SSB), both of which play important roles in forming SSB/protein complexes (6). Interaction between the SSB-Ct and cellular genome maintenance machinery is essential in E. coli and, given the conservation of the SSB-Ct sequence among diverse bacterial SSBs, such interactions are likely to be common among bacteria (3). Tools that allow biochemical dissection of SSB/protein interactions would greatly facilitate experiments probing the diverse roles played by SSB in genome maintenance pathways.
Fig. 1.

Small-molecule inhibitors disrupt ExoI/F-SSB-Ct complexes. (A) Schematic model depicting the effects of small molecules (Red) competing with SSB [tetramer (Yellow) with ssDNA (Orange)] for binding ExoI (Green). (B) Inhibitors [CFAM (Red), BCBP (Orange), BOTP (Green), or MPTA (Blue)] were incubated at indicated concentrations with ExoI/F-SSB-Ct complexes. Decreases in FP are attributed to inhibitor-mediated displacement of the F-SSB-Ct peptide. FP values are the mean of three measurements with errors bar depicting one standard deviation. In some instances, error bars are obscured by the symbols.
The x-ray crystal structure of E. coli Exonuclease I (ExoI) bound to a peptide comprising the SSB-Ct sequence has provided a molecular model of SSB/protein interactions (4). In this structure, the C-terminal-most Phe of the SSB-Ct sequence forms a critical contact with ExoI in which the Phe side chain is enveloped in a hydrophobic pocket and its α-carboxyl group is bound by an Arg side chain from ExoI (4). Intimate recognition of the SSB-Ct Phe appears to be a conserved feature in other SSB/protein interactions as well, and mutations that alter this residue in SSB are lethal to E. coli (4, 6–8). Roles for the acidic SSB-Ct residues in mediating interaction with ExoI have also been identified, leading to a model wherein SSB/ExoI association depends on multiple interactions for stability and specificity (6). The identification of this binding scheme has raised a number of questions as to the conservation of SSB-Ct binding sites among its many binding partners and the consequences of inhibiting interactions with SSB in reconstituted systems and in cells. To begin to answer these questions, we set out to develop a set of chemical tools to interrogate interactions between SSB and its protein partners.
Here, we identify four small-molecule inhibitors that disrupt SSB/ExoI complexes. Two of these compounds have chemical structures that closely resemble the critical C-terminal Phe from the SSB-Ct element, indicating that they could act as peptide mimetics. Each of the inhibitors disrupts ExoI/SSB-Ct peptide complexes and abrogates SSB stimulation of ExoI activity in nuclease reactions. Crystallographic and biochemical studies identify modes of inhibition for the compounds in which three of the molecules block SSB binding to ExoI by competitively binding to the SSB-Ct binding site on ExoI, whereas the fourth molecule appears to rely on allosteric effects to block SSB binding to ExoI. Remarkably, subsets of the compounds also dissociate complexes formed between the SSB-Ct element and two other SSB-interacting proteins (RecQ and PriA DNA helicases), indicating their utility as general SSB/protein complex inhibitors. Together, these studies provide unique biochemical tools for probing the roles of SSB/protein interactions.
Results
Identification of SSB/ExoI Interaction Inhibitors.
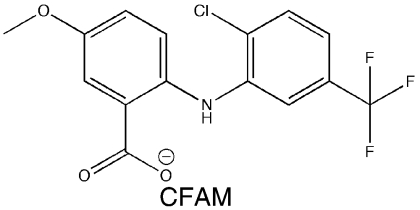
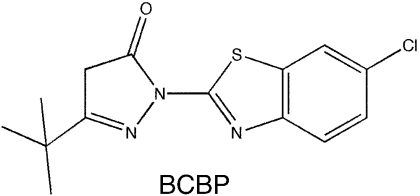
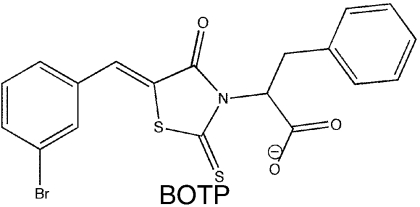
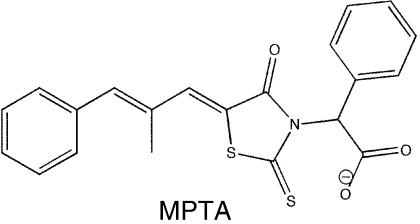
A library of 50,400 small-molecule compounds was screened using a high-throughput fluorescence polarization (FP) assay to identify inhibitors that dissociate the complex formed between SSB and ExoI, a well-studied SSB-binding partner. The assay monitored whether the addition of individual small molecules influences binding of a fluorescein-labeled SSB-Ct peptide (F-SSB-Ct) to ExoI by measuring the FP of F-SSB-Ct (FP is ∼200 mP when bound to ExoI and ∼40 mP when free) (4). Our screen identified more than 400 compounds that lowered FP values to ∼40 mP. The majority of these compounds were disregarded due to their intrinsic fluorescence or fluorescence-quenching properties or due to their common identification as false positive “hits” in other high-throughput FP screens. After testing the dose-dependent activity of the remaining compounds, four were pursued further (referred to as CFAM, BCBP, BOTP, and MPTA, Table 1). Interestingly, two of the compounds (BOTP and MPTA) had phenyl and carboxyl groups organized around chiral carbons in similar positions to analogous groups from the C-terminal-most Phe of the SSB-Ct. Previous experiments have shown that this Phe is critical for SSB/ExoI complex formation (4, 6, 7). These features could therefore be related to the abilities of the compounds to block ExoI/F-SSB-Ct complex formation.
Table 1.
Structures, IC50, Ki,app, 50% inhibition values for small-molecule inhibitors
| Small-molecule inhibitor (structure and name) |
ExoI |
Arg327Ala ExoI |
RecQ |
PriA |
||
| IC50, μM | 50% inhibition, μM | Ki,app, μM | IC50, μM (fold change) | IC50, μM | IC50, μM | |
 |
8 ± 1 | 34 ± 9 | 26 ± 4 | 12 ± 1 (1.5) | > 200 | ∼200 |
 |
23 ± 3 | 74 ± 34 | 32 ± 4 | 72 ± 11 (3.1) | > 200 | > 200 |
 |
28 ± 6 | 290 ± 170 | 126 ± 19 | 41 ± 3 (1.5) | ∼100 | ∼60 |
 |
∼80 | 310 ± 140 | 163 ± 33 | 85 ± 11 (1.1) | ∼100 | ∼100 |
Dose-Dependent Disruption of ExoI/SSB-Ct Peptide Complexes by Small-Molecule Inhibitors.
To quantify the potency of CFAM, BCBP, BOTP, and MPTA, the dose-dependent disruption of ExoI/F-SSB-Ct complexes was measured for each compound in competition-binding experiments. ExoI/F-SSB-Ct complexes were incubated with 0 to 200 μM concentrations of the compounds, and FP values were measured to monitor displacement of the F-SSB-Ct peptide (Fig. 1). In all cases, addition of the compounds led to lowered FP levels that were similar to that of the free F-SSB-Ct peptide. The concentrations required for disruption of 50% of the complexes (IC50) ranged from 8 ± 1 μM (CFAM) to ∼80 μM (MPTA) for the four compounds (Table 1). The IC50 range was similar to previously measured values for SSB-Ct peptides, which varied from 5 μM for the full-length SSB-Ct peptide to 250 μM for truncated peptide variants that bind to more limited portions of the SSB-Ct binding surface on ExoI (6).
Small-Molecule SSB-Interaction Inhibitors Abrogate SSB-Stimulated ExoI Activity.
ExoI nuclease activity is stimulated by SSB in a manner that requires direct interaction between the two proteins (4, 6, 7, 9), which allows SSB stimulation of ExoI activity to be used as a reporter for ExoI/SSB complex formation. To test the abilities of CFAM, BCBP, BOTP, and MPTA to disrupt ExoI/SSB complexes, the effects of the compounds on ExoI-specific activity were measured in ExoI and ExoI/SSB nuclease reactions. ExoI nuclease activity in the absence of SSB was not affected by the addition of the small molecules. However, in SSB-stimulated ExoI reactions, each of the inhibitors reduced ExoI-specific activity to SSB-free or near-SSB-free levels in a dose-dependent manner (Fig. 2). This behavior was consistent with the inhibitors binding at a site (or sites) on ExoI that is distinct from the enzyme substrate binding/active site and is similar to results obtained with SSB-Ct peptide inhibitors (6). The rank potency of the compounds paralleled their IC50 values, ranging from 34 ± 9μM (CFAM) to 310 ± 140μM (MPTA) (Table1), although higher concentrations of each compound were required for inhibition in the nuclease assay compared to the F-SSB-Ct competition-binding experiments (Fig. 1). This difference could reflect local concentration effects that arise from ExoI and SSB binding to common ssDNA molecules. In this situation the effective concentrations of SSB C termini and ExoI will be elevated relative to experiments in which the proteins are not colocalized by DNA binding.
Fig. 2.

Small-molecule inhibitors disrupt ExoI/SSB/ssDNA ternary complexes. Inhibitors (A) CFAM, (B) BCBP, (C) BOTP, or (D) MPTA were incubated at indicated concentrations in ExoI nuclease assays. Inhibition data are shown for assays in which 200 nM SSB was included (Circles) or omitted (Squares) from the reactions. Specific activity values are the mean of three measurements with errors bar depicting one standard deviation. In some instances, error bars are obscured by the symbols.
Kinetic studies were undertaken to determine the mechanism of inhibition of each small molecule and to measure their inhibitor dissociation constant (Ki) values. Previous studies showed that the SSB-Ct peptide acts as competitive inhibitor, increasing the apparent Michaelis constant (Km) of ExoI for SSB/ssDNA substrates without altering the turnover number (kcat) (6). Similar to these peptide results, the addition of each inhibitor to SSB-stimulated ExoI reactions led to concentration-dependent increases in apparent Km values with no measureable effect on kcat (Fig. S1). Statistical comparisons of global fits of the inhibition data for each compound to competitive, noncompetitive, or uncompetitive models showed that the probability that the compounds were acting by competitive inhibition was > 99.9% in all cases. The competitive Ki values for the compounds varied from 26 ± 4 μM for the most potent (CFAM) to 163 ± 33 μM for the least (MPTA) (Table 1). To determine whether the compounds could operate by mixed inhibition, in which the inhibitors can bind to either free ExoI or to the ExoI/substrate complex, the dissociation constants for the free ExoI/inhibitor complex (Kis) and ExoI/substrate/inhibitor complex (Kii) for each compound were estimated via global fits of the kinetic data to a mixed inhibition model (Fig. S1). The Kis values were found to be within error of the competitive Ki values for the compounds, whereas the Kii values were at least 28 times higher than the Kis values (Table S1). These data indicate that there is negligible inhibitor binding to the ExoI/substrate complex, consistent with the compounds acting as competitive inhibitors against ExoI.
X-ray Crystal Structures of SSB/Protein Interaction Inhibitors Bound to ExoI.
Two possible models could explain the inhibitory activities of the small molecules. First, the small-molecule inhibitors might directly compete with SSB for binding to a common site on ExoI. Second, the inhibitors could bind to an allosteric site on ExoI that influences the structure of the enzyme’s SSB binding site. Inhibitors operating by the latter model would still appear to be competitive in kinetic analyses if the allosteric site is exclusively available for inhibitor binding in free ExoI (i.e., if SSB binding to ExoI allosterically inactivates the inhibitor binding site).
To begin to test these models, crystallographic experiments were used to map the binding site(s) for the compounds. Attempts to cocrystallize ExoI bound to the inhibitors failed to produce crystals, which limited structural studies to experiments in which the compounds were soaked into apo-ExoI crystals. An earlier crystal structure showed that SSB-Ct peptides can bind to two sites on ExoI, referred to as A and B sites (Fig. 3A), although only the A site appears to be important for SSB binding and stimulation of ExoI in solution (4). Unfortunately, the arrangement of symmetrically related proteins in the crystal lattice of apo ExoI collapses the essential hydrophobic pocket used to bind the SSB-Ct peptide in the A site (compare Fig. 3A and B) (4). However, the B site is less impacted in the crystal lattice than the A site in the apo-ExoI crystals. Apo E. coli ExoI protein crystals were incubated with CFAM, BCBP, BOTP, or MPTA, and the resulting x-ray crystal structures were determined (Table S2). For crystals incubated with CFAM or BCBP, significant difference electron density was observed (Fig. 3C–F). Difference electron density was not observed for BOTP- or MPTA-soaked ExoI crystals.
Fig. 3.

Crystal structures of CFAM and BCBP bound to E. coli ExoI. (A) Crystal structure of E. coli ExoI bound to two SSB-Ct peptides (4). Biochemical experiments have shown that only the A site is used for SSB/ExoI association in solution; the B site is likely an artifact of high peptide concentrations used in crystallization (4). ExoI surface is stained according to electrostatics (Blue, electropositive; Red, electronegative; White, neutral) and peptides are colored by atoms. A bound Mg2+ ion is shown as a magenta sphere. (B) Crystal packing in apo-ExoI protein crystals. A symmetrically-related ExoI molecule (Yellow) binds to the SSB-Ct peptide-binding surface of an adjacent ExoI molecule, distorting the hydrophobic pocket that accommodates the SSB-Ct peptide in the A site but not the B site. (C) Crystal structure of CFAM bound to ExoI. ExoI is colored as in A and CFAM is colored by atoms (Magenta, carbon; Red, oxygen; Blue, nitrogen; Green, chlorine; Pale Blue, fluorine). (D) Contact map depicting ExoI residues and solution components that contact CFAM. Omit Fo-Fc electron-density contoured at 2.8σ is shown for CFAM. (E) Crystal structure of BCBP bound to ExoI. ExoI is colored as in A and BCBP is colored by atoms (Pink, carbon; Red, oxygen; Blue, nitrogen; Green, chlorine; Yellow, sulfur). (F) Contact map depicting ExoI residues and solution components that contact BCBP. Omit Fo-Fc electron-density contoured at 2.8σ is shown for BCBP. Distances between a water molecule (Red Circle) and BCBP and the closest nitrogen from the Arg327 guanidino group (+) are provided. All figures were made by using Pymol (20).
The 1.55 Å resolution structure of CFAM-bound ExoI showed that the compound binds to the B site of ExoI (Fig. 3C and D). The trifluoromethylanilino group of the inhibitor buries in the hydrophobic pocket that defines the B site, which is formed by Pro228, Trp245, Leu264, Cys330, Leu331, Leu334, and the hydrophobic portion of the Arg327 side chain. The chlorine from CFAM forms apparent van der Waals interactions with the guanidino group on the Arg327 side chain from ExoI and with DMSO present in solution (Fig. 3D). The remainder of the molecule associates with an electropositive element of ExoI referred to as the “basic ridge.” The basic ridge is important for ExoI complex formation with SSB in vitro (4).
The 1.6 Å resolution crystal structure of BCBP bound to ExoI showed that it also binds in the B site on ExoI (Fig. 3E and F). The same ExoI residues that provide the hydrophobic pocket for binding CFAM were also used in BCBP binding. Interestingly, BCBP and CFAM present very different chemical structures in the hydrophobic pocket, indicating that this pocket can accommodate a variety of groups. Similarly to CFAM, Arg327 provides important binding contacts through both the hydrophobic portion of its side chain and through interactions mediated by its guanidino group, although for BCBP the latter group uses apparent hydrogen bonds in water-mediated contacts with the compound. The remainder of BCBP does not bind to the basic ridge of ExoI but instead docks against a surface that is on the opposite side of the hydrophobic pocket. These different binding modes illustrate the potential for distinct inhibitor binding modes that could disrupt ExoI/SSB complexes.
Substituting Ala for Arg 327 in ExoI Has Only Minor Effects on Sensitivity to CFAM, BOTP, and MPTA but More Dramatic Effects with BCBP.
As described above, two models could explain inhibition by the small molecules: direct competition with SSB for binding to the A site on ExoI and indirect blocking of SSB binding through binding to an allosteric site on ExoI. Because the structural features that support SSB-Ct binding are very similar between the A and B sites (4), the observed B-site binding by the inhibitors may provide insights into how the molecules might bind to the A site. Alternatively, the structures might also support a model in which the compounds act by binding to the B site to allosterically alter A-site SSB-Ct binding. To distinguish between these models, the inhibitor sensitivity of an ExoI variant in which Arg327 was changed to Ala was assessed. Changing Arg327 to Ala has no measureable effect on SSB-Ct peptide binding (4) but would be predicted to have a major impact on B-site inhibitor binding given the prominent roles of Arg327 in binding to both CFAM and BCBP in the crystal structures (Fig. 3). We hypothesized that if inhibitor binding to the B site is needed for allosteric inhibition of SSB binding, then replacing Arg327 with Ala should reduce the inhibitor sensitivity of the ExoI variant.
Altering Arg327 to Ala in ExoI led to negligible reductions in CFAM, BOTP, and MPTA sensitivity. These compounds displaced F-SSB-Ct peptide from Arg327Ala ExoI with IC50 values that were only 1.1-fold to 1.5-fold higher than wild-type ExoI (Table 1), indicating that B-site binding does not appear to play a major role in their inhibitory activities. However, BCBP had a 3.1-fold higher IC50 with the Arg327Ala ExoI variant than with wild-type ExoI (Table 1). This change shows that B-site binding plays an important role in the inhibitory activity of BCBP, which distinguishes it from CFAM, BOTP, and MPTA. Similar experiments to test A-site ExoI variants in competition-binding studies could not be performed because structural changes that would be predicted to weaken inhibitor binding would also block SSB-Ct binding (4).
Small-Molecule Disruption of RecQ/SSB-Ct and PriA/SSB-Ct Complexes.
To examine the specificity of the small-molecules inhibitors, the dose-dependent disruption of SSB-Ct complexes formed with E. coli RecQ and PriA DNA helicases was examined in the peptide-binding assay. Direct SSB-Ct binding and SSB-stimulated DNA unwinding activity have been described for both RecQ and PriA (8, 10, 11). However, the fold that binds SSB in ExoI is not conserved in RecQ, nor is it predicted to be conserved in PriA, which leaves open the question of whether the inhibitors can act on these SSB/protein complexes. In addition, ExoI appears to bind the SSB-Ct element with higher affinity than RecQ or PriA (the ExoI, RecQ, and PriA dissociation constants for binding to SSB are 0.14, 6.4, and 2.4 μM, respectively) (4, 10, 11). These stability differences could reflect distinct SSB-Ct binding modes for each protein, which might manifest as differences in sensitivity to the small-molecule inhibitors.
For both RecQ and PriA, subsets of the inhibitors disrupted complexes formed with F-SSB-Ct (Table 1 and Fig. S2). BOTP and MPTA disrupted RecQ/F-SSB-Ct interactions with IC50 values of ∼100 μM, whereas neither CFAM nor BCBP appeared to disrupt the complex. These BOTP and MPTA IC50 values reflected 4-fold weaker, and similar activity of the respective compounds compared with ExoI. BOTP, MPTA, and CFAM disrupted PriA/F-SSB-Ct interactions with IC50 values of ∼60, ∼100, and ∼200 μM, respectively, whereas BCBP again failed to disrupt the complex (Table 1 and Fig. S2). BOTP and MPTA were nearly as potent against PriA/SSB-Ct complexes as they were with ExoI, but CFAM required over 20-fold higher compound concentrations to disrupt the complex than was needed with the ExoI/SSB-Ct complex. These data show that BOTP and MPTA (and CFAM, to a lesser extent) have broad SSB/protein complex disruption properties that could be useful in inhibiting complexes beyond those examined in this work, whereas BCBP appears to be exclusively active against the ExoI/SSB complex. Interestingly, BCBP was also the only inhibitor for which binding to the ExoI B-site was important for its activity, which could be related to its selective inhibition.
Discussion
By using a high-throughput FP screen, we have identified four small-molecule inhibitors that disrupt the complex formed between E. coli SSB and ExoI (Table 1). Each of the compounds dissociates ExoI/SSB-Ct peptide complexes and abrogates SSB stimulation of ExoI activity by blocking ExoI/SSB complex formation (Fig. 1 and 2). The accumulated data support a model in which three of the four compounds compete directly with SSB for binding to ExoI, and the fourth appears to use an allosteric mechanism to block ExoI/SSB complex formation. Subsets of the small molecules are also able to dissociate complexes formed between RecQ or PriA DNA helicases and SSB-Ct, indicating that some of the compounds specifically block the ExoI/SSB interaction, whereas others inhibit a broader set of SSB protein interactions.
Inhibitor Mechanisms for Blocking SSB Binding to ExoI.
For three of our compounds (CFAM, BOTP, and MPTA), the accumulated data point to a direct competition mode of inhibition in which the small molecules compete with SSB for binding to the A site on ExoI. The first indications that these compounds act by direct competition came from biochemical studies. Each compound reduced the specific activity of ExoI in reconstituted ExoI/SSB nuclease reactions but had no effect on ExoI activity in the absence of SSB (Fig. 2). CFAM reduced SSB-stimulated activity to that observed in the absence of SSB, whereas BOTP and MPTA reduced ExoI activity to near-SSB-free levels that were limited by the solubility of the compounds. The inability of the small molecules to inhibit ExoI activity in the absence of SSB showed that they are not general nuclease inhibitors; instead they specifically block SSB-stimulated ExoI activity. As further support for inhibition by direct competition, kinetic analyses showed that each of the small molecules acts by competitive kinetic mechanisms (Table 1, Fig. S1 and Table S1). These data show that the compounds can bind to the free ExoI enzyme but not to ExoI bound to its substrate (SSB/ssDNA). Parallel inhibitory behavior is observed when synthetic SSB-Ct peptides, which act by direct competition, are used as inhibitors in reconstituted ExoI/SSB nuclease reactions (6).
Structure-function studies provided additional critical support for competitive-binding inhibition mechanisms for CFAM, BOTP, and MPTA. Crystallographic studies mapped the CFAM binding site to a known SSB-Ct binding pocket on ExoI (Fig. 3C and D). Although this pocket (B site) does not appear to be important for SSB/ExoI complex formation in solution (4), it serves as a useful surrogate for defining how CFAM might bind to the A site on ExoI. Structural modeling of CFAM into the A site shows that this pocket could readily accommodate the compound by using an analogous binding strategy to that shown in Fig. 3 (Fig. S3). Importantly, mutagenesis results indicated that changing B-site residue Arg327, which interacts with CFAM in the structure, to Ala did not significantly alter CFAM’s IC50 (nor those of BOTP or MPTA) in peptide competition-binding experiments (Table 1). Therefore, B-site binding does not appear to play a role in CFAM, BOTP, or MPTA competition for SSB-Ct binding.
Finally, the broad activity of CFAM, BOTP, and MPTA against complexes formed between ExoI, RecQ, or PriA and the SSB-Ct peptide argues strongly that the small molecules must recognize structurally similar binding sites in the three proteins. As is described further below, ExoI, RecQ, and PriA lack known similarities beyond each protein’s ability to form complexes with SSB. It is therefore unlikely that structural features other than an SSB-Ct binding site would be found in all three proteins. Instead, it is far more likely that CFAM, BOTP, and MPTA act by binding to similar SSB-Ct binding sites in each protein. Consistent with this idea, the structures of two of the inhibitors (BOTP and MPTA) bear a striking similarity to one another and to a Phe amino acid with a free α-carboxyl group (Table 1). This is of interest because several studies have shown that the Phe residue from the SSB-Ct (an evolutionarily invariant residue among bacterial SSBs) participates in crucial interactions that drive SSB/ExoI complex formation (4, 6, 7). Given the similar placement of phenyl and carboxyl groups in BOTP, MPTA, and a C-terminal Phe, we suggest that these small molecules can bind to target proteins in an analogous manner to that of the SSB-Ct Phe.
In contrast to the other inhibitors identified in our screen, biochemical studies indicated that BCBP uses a distinct mechanism to block SSB/ExoI complex formation. First, changing B-site residue Arg327 to Ala led to a significant increase in BCBP’s IC50 in the peptide competition-binding assay (Table 1), indicating that B-site binding plays a role in the activity of BCBP (Table 1). A second notable difference is that BCBP is the only compound among the identified inhibitors that failed to block SSB-Ct binding to both RecQ and PriA. This selective inhibition likely stems from a distinct mechanism that BCBP uses to dissociate the ExoI/SSB complex. We propose that B-site binding by BCBP allosterically induces structural changes in the adjacent A site that disallow SSB binding, which explains the differences between BCBP and the other inhibitors identified in the high-throughput screen.
Subsets of the Compounds Act As General SSB/Protein Complex Inhibitors.
Three of the inhibitors (CFAM, BOTP, and MPTA) were able to dissociate SSB-Ct peptide complexes with RecQ and/or PriA complexes, whereas the remaining compound (BCBP) had no activity against complexes aside from ExoI/SSB-Ct (Table 1 and Fig. S2). Of the three broadly active inhibitors, BOTP and MPTA had similar activities against all of the tested SSB-binding proteins, whereas CFAM had more limited activity. As a corollary to this observation, these results also show that the SSB-Ct binding sites in ExoI, RecQ, and PriA share common features that allow binding of BOTP and MPTA, but that CFAM is better suited for binding ExoI than to the SSB-Ct binding sites in RecQ and PriA. Although the protein folds used by ExoI and RecQ to bind SSB are distinct, electrostatic similarity between the ExoI and RecQ SSB-Ct binding sites has been reported (8). The site of SSB-Ct binding on PriA has not been defined structurally. CFAM, BOTP, and MPTA may therefore prove to be useful general inhibitors of SSB/protein complexes and as small-molecule probes of SSB-Ct binding sites from diverse genome maintenance enzymes. Further experiments will be needed to assess the breadth of activities of the inhibitors report here, but both broad and selective SSB-interaction inhibitors provide useful tools for future dissection of the biochemical roles of numerous SSB/protein complexes found throughout bacteria.
Several observations argue that, in addition to their utility as tools for genome maintenance studies, the small-molecule inhibitors described in this work could form the basis of previously undescribed antibacterial therapeutics. First, protein complex formation with SSB is essential for bacterial cell viability, making SSB/protein interactions potential drug targets in bacteria (3). Second, the SSB-Ct sequence is well conserved among bacterial SSBs but is absent in eukaryotic Replication Protein A (3, 4). This distinction implies that compounds that can disrupt SSB/protein complexes could have the potential for broad-spectrum antibacterial activity but might not inhibit comparable eukaryotic systems. Third, because SSB interacts with at least a dozen different proteins in bacterial cells (3), multiple gain-of-function mutations could potentially be required for resistance acquisition against SSB-interaction inhibitors. Future experiments will be required to explore the possible antibacterial activities of CFAM, BCBP, BOTP, and MPTA.
Materials and Methods
Peptides, Proteins, and Small Molecules.
SSB-Ct peptide (Trp-Met-Asp-Phe-Asp-Asp-Asp-Ile-Pro-Phe) and F-SSB-Ct peptides (SSB-Ct with an N-terminal fluorescein) were prepared as described earlier (6). E. coli ExoI, the ExoI Arg327Ala variant, RecQ, PriA, and SSB were purified as described earlier (4, 12, 13). Small-molecule stocks were obtained from Chembridge or Maybridge: CFAM, 2-[2-chloro-5-(trifluoromethyl)anilino]-5-methoxybenzoic acid; BCBP, 3-(tert-butyl)-1-(6-chloro-1,3-benzothiazol-2-yl)-4,5-dihydro-1H-pyrazol-5-one; BOTP, 2-[5-(3-bromobenzylidene)-4-oxo-2-thioxo-1,3,thiazolidin-3-yl]-3-phenyl-propanoic acid; and MPTA, [5-(2-methyl-3-phenyl-2-propen-1-ylidene)-4-oxo-2-thioxo-1,3-thiazolidin-3-yl] (phenyl)acetic acid.
High-Throughput Interaction Inhibitor Screen.
E. coli ExoI (1 μM) and 10 nM F-SSB-Ct peptide were mixed in 20 mM Tris-HCl (pH 8.0), 100 mM NaCl, 1 mM MgCl2, 1 mM 2-mercaptoethanol for 5 min at room temperature. Individual small molecules [1 μL of 1 mM stock in 100% dimethyl sulfoxide (DMSO)] were added to 30 μL ExoI/F-SSB-Ct mixtures in black walled 384-well plates by using a liquid handling system. The FP of each reaction was measured by using a Tecan Saphire II. In total, 50,400 compounds from the Chemical Diversity, Maybridge, and Chembridge chemical libraries were screened. Control reactions using SSB-Ct peptide (positive control, 1 μL of 2 mM stock in 100% DMSO) or DMSO (negative control, 1 μL of 100% DMSO) were included on each plate.
Small-Molecule Competition-Binding Assay.
E. coli ExoI, PriA, or RecQ (1 μM) was incubated with 10 nM F-SSB-Ct peptide and 0–200 μM inhibitor compounds as described previously (6). Data are presented as the mean of three measurements with error bars depicting one standard deviation of the mean. IC50 values are the concentrations of inhibitors necessary for 50% dissociation of F-SSB-Ct complexes and as determined by fitting the data to a single binding-site model by using the GraphPad Prism software package (Prism). IC50 values for which reliable lower base line values were not observed are interpreted from trend lines and are presented without standard deviation estimates.
Exonuclease Inhibition Assay.
Nuclease assays were carried out as described previously (4). One unit of exonuclease activity is the amount of enzyme required to generate 1 μmol of acid-soluble products per min at 37 °C; specific enzyme activity is expressed as units per mg of protein. Data are presented as the mean of three measurements with Y-axis error bars depicting one standard deviation of the mean. Because BOTP and MPTA had limited solubility at 1 mM, their soluble concentrations in the assay conditions were determined by comparison with standard absorbance curves for each and are presented as the mean of three measurements with X-axis error indicating one standard deviation of the mean. Fifty percent inhibition values are the concentrations of inhibitors necessary to reduce ExoI nuclease activity with SSB/ssDNA substrate by half (relative to activity in the absence of SSB) and were determined by fitting to a single binding-site model by using the GraphPad Prism software package. Saturation kinetic data were globally fitted to competitive, noncompetitive, uncompetitive, and mixed inhibition models by using GraphPad Prism; in all cases the data fit best to a competitive model of inhibition. To compare competitive, noncompetitive, and uncompetitive models statistically, each pair was contrasted by using Akaike’s method (14) by using GraphPad Prism, which reports the relative percent chance that a model is correct. To determine best-fit Kis and Kii values for a mixed inhibition model, data were globally fitted to a mixed inhibition model with the program KinetAsyst (Intellikinetics), which uses the algorithm of Cleland (15).
Crystal Structures of CFAM/ExoI and BCBP/ExoI Complexes.
Apo E. coli ExoI crystals were formed as described previously (4). Crystals were incubated with individual small-molecule inhibitors (100 mM Tris-HCl, pH 8.0, 18% polyethylene glycol (PEG) 4000, 100 mM MgCl2, 1% DMSO, saturated inhibitor) for 2–5 d at room temperature, then transferred to a cryoprotectant solution (100 mM Tris-HCl, pH 8.0, 8% PEG 4000, 100 mM MgCl2, 1% DMSO, saturated inhibitor, 25% glycerol) prior to being flash frozen in liquid nitrogen. Diffraction data were indexed and scaled by using HKL2000 (16) and inhibitor-bound ExoI structures were determined by molecular replacement with Phaser (17) by using the apo E. coli ExoI structure (4) as a search model. Model building and refinement were carried out with Coot (18) and Refmac (19), respectively.
Supplementary Material
Acknowledgments.
We thank members of the Keck lab for critical reading of the manuscript. Small-molecule screening was supported by funds from the Lead Discovery Initiative (Wisconsin Alumni Research Foundation). National Institutes of Health Grant GM068061 (to J.L.K.) supported these studies.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0909191107/DCSupplemental.
References
- 1.Meyer RR, Laine PS. The single-stranded DNA-binding protein of Escherichia coli. Microbiol Rev. 1990;54:342–380. doi: 10.1128/mr.54.4.342-380.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lohman TM, Ferrari ME. Escherichia coli single-stranded DNA-binding protein: Multiple DNA-binding modes and cooperativities. Ann Rev Biochem. 1994;63:527–570. doi: 10.1146/annurev.bi.63.070194.002523. [DOI] [PubMed] [Google Scholar]
- 3.Shereda RD, Kozlov AG, Lohman TM, Cox MM, Keck JL. SSB as an organizer/mobilizer of genome maintenance complexes. Crit Rev Biochem Mol Biol. 2008;43:289–318. doi: 10.1080/10409230802341296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu D, Keck JL. Structural basis of Escherichia coli single-stranded DNA-binding protein stimulation of exonuclease I. Proc Natl Acad Sci USA. 2008;105:9169–9174. doi: 10.1073/pnas.0800741105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wold MS. Replication protein A: A heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Ann Rev Biochem. 1997;66:61–92. doi: 10.1146/annurev.biochem.66.1.61. [DOI] [PubMed] [Google Scholar]
- 6.Lu D, Windsor MA, Gellman SH, Keck JL. Peptide inhibitors identify roles for SSB C-terminal residues in SSB/Exonuclease I complex formation. Biochemistry. 2009;48:6764–6771. doi: 10.1021/bi900361r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Genschel J, Curth U, Urbanke C. Interaction of E. coli single-stranded DNA binding protein (SSB) with exonuclease I. The carboxy-terminus of SSB is the recognition site for the nuclease. Biol Chem. 2000;381:183–192. doi: 10.1515/BC.2000.025. [DOI] [PubMed] [Google Scholar]
- 8.Shereda RD, Reiter NJ, Butcher SE, Keck JL. Identification of the SSB binding site on E. coli RecQ reveals a conserved surface for binding SSB’s C terminus. J Mol Biol. 2009;386:612–625. doi: 10.1016/j.jmb.2008.12.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Molineux IJ, Gefter ML. Properties of the Escherichia coli DNA-binding (unwinding) protein interaction with nucleolytic enzymes and DNA. J Mol Biol. 1975;98:811–825. doi: 10.1016/s0022-2836(75)80012-x. [DOI] [PubMed] [Google Scholar]
- 10.Cadman CJ, McGlynn P. PriA helicase and SSB interact physically and functionally. Nucl Acids Res. 2004;32:6378–6387. doi: 10.1093/nar/gkh980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shereda RD, Bernstein DA, Keck JL. A central role for SSB in Escherichia coli RecQ DNA helicase function. J Biol Chem. 2007;282:19247–19258. doi: 10.1074/jbc.M608011200. [DOI] [PubMed] [Google Scholar]
- 12.Bernstein DA, Keck JL. Domain mapping of Escherichia coli RecQ defines the roles of conserved N- and C-terminal regions in the RecQ family. Nucl Acids Res. 2003;31:2778–2785. doi: 10.1093/nar/gkg376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lopper M, Boonsombat R, Sandler SJ, Keck JL. A hand-off mechanism for primosome assembly in replication restart. Mol Cell. 2007;26:781–793. doi: 10.1016/j.molcel.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akaike H. A new look at the statistical model identification. IEEE Transactions on Automatic Control. 1974;19:716–723. [Google Scholar]
- 15.Cleland WW. Statistical analysis of enzyme kinetic data. In: Purich DL, editor. Enzyme Kinetics and Mechanisms, Methods in Enzymology. Vol 63. New York: Academic; 1979. pp. 103–137. [DOI] [PubMed] [Google Scholar]
- 16.Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in oscillation mode. In: Carter CW, Sweet RM, editors. Methods in Enzymology. Vol 276A. New York: Academic; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 17.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Cryst D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 19.Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Cryst D. 2001;57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 20.Delano WL. The PyMol molecular graphics system. San Carlos, CA: DeLano Scientific; 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.