

Abstract
The structure-activity relationship (SAR) for the N-benzyl group in the clinical antiepileptic agent (R)-lacosamide ((R)-N-benzyl 2-acetamido-3-methoxypropionamide, (R)-3) has been explored. Forty-three compounds were prepared and then evaluated at the National Institute of Neurological Disorders and Stroke Anticonvulsant Screening Program for seizure protection in the maximal electroshock (MES) and subcutaneous Metrazol models. Comparing activities for two series of substituted aryl regioisomers (2′, 3′, 4′) showed that 4′-modified derivatives had the highest activity. Significantly, structural latitude existed at the 4′-site. The SAR indicated that non-bulky 4′-substituted (R)-3 derivatives exhibited superb activity, independent of their electronic properties. Activities in the MES test of several compounds were either comparable with or exceeded that of (R)-3, and surpassed the activities observed for the traditional antiepileptic agents phenytoin, phenobarbital, and valproate.
Epilepsy, a major neurological disorder that affects all populations,1 describes the types of recurrent seizures produced by paroxysmal, excessive, synchronous neuronal discharges in the brain.2,3 In the United States alone, over 2 million people suffer from epilepsy and its sequelae; 340,000 are children.4 For many of these individuals, the disabilities and associated neuropsychological and behavioral factors adversely affect their quality of life. The lifestyle restrictions plus the large expense for treatment, lost productivity, and rehabilitation result in a huge cost to society.4
The treatment mainstay for patients with epileptic disorders has been the long-term and consistent administration of anticonvulsant drugs.5,6 Unfortunately, current medications are ineffective for approximately one-third of these patients. 7 Many continue to have seizures, while others experience disturbing side effects (e.g., drowsiness, dizziness, nausea, liver damage).8 Thus, there is a need for more efficacious drugs that function by different pharmacological pathways.
In 1985, we discovered a novel class of anticonvulsant agents, termed functionalized amino acids (FAA,a 1).9 We subsequently synthesized more than 250 FAAs.10–18 Each of the newly synthesized molecules were evaluated for activity and toxicity in a series of in vivo animal models. Reports from our investigations have led other researchers to further pursue the pharmacological benefits of FAAs.19 Most sites in 1 (e.g., R1, R2, R3) have been modified. We learned that acetyl (CH3C(O)) showed the highest anticonvulsant activity in an induced rodent seizure model (maximal electroshock (MES)) for R 1. The MES test is a well established, commonly used animal model to identify new drug candidates having potential human efficacy to treat generalized and partial seizures that are secondarily generalized. We also determined that the optimal R3 substituent was a benzyl (PhCH2)-type moiety. Finally, isoelectronic substitution of the amide carbonyl (site a) with a thiocarbonyl group, insertion of an alkyl unit or deletion of the chiral center in 1 (site b), and replacement of the amide hydrogen with an alkyl group (site c) reduced anticonvulsant activity.9–18
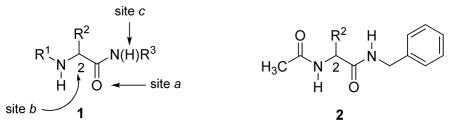
The stringent structural requirements for 1 (R1, R3, sites a-c) led us to focus on molecular template 2. We observed impressive anticonvulsant activities in both mice and rats for smaller sized R2 groups (e.g., 2-furanyl, 2-pyrrolyl, 1-pyrazolyl, 2-oxazolyl, 2-thiazolyl, 2-pyridyl, 2-pyrimidyl, 2-pyrazinyl, O-methylhydroxylamino, N,O-dimethylhydroxylamino) that contained a substituted heteroatom, that is, one atom removed from the C(2) chiral carbon. 13–16,18 These compounds exhibited activity in the MES-induced seizure test (mice, ip) that were either comparable with or exceeded that of the standarded antiepileptic agent phenytoin (MES ED50 = 9.5 mg/kg) in the MES-induced seizure test.20
A single 1 structure-activity relationship (SAR) feature dominated all others. We demonstrated that the principal anticonvulsant activity of the test candidate resided in the D-amino acid configuration.11–13,18 The potency ratio of the more active to the less active isomer21 ranged from 10 to >22. These differences are among the highest, if not the highest, reported for MES-selective anticonvulsants.
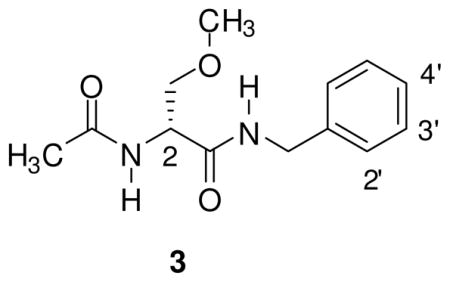
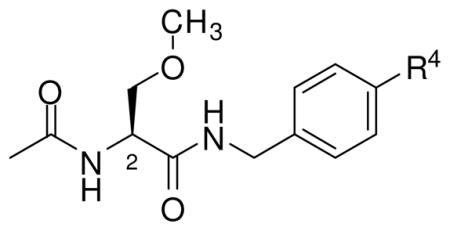
(R)-Lacosamide ((R)-3, (R)-N-benzyl 2-acetamido-3-methoxypropionamide 18) emerged as the lead compound, 1, and has been successfully marketed in the United States and Europe for the adjunctive treatment of partial-onset seizures in adults.22 Whole animal pharmacology studies have revealed a distinctive profile for (R)-3 and other 2s.11–18,23 Accordingly, we have used agents containing “affinity bait” (AB) and “chemical reporter” (CR) groups to initiate a chemical biology–based study to search the brain proteome for (R)-3 binding targets.24 The AB moiety is designed to irreversibly react with the target, and the CR group permits protein detection and capture. In an effort to facilitate these studies, we have determined the SAR for the C(2) side chain oxygen substituent in (R)-325 that identifes the structural parameters for the AB and CR moieties at this site. We report herein that substitution at the N-benzyl 4′ position in (R)-3 provided compounds that exhibit superb anticonvulsant activities and safety profiles. This finding was surprising since preliminary studies with other 2s suggested that substitutions made at this specific site were not very promising candidates. 15,26,27
RESULTS AND DISCUSSION
Choice of Compounds
Earlier studies of 2 indicated that substituting a N-benzyl group led to compounds with diminished anticonvulsant profiles.15,26,27 When the 4′-substituted 2 compounds were compared with the 3′-substituted isomers, the 4′-substituted derivatives provided significantly greater seizure protection as defined in the rodent MES seizure model.18,26 To confirm this finding, we prepared two different series of compounds in which we placed the N-benzyl amide substituent at either the 4′ or 3′ sites (Table 1). In addition, we prepared the 2′ regioisomer. The fluoro ((R)-4–(R)-6) and the trifluoromethoxy ((R)-7–(R)-9) substituents were selected for this study. In both series, the order of favorable anticonvulsant activity in the MES test (mice, ip) was 4′ > 3′ > 2′ (see Pharmacological Activity). Accordingly, we focused our SAR study on the 4′ position in (R)-3.
Table 1.
Effect of Site of N-Benzyl Substitution on the Anticonvulsant Activities of (R)-(N)-(4-Substituted)benzyl 2-Acetamido-3-methoxypropionamides Derivativesa
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Mice (ip)b |
Rat (po)c |
||||||||
| No. | R4 | Aryl Site | Mp (°C) | MES,d ED50 | Tox,e TD50 | PI f | MES,d ED50 | Tox,g TD50 | PI f |
| (R)-3h | H | 143–144 | 4.5 [0.5] (3.7 – 5.5) | 27 [0.25] (26 – 28) | 6.0 | 3.9 [2] (2.6 – 6.2) | >500 [0.5] | >130 | |
| (R)-4 | F | 2′ | 173–175 | >10, <30 [0.5] | >100, <300 [0.5] | 11 [0.25] (9.5 – 13) | >30 [0.5] | >2.7 | |
| (R)-5h | F | 3′ | 150–151 | 6.9 [0.25] (6.1 – 8.0) | 46 [0.25] (40 – 55) | 6.7 | 6.9 [0.5] (4.3 – 9.9) | >400 | >58 |
| (R)-6h | F | 4′ | 144–145 | 4.2 [0.5] (3.5 – 5.1) | 28 [0.25] (22 – 34) | 6.7 | 2.6 [2] (1.9 – 3.6) | >125, <250 | >48 |
| (R)-7 | OCF3 | 2′ | 130–131 | >30, <100 [0.25] | >100, <300 [0.5] | 23 [0.25] (16–35) | >500 | >22 | |
| (R)-8 | OCF3 | 3′ | 147–148 | >10, <30 [0.5] | >30, <100 [0.5] | ||||
| (R)-9 | OCF3 | 4′ | 134–135 | 3.6 [0.25] (3.0 – 4.3) | 13 [0.25] (9.2 – 19) | 3.6 | 1.7 [0.5] (1.4 – 2.2) | 63 [0.5] (47 – 76) | 37 |
| phenytoin i | 9.5 [2] (8.1 – 10) | 66 [2] (53 – 72) | 6.9 | 30 [4] (22 – 39) | j | >100 | |||
| phenobarbital i | 22 [1] (15 – 23) | 69 [0.5] (63 – 73) | 3.2 | 9.1 [5] (7.6 – 12) | 61 [0.5] (44 – 96) | 6.7 | |||
| valproate i | 270 [0.25] (250 – 340) | 430 [0.25] (370 – 450) | 1.6 | 490 [0.5] (350 – 730) | 280 [0.5] (190 – 350) | 0.6 | |||
The compounds were tested through the auspices of the NINDS ASP.
The compounds were administered intraperitoneally. ED50 and TD50 values are in milligrams per kilogram.
The compounds were administered orally. ED50 and TD50 values are in mg/kg.
MES = maximal electroshock seizure test.
TD50 value determined from the rotorod test.
PI = protective index (TD50/ED50).
Tox = behavioral toxicity.
Choi, D.; Stables, J. P.; Kohn, H. J. Med. Chem. 1996, 39, 1907–1916.
Porter, R. J.; Cereghino, J J.; Gladding, G. D.; Hessie, B. J.: Kupferberg, H. J.; Scoville, B.; White, B. G.; Cleveland Clin. Q. 1984, 51, 293–305.
No ataxia observed up to 3000 mg/kg.
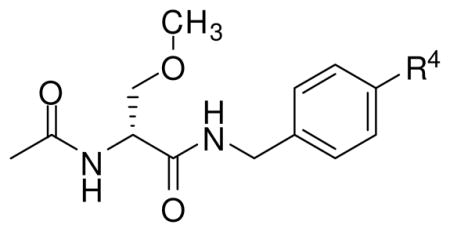
Table 2 lists the prepared N-(4′-substituted)benzyl (R)-3 derivatives ((R)-6, (R)-9–(R)-39). The first set of compounds contained a hydrocarbon group attached at the 4′-benzyl site. We determined the effect of structural size of the N-benzyl 4′-substituent on (R)-3 anticonvulsant activity by preparing alkyl derivatives (R)-10–(R)-14. Next, the effect of a two-carbon hydrocarbon substituent at the 4′-site shape was evaluated by systematically changing it from tetrahedral (sp3) ((R)-11) to planar (sp2) ((R)-21) to linear (sp) ((R)-2324). Then, to gauge the importance of size, electronic effects, hydrophobic interactions, and hydrogen bonding interactions on anticonvulsant activity, we synthesized 4′-substituted hydrocarbon derivatives (R)-15–(R)-20, (R)-22, and (R)-24–(R)-27 in which additional groups were appended to the hydrocarbon moiety. A second set of compounds prepared were N-(4′-substituted)benzyl (R)-3 analogs, (R)-6, (R)-9, and (R)-28–(R)-39, that contained a substituent directly attached to the N-benzyl moiety that either could withdraw or donate electrons to the aromatic ring. Here, we synthesized the cyano ((R)-28), aldehyde ((R)-29), carboxylic acid ((R)-30), methyl ester ((R)-31), nitrogen-substituted ((R)-32–(R)-34), oxygen-containing ((R)-9, (R)-35), halogen-substituted ((R)-6, (R)-36–(R)-38), and sulfamide ((R)-39) (R)-3 analogs.
Table 2.
Selected Physical and Pharmacological Data for (R)-(N)-(4-Substituted)benzyl 2-Acetamido-3-methoxypropionamide Derivativesa
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Mice (ip)b |
Rat (po)c |
|||||||
| No. | R4 | Mp (°C) | MES,d ED50 | Tox,e TD50 | PI f | MES,d ED50 | Tox,g TD50 | PI f |
| (R)-3h | H | 143–144 | 4.5 [0.5] (3.7 – 5.5) | 27 [0.25] (26 – 28) | 6.0 | 3.9 [2] (2.6 – 6.2) | >500 [0.5] | >130 |
| (R)-10 | CH3 | 128–129 | 11 [0.25] (7.3 – 15) | 31 [0.25] (22 – 38) | 3 | 8.1 [0.25] (5.6 – 10) | >500 | >62 |
| (R)-11 | CH2CH3 | 132–133 | >10, <30 [0.5] | >30, <100 [0.5] | 18 [0.25] (13 – 22) | >250 | >14 | |
| (R)-12 | (CH2)2CH3 | 126–127 | 8.5 [0.25] (6.4 – 10) | 13 [0.5] (12 – 15) | 1.5 | <30 [0.25 to 0.5] | >30 [0.25 to 4.0] | |
| (R)-13 | CH(CH3)2 | 95–97 | >10, <30 [0.5] | >100, <300 [0.5] | >30 [0.25 to 4.0] | >30 [0.25 to 4.0] | ||
| (R)-14 | C(CH3)3 | 125–126 | >100, <300 [0.5] | >100, <300 [0.5] | >30 [0.25 to 4.0] | >30 [0.25 to 4.0] | ||
| (R)-15 | CH2NH2·HCl | > 210 | >300 [0.5] | >300 [0.5] | ||||
| (R)-16 | CH2NH-t-Boc | 153–154 | >300 [0.5] | >300 [0.5] | ||||
| (R)-17 | CH2OCH3 | 119–120 | 73 [0.25] (62 – 88) | 180 [0.25] (160 – 210) [0.25] | 2.3 | 45 [0.5] (29 – 76) | >500 [0.25 to 4.0] | >11 |
| (R)-18 | CF3 | 160–161 | >10, <30 [0.5] | >100, <300 [0.5] | 4.9 [1] (2.7 – 7.6) | > 280 | >57 | |
| (R)-19 | (CH2)3OH | 118 | >300 [0.5] | >300 [0.5] | ||||
| (R)-20 | (CH2)3OCH3 | 105–107 | 20 [0.25] (18 – 23) | 62 [0.25] (57 – 74) | 3.1 | 16 [0.5] (8.8 – 25) | >500 [0.5 to 4.0] | >32 |
| (R)-21 | CH=CH2 | 148–149 | 3.5 [0.25] (2.8 – 4.6) | 16 [0.25] (11 – 19) | 4.6 | 7.6 [0.5] (2.9 – 12) | >150, <225 | >20 |
| (R)-22 | C6H5 | 178–180 | 8.0 [0.5] (5.3 – 12) | 11 [0.5] | 2.0 [0.5] (0.8 –3.4) | 49 [0.5] (34 – 77) | >25 | |
| (R)-23i | C≡CH | 161–162 | >3, <10 [0.5] | >10, <30 [0.5] | 3.4 [0.5] (2.0 – 6.1) | > 250 | >74 | |
| (R)-24 | C≡C-CH3 | 178–180 | >10, <30 [0.5] | >30, <100 [0.5] | ||||
| (R)-25 | C≡C-C(CH3)3 | 120–121 | >300 | >30, <100 [0.5] | ||||
| (R)-26 | C≡C-Si(CH3)3 | 126–127 | >3 [0.5] | >3, <10 [0.5] | ||||
| (R)-27 | C≡CCH2OCH3 | 141–142 | 10 [0.25] (7.7 – 12) | 15 [0.25] (13 – 17) | 1.5 | 18 [1] (8.6 – 33) | 100 [0.5] (86 – 120) | 5.5 |
| (R)-28 | CN | 168–169 | 150 [1.0] (140 – 170) | >500 | >3.3 | >30 | >30 | |
| (R)-29 | C(H)O | 132–133 | >300 | >300 | ||||
| (R)-30 | CO2H | 197–198 | >300 [0.5] | >300 [0.5] | ||||
| (R)-31 | CO2CH3 | 167–168 | >100, <300 | >300 [0.5] | ||||
| (R)-32 | NH2 | 151–152 | >300 [0.5] | >300 [0.5] | ||||
| (R)-33 | N(H)C(O)CF3 | 202–204 | >300 [0.5] | >100, <300 [0.5] | >30 | >30 | ||
| (R)-34 | N3 | 149–150 | 8.4 [0.25] (5.7 – 12) | 46 | 5.5 | 3.9 [0.5] (2.5 – 6.2) | >250 | >64 |
| (R)-35 | OCH3 | 146–147 | >30, <100 [0.5] | >100, <300 [0.5] | 28 [0.5] (21 – 35) | >500 [0.25 to 4.0] | >188 | |
| (R)-9 | OCF3 | 134–135 | 3.6 [0.25] (3.0–4.3) | 13 [0.25] (9.2 – 19) | 3.6 | 1.7 [0.5] (1.4 – 2.2) | 63 [0.5] (48–76) | 37 |
| (R)-6h | F | 144–145 | 4.2 [0.25] (3.5 – 5.1) | 28 [0.25] (22 – 34) | 6.7 | 2.6 [2] (1.9 – 3.6) | >125, <250 | >48 |
| (R)-36 | Cl | 155 | 5.0 [0.25] (4.4 – 5.6) | 28 [0.25] (22 – 33) | 5.6 | 1.0 [0.5] (0.4 – 1.8) | >200 | >200 |
| (R)-37 | Br | 159–161 | 8.7 [0.25] (7.2 – 10) | 30 [0.25] (24 – 36) | 3.5 | 4.9 [0.5] (3.0 – 7.2) | 300 [0.25] (217 – 347) | 61 |
| (R)-38 | I | 159–160 | 16 [0.25] (14 – 18) | 41 [0.25] (28 – 58) | 2.6 | 12 [0.25] (7.0 – 19) | >500 [0.25 to 4.0] | >43 |
| (R)-39 | SO2NH2 | 177–179 | >300 [0.5] | >300 [0.5] | ||||
| phenytoin j | 9.5 [2] (8.1 – 10) | 66 [2] (53 – 72) | 6.9 | 30 [4] (22 – 39) | k | >100 | ||
| phenobarbital j | 22 [1] (15 – 23) | 69 [0.5] (63 – 73) | 3.2 | 9.1 [5] (7.6 – 12) | 61 [0.5] (44 – 96) | 6.7 | ||
| valproate j | 270 [0.25] (250 – 340) | 430 [0.25] (370 – 450) | 1.6 | 490 [0.5] (350 – 730) | 280 [0.5] (190 – 350) | 0.6 | ||
The compounds were tested through the auspices of the NINDS ASP.
The compounds were administered intraperitoneally. ED50 and TD50 values are in milligrams per kilogram.
The compounds were administered orally. ED50 and TD50 values are in mg/kg.
MES = maximal electroshock seizure test.
TD50 value determined from the rotorod test.
PI = protective index (TD50/ED50).
Tox = behavioral toxicity.
ref 18.
ref 24.
Porter, R. J.; Cereghino, J J.; Gladding, G. D.; Hessie, B. J.: Kupferberg, H. J.; Scoville, B.; White, B. G.; Cleveland Clin. Q. 1984, 51, 293–305.
No ataxia observed up to 3000 mg/kg.
Finally, for 8 N-(4′-substituted)benzyl (R)-3 derivatives we prepared the corresponding (S)-stereoisomer (Table 3). Structural characterization of the (S)-derivatives ((S)-11, (S)-21, (S)-23, (S)-26, (S)-28, (S)-29, (S)-34, (S)-38) confirmed that the syntheses routinely gave stereospecific products. Determining their pharmacological evaluation permitted us to verify that the principal activity for the N-(4′-substituted)benzyl 2-acetamido-3-methoxypropionamides resided in the (R)-stereoisomer (D-configuration).
Table 3.
Effect of C(2) (S)-Stereochemistry on the Anticonvulsant Activities of (4-Substituted)benzyl Analogs of (N)-Benzyl 2-Acetamido-3-methoxypropionamide Derivativesa
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Mice (ip)b |
Rat (po)c |
|||||||
| no. | R4 | mp (°C) | MES,d ED50 | Tox,e TD50 | PI f | MES,d ED50 | Tox,g TD50 | PI f |
| (S)-3h | H | 143–144 | >100, <300 | >300 | >30 | >30 | ||
| (S)-11 | CH2CH3 | 132–133 | >100, <300 [0.5] | >100, <300 [0.5] | >30 [0.25 to 4] | >30 [0.25 to 4] | ||
| (S)-21 | CH=CH2 | 140–142 | >100, <300 [0.5] | >300 | ||||
| (S)-23i | C≡CH | 159–160 | >300 | >300 | ||||
| (S)-26 | C≡C- Si(CH3)3 | 126–127 | >30, <100 [0.5] | >100, <300 [0.5] | >30 | >30 | ||
| (S)-28 | CN | 168–169 | >100, <300 [0.5] | >300 [0.5] | >30 | >30 | ||
| (S)-29 | C(H)O | 132–133 | >300 | >300 | ||||
| (S)-34 | N3 | 149–150 | >100, <300 | >300 | >30 | >30 | ||
| (S)-38 | I | 159–160 | >100, <300 [0.5] | >300 | >30 | >30 | ||
| phenytoin j | 9.5 [2] (8.1 – 10) | 66 [2] (53 – 72) | 6.9 | 30 [4] (22 – 39) | k | >100 | ||
| phenobarbital j | 22 [1] (15 – 23) | 69 [0.5] (63 – 73) | 3.2 | 9.1 [5] (7.6 – 12) | 61 [0.5] (44 – 96) | 6.7 | ||
| valproate j | 270 [0.25] (250 – 340) | 430 [0.25] (370 – 450) | 1.6 | 490 [0.5] (350 – 730) | 280 [0.5] (190 – 350) | 0.6 | ||
The compounds were tested through the auspices of the NINDS ASP.
The compounds were administered intraperitoneally. ED50 and TD50 values are in milligrams per kilogram.
The compounds were administered orally. ED50 and TD50 values are in mg/kg.
MES = maximal electroshock seizure test.
TD50 value determined from the rotorod test.
PI = protective index (TD50/ED50).
Tox = behavioral toxicity.
ref 18.
ref 24
Porter, R. J.; Cereghino, J J.; Gladding, G. D.; Hessie, B. J.: Kupferberg, H. J.; Scoville, B.; White, B. G.; Cleveland Clin. Q. 1984, 51, 293–305.
No ataxia observed up to 3000 mg/kg.
Chemistry
Synthesis of (R)-4–(R)-39 followed a standard protocol (Scheme 1). Beginning with either N-t-Boc (40)– or N-Cbz (41)–protected D-serine, the acid was coupled with the N-(4′-substituted)benzylamine 42, using either the mixed anhydride (isobutyl chloroformate (IBCF), N-methylmorphline (NMM))28a or the 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM)28b procedure to give amides 43 and 44, respectively. The 4′-substituted benzyl amine was either commercially available or synthesized using previously reported methods (see Supporting Information). We observed that amide coupling proceeded with no racemization of the C(2) chiral center. After purification, methylation of the serine hydroxy group with CH3I and Ag2O provided the optically pure ethers 45 and 46. Removing the N-protecting group (i.e., acid, H2/Pd-C) followed by acetylation (acetyl chloride, Et3N) gave the desired N-(4′-substituted)benzyl (R)-3 derivative 47, in most cases.
Scheme 1.

General Procedure for the Preparation of (R)- and (S)-N-(4′-Substituted)benzyl 2-Acetamido-3-methoxypropionamide Derivatives
For the (R)-stereoisomer of 12, 15, 19, 20, 23–27, 29, 30, and 32–34, additional steps were required to generate the sought-after compound. 4′-Iodo derivative (R)-38 served as a precursor to (R)-12, (R)-23, (R)-24, (R)-25, (R)-26 and (R)-27 (Scheme 2). Using Sonogashira coupling conditions (CuI, PdCl2(PPh3)2, alkyne),29 (R)-38 was treated with 1-propyne, 3,3-dimethylbut-1-yne, trimethylsilylacetylene, and 3-methoxyprop-1-yne to give alkynes (R)-24, (R)-25, (R)-26, and (R)-27, respectively. In most cases, the Pd-based impurities were removed from the coupled products with a scavenger resin (PhosPhonics, cat# SPM32). Treating (R)-26 with TBAF gave the parent acetylene (R)-23, while hydrogenation (PtO2) of (R)-24 gave (R)-12. Catalytic reduction (PtO2, H2) of (R)-27 provided (R)-20 (Scheme 3). Similarly, the 4′-nitro derivative (R)-4826 served as a precursor to the aniline-derivative (R)-32, and trifluoroacetamide (R)-33. Catalytic reduction (PtO2, H2) of (R)-48 gave (R)-32, which was then treated with ethyltrifluorothioacetate30 to yield (R)-33. Synthesis of azide (R)-34 began with (R)-serine ((R)-49) (Scheme 4). Acetylation followed by mixed anhydride coupling28a with 4-aminobenzylamine (50) gave (R)-51. The aniline derivative (R)-51 was directly converted to the azide (R)-52 by sequential addition of t-butyl nitrite and trimethylsilyl azide 31, which was then treated with CH3I and Ag2O to give (R)-34. We used different deprotection methods to prepare (R)-15, (R)-29, and (R)-30 (Scheme 5). Compound (R)-15 was prepared from (R)-16 upon treatment with HCl in dioxane. Similarly, deprotection (HCl in THF/H2O) of acetal (R)-53 gave (R)-29. Finally, methyl ester (R)-31 was converted to acid (R)-30 with LiOH, followed by workup without racemization. We employed a different method to prepare (R)-19 (Scheme 6). For this compound, we prepared optically pure (R)-5825 and then coupled (DMTMM28b) it with benzylamine 57.
Scheme 2.

Sonogashira Coupling of Compound 38 to give Derivatives 12, 23, 24, 25, 26 and 27
Scheme 3.

Catalytic Reduction Procedures to Prepare Compounds 20, 32, 33
Scheme 4.

Preparation of Compounds (R)-34 and (S)-34
Scheme 5.

Deprotection Procedures Used to Prepare Compounds 15, 29 and 30
Scheme 6.

Preparation of Compound (R)-19
The (S)-3 analogs listed in Table 3 were prepared using the same methods employed for the corresponding (R)-stereoisomer.
Three criteria were used to assess the enantiopurity of (R)-4–(R)-39 and (S)-11, (S)-21, (S)-23, (S)-26, (S)-28, (S)-29, (S)-34, and (S)-38. These were melting point, optical rotation, and the detection of only a single acetyl methyl and O-methyl signal in the 1H NMR spectrum for each compound when a saturated solution of (R)-(−)-mandelic acid was added.32
We report in the Experimental Section the details (synthetic procedure, characterization) for the final step for all compounds evaluated in the seizure models. In the Supporting Information, we provide asynthetic scheme for each compound tested, and for all the synthetic compounds prepared in this study the experimental procedures that were utilized, and their physical and spectroscopic properties.
Pharmacological Activity
Compounds (R)-4–(R)-39, (S)-11, (S)-21, (S)-23, (S)-26, (S)-28, (S)-29, (S)-34, and (S)-38 were tested for anticonvulsant activity at the National Institute of Neurological Disorders and Stroke’s (NINDS) Anticonvulsant Screening Program (ASP) using the procedures described by Stables and Kupferberg.33 The pharmacological data from the MES test are summarized in Tables 1–3, along with similar results obtained for (R)-3 and the clinical antiepileptic agents phenytoin, 34 valproate,34 and phenobarbital.34 All compounds were administered intraperitoneally (ip) to mice and orally (po) to rats. The tables list the values that were determined to be protective in blocking hind limb extension induced in the MES seizure model from the rodent identification studies. For compounds that showed significant activity, we report the effective dose (50%) (ED50) values obtained in quantitative screening evaluations. Also provided are the median doses for neurological impairment (50%) (TD50) in mice, using the rotorod test,35 and the behavioral toxicity effects observed in rats. TD50 values were determined for those compounds exhibiting significant activity in the MES test. The protective index (PI = TD50/ED50) for these analogs are also listed. Although all of the compounds were evaluated in the subcutaneous Metrazol (scMet) seizure model none provided any protection at the doses (typically 300 mg/kg) and times (0.5 and 4 h) tested (data not shown). The absence of seizure protection in this assay is a hallmark of FAA activity and this class of compounds.9–18
Table 1 provides the comparative anticonvulsant activities for N-benzyl 2-acetamido-3-methoxypropionamide derivatives in which we systematically placed either a fluoro or a trifluoromethoxy group at the 2′-, 3′-, and 4′-positions of the N-benzylamide moiety. We prepared only the (R)-stereoisomer for these studies since previous investigations showed that the principal anticonvulsant activity resided in this stereoisomer. 11–13,18 In Table 3, we present additional data consistent with this finding. In the two series of compounds listed in Table 1, the 4′-isomers ((R)-6, (R)-9) were more active than the corresponding 3′-regioisomers ((R)-5, (R)-8)), a result in agreement with earlier FAA studies.18,26 Previously, our laboratory had not prepared any N-(2′-substituted)benzylamides 2s. Thus, it was important for us to determine the effect on anticonvulsant activity of substitution at this site. Using the trifluoromethoxy series as an example, the ED50 values for the 2′ ((R)-7), 3′ ((R)-8), and 4′ ((R)-9) isomers in mice were 30–100, 10–30, and 3.6 mg/kg, respectively. A similar trend was observed for the 4′-fluoro regioisomers (R)-4–(R)-6. These findings led us to focus our SAR studies on N-(4′-substituted)benzyl (R)-3 derivatives.
Table 2 reports the observed anticonvulsant activities for 32 N-(4′-substituted)benzyl (R)-3 analogs. Listed first are the 4′-modified alkyl (R)-3 analogs, (R)-10–(R)-14, where the size of the 4′ alkyl group progressively increases. The MES seizure protection in mice for the 4′-substituted methyl ((R)-10, 11 mg/kg), ethyl ((R)-11, 10–30 mg/kg), and propyl ((R)-12, 8.5 mg/kg) derivatives were only slightly lower than the parent compound (R)-3 (4.5 mg/kg).18 A similar finding was observed in the model but using the Sprague Dawley Rat. This finding was surprising since earlier results suggested that substituents placed at the 4′-position of the N-benzyl amide moiety in (R)-3 resulted in a significant loss of MES seizure protection upon administration to mice.15,26,27 For example, we reported previously that the MES ED50 values for the 4′-NO2 ((R)-48) and the 4′-NCS ((R)-59) analogs were 100–300 and 24 mg/kg, respectively.26 However, when the n-propyl moiety in (R)-12 was changed to the isopropyl group to give (R)-13 (mice: MES ED50 = 10–30 mg/kg, rat: MES ED50 = >30 mg/kg) and then to a t-butyl moiety to provide (R)-14 (mice: MES ED50 = 100–300 mg/kg, rats: MES ED50 = >30 mg/kg) we saw a progressive loss of anticonvulsant activity. These findings indicated that inclusion of bulky groups at the 4′ position interfered with drug function. Conversely, non-bulky hydrophobic groups retained excellent activity in both species.
Next, we evaluated six modified 4′-alkyl (R)-3 derivatives, (R)-15–(R)-20, that contained a functional group(s) attached to the 4′-substituted alkyl side chain. Incorporating an amino ((R)-15), a carbamate ((R)-16), and an alcohol ((R)-19) moiety in the alkyl side chain resulted in compounds with no anticonvulsant activity in mice at 300 mg/kg. Inclusion of a methoxy group to give (R)-17 (mice: MES ED50 = 73 mg/kg, rats: MES ED50 = 45 mg/kg), and (R)-20 (mice: MES ED50 = 20 mg/kg, rats: MES ED50 = 16 mg/kg) gave compounds that provided significant seizure protection. Nonetheless, the activity of (R)-17 was noticeably less than its isosteric propyl analog (R)-12 (mice: MES ED50 = 8.5 mg/kg, rats: MES ED50 = <30 mg/kg). Finally, the 4′-trifluoromethyl derivative (R)-18 exhibited significant activity in mice (MES ED50 = 10–30 mg/kg) and exceptional anticonvulsant activity in rats (MES ED 50 = 4.9 mg/kg). The activity of (R)-18 in the rat closely matched that found for (R)-3 (MES ED50 = 3.9 mg/kg), was slightly better than the 4′-methyl derivative (R)-10 (MES ED50 = 8.1 mg/kg), and exceeded the values reported for the established anticonvulsants phenytoin,20 phenobarbital,34 and valproate.20 Taken together, these findings show that additional attachment of specific functional groups (i.e., alcohol, amino, carbamate) on the 4′-alkyl substituent led to compounds with diminished anticonvulsant activities.
The next set of compounds listed in Table 2 were the 4′-ethenyl ((R)-21), 4′-phenyl ((R)-22), and 4′-alkynyl ((R)-23–(R)-27) (R)-3 derivatives. In this series the spatial disposition of the 4′ group was changed from the tetrahedral (sp3) arrangement found in (R)-10–(R)-14 to either trigonal (sp2) or linear (sp) arrangements. Superb activities in the primary models were observed for the non-bulky derivatives (R)-21, (R)-22, (R)-23, (R)-24, and (R)-27. The MES ED50 values in mice ranged from 3–10 mg/kg. Similarly, (R)-21 (MES ED50 = 7.6 mg/kg) and (R)-23 (ED50 = 3.4 mg/kg) showed excellent seizure protection in the rat. Comparing the anticonvulsant activities for the 4′-substituted ethyl ((R)-11), ethenyl ((R)-21), and ethynyl ((R)-23) derivatives, we observed a modest improvement in anticonvulsant activities in both mice and rats as we progressively decreased the spatial size of the 4′-group. This finding is consistent with the pattern observed for the alkyl derivatives (R)-10–(R)-14. The effect of the 4′-group steric size on anticonvulsant activity was further reinforced by the activities of (R)-25 and (R)-27. Attachment of a t-butyl on the terminal end of the 4′-ethynyl group in (R)-23 to give (R)-25 led to a precipitous drop in anticonvulsant activity in mice (MES ED50 = >300 mg/kg), while adding the straight chain methoxymethylene unit to (R)-23 to give (R)-27 did not appreciably affect anticonvulsant activity ((R)-23, mice: MES ED50 = 3–10 mg/kg, rat: MES ED 50 = 3.4 mg/kg; (R)-27, mice: MES ED50 = 10 mg/kg, rat: MES ED50 = 18 mg/kg). When the trimethylsilylacetylenic derivative (R)-26 was evaluated in the MES seizure model, significant toxicity was observed at a 3 mg/kg dose, thereby preventing us from assessing the trimethylsilyl group’s impact on anticonvulsant activity. The pronounced anticonvulsant activity of the 4′-phenyl analog (R)-22 (mice: MES ED50 = 3–10 mg/kg, rat: MES ED50 = 2.0 mg/kg) was surprising given that the lowest energy conformer for biphenyl in the gaseous state is twisted, and the dihedral angle between the two phenyl rings is close to 44°.36 The progressive loss of activity observed as we proceeded from (R)-12–(R)-14 suggested that increases in the three-dimensional steric size of the 4′ site would result in a loss in activity, and thus the attachment of the 4′-phenyl group to give (R)-22 would lead to a reduction in anticonvulsant activity. The excellent seizure protection of (R)-22 can be rationalized by the low barrier for rotation around the central biphenyl bond (est. 1.4 kcal/mol).37 Accordingly, the energy penalty for a near planar arrangement could be compensated by beneficial drug-receptor interactions (e.g., hydrophobic, aromatic interactions).
The final set of 4′-modified (R)-3 analogs was the largest and comprised 14 derivatives each of which contained a functional group directly attached to the N-benzyl moiety ((R)-6, (R)-9, (R)-28–(R)-39). This group of compounds together with the alkyl ((R)-10–(R)-12), trifluoromethyl ((R)-18), ethenyl ((R)-21), alkynyl ((R)-23, (R)-24, (R)-27), and aryl ((R)-22) derivatives documented that attachment of functional groups at the 4′-site can provide strong seizure protection. Testing results demonstrated a highly significant anticonvulsant effect for the 4′-azido ((R)-34), 4′-trifluoromethoxy ((R)-9), 4′-F ((R)-6), and 4′-Cl ((R)-36) derivatives in mice (MES ED50 = 3.6–8.4 mg/kg) as well as in the rat (MES ED50 = 1.0–3.9 mg/kg). These values were either comparable with or exceeded that of (R)-3 (mice: MES ED50 = 4.5 mg/kg; rat: MES ED50 = 3.9 mg/kg).18 As a group, all the 4′-halogen–substituted (R)-3 derivatives ((R)-6, (R)-36–(R)-38) showed pronounced activity. In addition, the smaller halogens (F, Cl) exhibited slightly better seizure protection than the larger halogen derivatives (Br, I). We noticed a significant loss in seizure protection in mice when proceeding from 4′-ethynyl derivative (R)-23 (MES ED50 = 3–10 mg/kg) to the 4′-cyano analog (R)-28 (MES ED 50 = 150 mg/kg). This finding was surprising given their similar size. Finally, attaching a 4′-substituent that could either ionize at physiological pH values or donate a hydrogen bond (i.e., 4′-CO2H ((R)-30), 4′-NH2 ((R)-32), 4′-NHC(O)CF3 ((R)-33), 4′-SO2NH2 ((R)-39) provided compounds that were inactive in the MES test (mice) at the highest doses utilized (300 mg/kg).
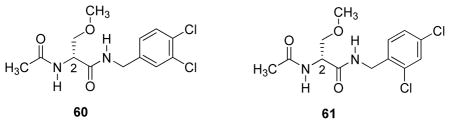
The anticonvulsant activity observed for the 4′-chloro derivative (R)-36 in rats (MES ED50 = 1 mg/kg) made this compound the most potent 2 prepared to date; it was nearly four times more active than (R)-3. Using the methodology advanced by Topliss,38 we explored whether the improved activity of (R)-36 over (R)-3 was due to an electronic effect provided by the 4′-chloro substituent. Accordingly, we prepared the 3′,4′-dichloro derivative (R)-60. When (R)-60 was evaluated in mice, protection in the MES-induced seizure test was between 30–100 mg/kg, demonstrating that this derivative was considerably less active than (R)-36. Following the Topliss methodology, we compared the activity of (R)-36 (MES ED50 = 5 mg/kg) with 4′-CF3 ((R)-18, MES ED50 = 10–30 mg/kg), 4′-Br ((R)-37, MES ED50 = 8.7 mg/kg), 4′-I ((R)-38, MES ED50 = 16 mg/kg), 2′,4′-dichloro ((R)-61, MES ED50 = 30–100 mg/kg), and 4′-NO2 ((R)-48, MES ED50 = 100–300 mg/kg26) (R)-3 derivatives in mice and found that (R)-36 was the most potent. Collectively, these findings suggest that the improved activity observed for (R)-36 over (R)-3 is not due to electronic effects provided by the 4′-chloro substituent.38
Finally, we evaluated eight (S)-N-(4′-substituted)benzyl 2-acetamido-3-methoxypropionamide derivatives (Table 3). Included in this set were 4′-substituents (i.e., I, CH 2CH3, C(H)CH2, CCH, N3) that displayed superb anticonvulsant activities when incorporated in the (R)-enantiomer. Most (S)-stereoisomers exhibited very little seizure protection in the MES test in mice with activities either between 100–300 mg/kg or greater than 300 mg/kg. Only (S)-26 displayed protection but it was modest (MES ED50 = 30–100 mg/kg). Our inability to determine the anticonvulsant activity for (R)-26 did not allow us to further explore the pharmacological basis for this finding. The minimal anticonvulsant activities observed for these (S)-stereoisomers when compared with their N-(4′-substituted)benzyl (R)-3 counterparts were in agreement with previous findings for chiral 2s.11–13,18
Several of the more active N-(4′-substituted)benzyl (R)-3 derivatives were also evaluated in the rapid hippocampal kindled rat model39 (Table 4). This assay is considered a model of partial complex seizures, which are the most common type of seizures in humans. They also represent the subgroup of patients with the highest proportion of drug resistance.40 Administration of (R)-17, (R)-20, (R)-21, (R)-34, (R)-36, and (R)-38 led to a significant decrease in seizures (Racine score proceeding from 5 to 0–2) and a marked corresponding reduction in the after-discharge duration. The ED50 value for (R)-34 and (R)-38 in the hippocampal kindled test (rat, ip) were calculated to be 6 and 12 mg/kg, respectively, surpassing the ED 50 value for (R)-3 (14 mg/kg) and that of the standard antiepileptic drugs phenytoin, phenobarbital, and valproate.23,41
Table 4.
Evaluation of Selected (R)-N-(4-Substituted)benzyl 2-Acetamido-3-methoxypropionamide Derivatives in the Rat Hippocampal Kindled Seizure Model Assaya
 | ||||||
|---|---|---|---|---|---|---|
| Preliminary Hippocampal Kindling Test (Rat) |
Hippocampal Kindled Rat (0.25 h) (ip) |
|||||
| Seizure Score |
After discharge duration (sec) |
|||||
| No. | R4 | Pre-drug | Drug | Pre-drug | Drug | ED50 (mg/kg) (interval) |
| (R)-17 | CH2OCH3 | 5 | 0 | 40 – 69 | 12 | 87 (58 – 129) |
| (R)-20 | (CH2)3OCH3 | 4 – 5 | 2 | 21 – 40 | 21 | ND |
| (R)-21 | CH=CH2 | 4 – 5 | 0 | 27 – 43 | 0–11 | ND |
| (R)-34 | N3 | 4 | 0 | 29 – 65 | 0 | 6 (3 – 9) |
| (R)-36 | Cl | 4 – 5 | 0 | 25 – 159 | 2 | ND |
| (R)-38 | I | 5 | 0 | 17 – 36 | 0–4 | 12 (7.5 – 18) |
| (R)-3b,c | H | 5 | 0 | 33 – 53 | 0 | 14 (9.1 – 18) |
| phenytoinc | 4 – 5 | 3 | 42 – 72 | 51 | 34 (21 – 45) | |
| phenobarbitalc | 20 (14 – 28) | |||||
| valproatec | 210 (150 – 280) | |||||
The compounds were tested through the auspices of the NINDS ASP.
Ref 23.
NINDS ASP internal control data.
CONCLUSIONS
The SAR for the N-benzyl group in the clinically available antiepileptic agent (R)-3 has been explored. We prepared and characterized 43 compounds, which were evaluated at the NINDS ASP for seizure protection in MES and scMet rodent models. Significant anticonvulsant protection against MES-induced seizures was observed for many of these compounds. Comparison of the protective actions for two series of substituted aryl regioisomers (2′, 3′, 4′) demonstrated that the 4′-modified derivatives exhibited the highest level of seizure protection. It was determined that some degree of structural latitude existed at this site. The SAR indicated that non-bulky 4′-substituted (R)-3 derivatives exhibited superb activity independent of their electronic properties. The anticonvulsant activities of (R)-6, (R)-9, (R)-21, (R)-22, (R)-23, (R)-34, (R)-36 and (R)-37 were either comparable with or exceeded that of (R)-318 in rodent MES tests, and they surpassed the activities observed for the traditional antiepileptic agents, phenytoin,34 phenobarbital,34 and valproate.34
The current study complements an earlier report that explored the effect of structural replacement of the C(2)-methoxy group in (R)-3 on anticonvulsant activity.25 In this investigation, we similarly observed that small, non-bulky 3-alkoxy groups provided the highest seizure protection. Taken together, the SAR results provide clues concerning the topography and binding properties of the receptor binding site(s) that elicit (R)-3 function. Furthermore, the finding that structural modifications can be made at the C(2) methoxy side chain and at the 4′ benzyl amide site in (R)-3 without loss of significant anticonvulsant activity sets the stage for the construction of AB&CR agents24 designed to interrogate the brain proteome for drug binding sites. Identification of the (R)-3 receptor sites will advance future antiepileptic drug discovery efforts.
EXPERIMENTAL SECTION
General Methods
Melting points were determined in open capillary tubes using a Thomas-Hoover melting point apparatus and are uncorrected. Infrared spectra (IR) were run on an ATI Mattson Genesis FT-IR spectrometer. Absorption values are expressed in wavenumbers (cm−1). Optical rotations were obtained on a Jasco P-1030 polarimeter at the sodium D line (589 nm) using a 1 dm path length cell. NMR spectra were obtained at 300 MHz or 400 MHz (1H) and 75 MHz or 100 MHz (13C) using TMS as an internal standard. Chemical shifts (d) are reported in parts per million (ppm) from tetramethylsilane. Low-resolution mass spectra were obtained with a BioToF-II-Bruker Daltonics spectrometer by Drs. Matt Crowe and S. Habibi at the University of North Carolina Department of Chemistry. The high- resolution mass spectra were performed on a Bruker Apex-Q 12 Telsa FTICR spectrometer by Drs. MattCrowe and S. Habibi. Microanalyses were performed by Atlantic Microlab, Inc. (Norcross, GA). Reactions were monitored by analytical thin-layer chromatography (TLC) plates (Aldrich, Cat # Z12272-6 or Dynamic Adsorbents Inc., Cat # 84111) and analyzed with 254 nm light. The reactions were purified by MPLC (CombiFlash Rf) with self-packed columns (silica gel from Dynamic Adsorbents Inc., Cat # 02826-25) or by flash column chromatography using silica gel (Dynamic Adsorbents Inc., Cat # 02826-25). All chemicals and solvents were reagent grade and used as obtained from commercial sources without further purification. THF was distilled from blue sodium benzophenone ketyl. Yields reported are for purified products and were not optimized. All compounds were checked by TLC, 1H and 13C NMR, MS, and elemental analyses. The analytical results are within +0.40% of the theoretical value. The TLC, NMR and the analytical data confirmed the purity of the products was ≥95%.
General Procedure for the Mixed Anhydride Coupling Reaction (Method A)
To a cooled THF solution (−78 °C, dry ice acetone bath) of acid (R)/(S)-43 or 44 ([C] ~ 0.1 M) was added NMM (1.0–1.2 equiv), stirred for 2 min, IBCF (1.0–1.2 equiv), stirred for 5 min, and then the desired benzylamine (1.0–1.2 equiv). Upon addition the reaction was allowed to warm to room temperature and further stirred (2–3 h). The salts were filtered and rinsed with THF and the filtrate was concentrated in vacuo. The residue obtained was purified by flash chromatography, and/or by recrystallization from EtOAc when necessary.
General Procedure for the Preparation of 3-Methoxy-2-amidopropionamide Derivatives (Method B)
To a CH3CN solution of alcohol ([C] ~0.05–0.5 M) was successively added Ag2O (5 equiv) and MeI (10 equiv) at room temperature. The reaction mixture was maintained at room temperature (2–4 d) and filtered through Celite®, and the solvent was evaporated in vacuo. The residue was purified by column chromatography on SiO2.
General Procedure for the Preparation Cbz-Deprotection (Method C)
An EtOH or MeOH solution ([C] ~0.01–0.2 M) of (R)- or (S)-46 was treated with H2 (1 atm) in presence of 10% Pd/C (10–20% w/w) at room temperature (16 h). The mixture was carefully filtered through a bed of Celite®. The pad was washed with MeOH and CH2Cl2, and the washings were collected and evaporated in vacuo. The compounds were used without further purification.
General Procedure for the N-Acetylation (Method D)
The 2-aminopropionamide residue was dissolved in CH2Cl2 (0.05–0.3 M) and then triethylamine (1.2–6 equiv) and acetyl chloride (1.2–3 equiv) were carefully added at 0 °C and the resulting solution was stirred at room temperature (2–4 h). An aqueous 10% citric acid solution was added and the reaction mixture was extracted with CH2Cl2. The organic layers were combined, washed with a saturated NaHCO3 solution, dried (Na2SO4), and concentrated in vacuo. The residue was recrystallized with EtOAc and/or purified by column chromatography on SiO2.
Preparation of (R)-N-(2′-Fluoro)benzyl 2-Acetamido-3-methoxypropionamide ((R)-4)
TFA (10 mL) was added to a CH2Cl2 solution (200 mL) of (R)-N-(2′-fluoro)benzyl 2-N-(tert-butoxycarbonyl)amino-3-methoxypropionamide (2.90 g, 8.9 mmol), and the solution was stirred at room temperature (1 h). A saturated aqueous NaHCO3 solution was added until pH ~ 9. The layers were separated and the aqueous layer was washed with CH2Cl2 (2 × 100 mL). The organic layers were combined, dried (MgSO4), and concentrated in vacuo.
Using Method D, triethylamine (3.7 mL, 26.7 mmol) and acetyl chloride (1.3 mL, 17.8 mmol) gave 1.12 g (88%) of (R)-4 as a white solid after purification by flash column chromatography on silica gel with EtOAc/MeOH (10/0 to 9/1) as the eluant: Rf = 0.52 (EtOAc); mp 173–175 °C; [α]25.2D − 23.0° (c 1.0, CHCl3); IR (nujol) 3289, 2925, 2858, 1638, 1550, 1457, 1376, 1237, 1135, 977, 842, 754, 610 cm−1; 1H NMR (CDCl3) δ 2.00 (s, CH3C(O)), 3.35 (s, OCH3), 3.43 (dd, J = 7.2, 9.0 Hz, CHH′O), 3.76 (dd, J = 4.5, 9.0 Hz, CHH′O), 4.46–4.63 (m, CH2NH, CH), 4.84–4.91 (br s, NHCH2), 6.64 (d, J = 7.2 Hz, NHC(O)CH3), 7.00–7.13 (m, 2 ArH), 7.23–7.32 (m, 2 ArH), addition of excess of (R)-(−)-mandelic acid to a CDCl3 solution of (R)-4 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 23.0 (CH3C(O)), 37.4 (d, J = 4.0 Hz, NHCH2), 52.4 (CHCH2), 58.9 (OCH3), 71.8 (CH2OCH3), 115.1 (d, J = 21.0 Hz, C3), 124.2 (d, J = 3.4 Hz, C5), 124.8 (d, J = 14.5 Hz, C4 or C6), 129.1 (d, J = 8.2 Hz, C6 or C4), 129.6–129.7 (br d, C1), 160.8 (d, J = 244.7 Hz, CF), 170.1, 170.4 (2 C(O)); HRMS (M + Cs+)(ESI+) 401.0278 [M + Cs+] (calcd for C13H17FN2O3Cs+ 401.0274). Anal. (C13H17FN2O3): C, H, F, N.
Preparation of (R)-N-(2′-Trifluoromethoxy)benzyl 2-Acetamido-3-methoxypropionamide ((R)-7)
An EtOH solution (200 mL) of (R)-N-(2′-trifluoromethoxy)benzyl 2-N-(benzyloxycarbonyl)amino-3-methoxypropionamide (3.40 g, 8.0 mmol) was treated with H 2 (1 atm) in presence of 10% Pd/C (340 mg) at room temperature (16 h). The mixture was carefully filtered through a bed of Celite®. The pad was washed with MeOH and CH2Cl2, and the washings were collected and evaporated in vacuo.
Using Method D, triethylamine (3.3 mL, 24.0 mmol) and acetyl chloride (1.2 mL, 16.0 mmol) gave 3.50 g (88%) of (R)-7 as a white solid after purification by flash column chromatography on silica gel with EtOAc/MeOH (10/0 to 9/1) as the eluant: Rf = 0.36 (EtOAc); mp 130–131 °C; [α]24.8D −15.1° (c 1.0, CHCl3); IR (nujol) 2919, 2858, 1640, 1547, 1458, 1272, 1203, 1164, 767, 712, 606 cm−1; 1H NMR (CDCl3) δ 1.98 (s, CH3C(O)), 3.35 (s, OCH3), 3.45 (br dd, J = 7.2, 9.3 Hz, CHH′O), 3.76 (dd, J = 4.2, 9.3 Hz, CHH′O), 4.46–4.63 (m, CH2NH, CH), 6.71 (br d, J = 6.3 Hz, NHC(O)CH3), 7.22–7.40 (m, CH2NH, 4 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-7 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 22.9 (CH3C(O)), 38.1 (CH2NH), 52.4 (CHCH2), 58.9 (OCH3), 71.7 (CH2OCH3), 120.4 (1 ArC), 120.5 (q, J = 256.1 Hz, OCF3), 127.0, 128.8, 129.7, 130.3, 147.1 (5 ArC), 170.2, 170.4 (2 C(O)); HRMS (M+Na+)(ESI+) 357.1038 [M + Na+] (calcd for C 14H17F3N2O4Na+ 357.1038). Anal. (C14H17F3N2O4): C, H, F, N.
Preparation of (R)-N-(3′-Trifluoromethoxy)benzyl 2-Acetamido-3-methoxypropionamide ((R)-8)
An EtOH solution (200 mL) of (R)-N-(3′-trifluoromethoxy)benzyl 2-N-(benzyloxycarbonyl)amino-3- methoxypropionamide (1.80 g, 4.2 mmol) was treated with H2 (1 atm) in presence of 10% Pd/C (180 mg) at room temperature (16 h). The mixture was carefully filtered through a bed of Celite® and the filtrate was evaporated in vacuo to obtain a yellow oil.
Using Method D, triethylamine (0.7 mL, 5.0 mmol) and acetyl chloride (0.35 mL, 5.0 mmol) gave 750 mg (54%) of (R)-8 as a white solid after recrystallization with EtOAc: Rf = 0.33 (EtOAc); mp = 147–148 °C; [α]25.0D −12.1° (c 1.0, CHCl3); IR (nujol) 3287, 3041, 2859, 2355, 1637, 1552, 1456, 1377, 1272, 1214, 1150, 715, 610 cm−1; 1H NMR (CDCl3) δ 2.04 (s, CH3C(O)), 3.39 (s, OCH3), 3.44 (dd, J = 7.6, 9.0 Hz, CHH′O), 3.83 (dd, J = 4.2, 9.0 Hz, CHH′O), 4.43–4.60 (m, CH2NH, CH), 6.38–6.46 (br d, NHC(O)CH3), 6.82–6.91 (br t, CH2NH), 7.11–7.15 (m, 2 ArH), 7.19 (d, J = 7.8 Hz, 1 ArH), 7.36 (dt, J = 1.9, 7.8 Hz, 1 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-8 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 23.1 (CH3C(O)), 42.8 (CH2NH), 52.5 (CHCH2), 59.0 (OCH3), 71.6 (CH2OCH3), 120.4 (q, J = 255.6 Hz, OCF3), 119.7, 119.8, 125.6, 130.0, 140.4 (5 ArC), 149.5 (COCF3), 170.2, 170.4 (2 C(O)); LRMS (M + Na+) (ESI+) 357.1 [M + Na +] (calcd for C14H17F3N2O4 Na+ 357.1). Anal. (C14H17F3N2O4): C, H, F, N.
Preparation of (R)-N-(4′-Trifluoromethoxy)benzyl 2-Acetamido-3-methoxypropionamide ((R)-9)
An EtOH solution (400 mL) of (R)-N-(4′-trifluoromethoxy)benzyl 2-N-(benzyloxycarbonyl)amino-3-methoxypropionamide (3.90 g, 9.2 mmol) was treated with H2 (1 atm) in presence of 10% Pd/C (390 mg) at room temperature (16 h). The mixture was carefully filtered through a bed of Celite® and the filtrate was evaporated in vacuo to obtain a brown oil: 1H NMR (CDCl3) δ 1.44–1.95 (br s, NH2), 3.38 (s, OCH3), 3.50–3.67 (br m, CH2, CH), 4.46 (d, J = 5.7 Hz, NCH2), 7.17 (d, J = 8.0 Hz, 2 ArH), 7.31 (d, J = 8.0 Hz, 2 ArH), 7.80–8.00 (br s, NHC(O)).
Using Method D, triethylamine (1.5 mL, 11.0 mmol) and acetyl chloride (0.78 mL, 11.0 mmol) gave 2.50 g (83%) of (R)-9 as a white solid after recrystallization with EtOAc: Rf = 0.49 (EtOAc); mp 134–135 °C; [α]24.9D −17.6° (c 0.5, CHCl3); IR (nujol) 3279, 3088, 2958, 2858, 1638, 1553, 1456, 1377, 1285, 1221, 1148, 988, 918, 841, 725 cm−1; 1H NMR (CDCl3) δ 2.04 (s, CH3C(O)), 3.39 (s, OCH3), 3.44 (dd, J = 7.5, 9.0 Hz, CHH′O), 3.82 (dd, J = 4.2, 9.0 Hz, CHH′O), 4.44–4.52 (m, CH2NH), 4.52–4.59 (m, CH), 6.41 (br d, J = 6.6 Hz, NHC(O)CH3), 6.78–6.89 (br t, CH2NH), 7.18 (d, J = 8.1 Hz, 2 ArH), 7.29 (d, J = 8.1 Hz, 2 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-9 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (CDCl3) δ23.1 (CH3C(O)), 42.7 (CH2NH), 52.5 (CHCH2), 59.1 (OCH3), 71.7 (CH2OCH3), 120.4 (q, J = 255.5 Hz, CF3), 121.2, 128.7, 136.7 (3 ArC), 148.4 (d, J = 1.7 Hz, COCF3), 170.1, 170.4 (2 C(O)); HRMS (M + H+)(ESI+) 335.1219 [M + H+] (calcd for C14H17F3N2O4H+ 335.1218). Anal. (C14H17F3N2O4): C, H, F, N.
Preparation of (R)-N-(4′-Methyl)benzyl 2-Acetamido-3-methoxypropionamide ((R)-10)
An EtOH solution (250 mL) of (R)-N-(4′-methyl)benzyl 2-N-(benzyloxycarbonyl)amino-3-methoxypropionamide (3.20 g, 9.0 mmol) was treated with H2 (1 atm) in presence of 10% Pd/C (320 mg) at room temperature (16 h). The mixture was carefully filtered through a bed of Celite ® and the filtrate was evaporated in vacuo to obtain a colorless oil: 1H NMR (CDCl3) δ 1.60–1.65 (br s, NH2), 2.33 (s, PhCH3), 3.37 (s, OCH3), 3.56–3.67 (m, CH2, CH), 4.34–4.43 (m, HNCH2), 7.10–7.79 (m, 4 ArH), 7.70–7.77 (br s, NHC(O)); 13C NMR (CDCl3) δ 21.0 (PhCH3), 42.9 (CH2NH), 54.8 (CH), 58.8 (OCH3), 74.5 (CH2), 127.6, 129.3, 135.3, 137.0 (4 ArC), 172.5 (C(O)).
Using Method D, triethylamine (1.5 mL, 10.8 mmol) and acetyl chloride (766 μL, 10.8 mmol) gave 1.70 g (72%) of (R)-10 as a white solid after 2 recrystallizations with EtOAc: Rf = 0.50 (EtOAc); mp 128–129 °C; [α]25D −22.4° (c 1.0, CHCl3); IR (nujol) 3285, 3062, 1637, 1548, 1458, 1375, 1311, 1105, 915, 808, 724 cm−1; 1H NMR (CDCl3) δ 1.98 (s, CH3C(O)), 2.32 (PhCH3), 3.35 (s, OCH3), 3.45 (dd, J = 6.9, 9.3 Hz, CHH′O), 3.75 (dd, J = 4.2, 9.3 Hz, CHH′O), 4.36–4.43 (m, CH2NH), 4.57–4.62 (m, CH), 6.71 (br d, J = 6.9 Hz, NHC(O)CH3), 6.98–7.04 (br t, CH2NH), 7.09–7.16 (m, 4 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-10 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 21.0 (PhCH3), 23.1 (CH3C(O)), 43.3 (CH2NH), 52.4 (CHCH2), 59.0 (OCH3), 71.7 (CH2OCH3), 127.4, 129.3, 134.8, 137.1 (4 ArC), 169.9, 170.3 (2 C(O)); HRMS (M + H+)(ESI+) 265.1552 [M + H+] (calcd for C14H20N2O3H+ 265.1552). Anal. (C14H20N2O3): C, H, N.
Preparation of (R)-N-(4′-Ethyl)benzyl 2-Acetamido-3-methoxypropionamide ((R)-11)
A MeOH solution (250 mL) of (R)-N-(4′-ethyl)benzyl 2-N-(benzyloxycarbonyl)amino-3-methoxypropionamide (1.20 g, 3.0 mmol) was treated with H2 (1 atm) in presence of 10% Pd/C (120 mg) at room temperature (3 d). The mixture was carefully filtered through a bed of Celite®. The pad was washed with MeOH and CH2Cl2, and the washings were collected and evaporated in vacuo to obtain a yellow solid.
Using Method D, triethylamine (0.5 mL, 3.5 mmol) and acetyl chloride (250 μL, 3.5 mmol) gave (R)-11 as a white solid after recrystallization with EtOAc: mp 132–133 °C; IR (nujol) 3413, 3305, 3057, 2968, 2932, 1693, 1528, 1266, 1116 cm −1; 1H NMR (CDCl3) δ 1.22 (t, J = 7.5 Hz, CH3CH2), 2.02 (s, CH3C(O)), 2.63 (q, J = 7.5 Hz, CH3CH2), 3.37 (s, OCH3), 3.43 (dd, J = 7.2, 9.0 Hz, CHH′O), 3.79 (dd, J = 4.2, 9.0 Hz, CHH′O), 4.43 (d, J = 6.0 Hz, CH2NH), 4.50–4.58 (m CH), 6.47 (br d, J = 6.9 Hz, NHC(O)CH3), 6.71–6.82 (br t, CH2NH), 7.15–7.18 (m, 4 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-11 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 15.5 (CH2CH3), 23.1 (CH3C(O)), 28.5 (CH2CH3), 43.3 (CH2NH), 52.4 (CHCH2), 59.0 (OCH3), 71.7 (CH2OCH3), 127.5, 128.1, 135.0, 143.5 (4 ArC), 169.8, 170.3 (2 C(O)); HRMS (M + H+)(ESI+) 279.1708 [M + H+] (calcd for C15H22N2O3H+ 279.1705). Anal. (C15H22N2O3): C, H, N.
Preparation of (S)-N-(4′-Ethyl)benzyl 2-Acetamido-3-methoxypropionamide ((S)-11)
A MeOH solution (250 mL) of (S)-N-(4′-ethyl)benzyl 2-N-(benzyloxycarbonyl)amino-3-methoxypropionamide (1.20 g, 3.0 mmol) was treated with H2 (1 atm) in presence of 10% Pd/C (120 mg) at room temperature (3 d). The mixture was carefully filtered through a bed of Celite®. The pad was washed with MeOH and CH2Cl2, and the washings were collected and evaporated in vacuo to obtain a yellow solid.
Using Method D, triethylamine (0.5 mL, 3.5 mmol) and acetyl chloride (250 μL, 3.5 mmol) gave (S)-11 as a white solid after recrystallization with EtOAc: mp 132–133 °C; IR (nujol) 3286, 2932, 2928, 1637, 1554, 1458, 1375, 1311, 1197, 1102, 1051, 909, 821, 724 cm−1; 1H NMR (CDCl3) δ 1.22 (t, J = 7.5 Hz, CH3CH2), 2.02 (s, CH3C(O)), 2.62 (q, J = 7.5 Hz, CH3CH2), 3.37 (s, OCH3), 3.43 (dd, J = 7.2, 9.0 Hz, CHH′O), 3.79 (dd, J = 4.5, 9.0 Hz, CHH′O), 4.43 (d, J = 5.7 Hz, CH2NH), 4.43–4.58 (m CH), 6.48 (br d, J = 6.3 Hz, NHC(O)CH3), 6.71–6.82 (br t, CH2NH), 7.15–7.18 (m, 4 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (S)-11 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 15.8 (CH2CH3), 23.4 (CH3C(O)), 28.7 (CH2CH3), 43.5 (CH2NH), 52.6 (CHCH2), 59.3 (OCH3), 72.0 (CH2OCH3), 127.7, 128.4, 135.2, 143.8 (4 ArC), 170.1, 170.5 (2 C(O)); HRMS (M + H+)(ESI+) 279.1708 [M + H+] (calcd for C15H22N2O3H+ 279.1705). Anal. (C15H22N2O3): C, H, N.
Preparation of (R)-N-(4′-Propyl)benzyl 2-Acetamido-3-methoxypropionamide ((R)-12)
PtO2 (100 mg) was added to an EtOH solution of (R)-24 (700 mg, 2.25 mmol), and the mixture was stirred at room temprerature under H2 (1 atm) (24 h). The reaction mixture was filtered through a pad of Celite®, and the pad was washed successively with EtOH and CH2Cl2. The filtrate was concentrated under vacuum and the residue was purified by flash chromatography on silica gel with EtOAc/hexanes (8/2 to 10/0) as the eluant to obtain (R)-12 (560 mg, 79%) as a white solid: Rf = 0.37 (EtOAc); mp 126–127 °C; [α]25.4D = −27.2° (c 0.5, CHCl3); IR (nujol) 3439, 3374, 3140, 2949, 2859, 1637, 1548, 1457, 1374, 1305, 1137, 1098, 972, 832, 725 cm−1; 1H NMR (CDCl3) δ 0.93 (t, J = 7.5 Hz, CH3CH2), 1.57–1.64 (m, CH2CH2CH3), 2.04 (s, CH3C(O)), 2.57 (t, J = 7.8 Hz, CH2CH2CH3), 3.34–3.45 (m, CHH′O, OCH3), 3.81 (dd, J = 4.2, 9.0 Hz, CHH′O), 4.44 (d, J = 5.7 Hz, CH2NH), 4.50–4.57 (m, CH), 6.38–6.46 (br m, CHNH), 6.63–6.73 (br m, CH2NH), 7.12–7.19 (m, 4 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-12 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 13.8 (CH2CH3), 23.1 (CH3C(O)), 24.5 (CH2CH3), 37.6 (CH2CH2CH3), 43.3 (CH2NH), 52.4 (CHCH2), 59.0 (OCH3), 71.8 (CH2OCH3), 123.4, 128.7, 135.0, 142.0 (4 ArC), 169.9, 170.3 (2 C(O)); HRMS (M + H+)(ESI+) 293.1865 [M + H+] (calcd for C16H24N2O3H+ 293.1865). Anal. (C16H24N2O3): C, H, N.
Preparation of (R)-N-(4′-iso-Propyl)benzyl 2-Acetamido-3-methoxypropionamide ((R)-13)
An EtOH solution (250 mL) of (R)-N-(4′-iso-propyl)benzyl 2-N-(benzyloxycarbonyl)amino-3- methoxypropionamide (1.80 g, 4.7 mmol) was treated with H2 (1 atm) in presence of 10% Pd/C (180 mg) at room temperature (16 h). The mixture was carefully filtered through a bed of Celite® and the filtrate was evaporated in vacuo to obtain a brown oil: 1H NMR (CDCl3) δ 1.24 (d, J = 6.9 Hz, CH(CH3)2), 2.89 (sept., J = 6.9 Hz, CH(CH3)2), 3.38 (s, OCH3), 3.59–3.66 (m, CH2, CH), 4.35–4.49 (m, HNCH2), 7.16 (m, 4 ArH), 7.69–7.82 (br s, NHC(O)); 13C NMR (CDCl3) δ 23.9 (CH(CH3)2), 33.8 (CH(CH3)2), 42.9 (CH2NH), 54.8 (CHCH2), 58.8 (OCH3), 74.5 (CH2), 126.7, 127.7, 135.6, 148.1 (4 ArC), 172.5 (C(O)).
Using Method D, triethylamine (0.79 mL, 5.6 mmol) and acetyl chloride (0.40 mL, 5.6 mmol) gave 2.40 g (62%) of (R)-13 as a white solid after purification by flash column chromatography on silica gel with EtOAc/hexanes (80/20 to 100/0) as the eluant: Rf = 0.39 (EtOAc/hexanes 80/20); mp 95–97 °C; [α]27.0 D −10.5° (c 0.5, CHCl3); IR (nujol) 3289, 2921, 2858, 1635, 1550, 1457, 1376, 1312, 1193, 1101, 1048, 913, 811, 723 cm−1; 1H NMR (CDCl3) δ 1.24 (d, J = 6.9 Hz, CH(CH3)2), 2.03 (s, CH3C(O)), 2.90 (sept., J = 6.9 Hz, CH(CH3)2), 3.38 (s, OCH3), 3.43 (dd, J = 7.8, 9.0 Hz, CHH′O), 3.81 (dd, J = 4.2, 9.0 Hz, CHH′O), 4.44 (d, J = 5.7 Hz, CH2NH), 4.50–4.56 (m, CH), 6.44 (br d, J = 6.3 Hz, NHC(O)CH3), 6.65–6.74 (br t, CH2NH), 7.19 (s, 4 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-13 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 23.2 (CH3C(O)), 24.0 (CH(CH3)2), 33.8 (CH(CH3)2), 43.3 (CH2NH), 52.4 (CHCH2), 59.0 (OCH3), 71.7 (CH2OCH3), 126.7, 127.5, 135.1, 148.2 (4 ArC), 169.9, 170.3 (2 C(O)); MS (M + Na+)(ESI+) 315.2 [M + H+] (calcd for C16H22N2O3Na+ 315.2). Anal. (C16H22N2O3): C, H, N.
Preparation of (R)-N-(4′-tert-Butyl)benzyl 2-Acetamido-3-methoxypropionamide ((R)-14)
An EtOH solution (250 mL) of (R)-N-(4′-tert-butyl)benzyl 2-N-(benzyloxycarbonyl)amino-3- methoxypropionamide (4.00 g, 10.0 mmol) was treated with H2 (1 atm) in presence of 10% Pd/C (400 mg) at room temperature (16 h). The mixture was carefully filtered through a bed of Celite® and the filtrate was evaporated in vacuo to obtain a brown oil: 1H NMR (CDCl3) δ 1.31 (s, C(CH3)3), 1.58–1.62 (br s, NH2), 3.38 (s, OCH3), 3.59–3.66 (m, CH2, CH), 4.36–4.49 (m, HNCH2), 7.21 (d, J = 8.4 Hz, 2 ArH), 7.36 (d, J = 8.4 Hz, 2 ArH), 7.69–7.81 (br s, NHC(O)); 13C NMR (CDCl3) δ 31.3 (C(CH3)3), 34.5 (C(CH3)3), 42.8 (CH2NH), 54.9 (CH), 58.8 (OCH3), 74.5 (CH2), 125.5, 127.4, 135.2, 150.3 (4 ArC), 172.5 (C(O)); HRMS (M + H+)(ESI+) 265.1916 [M + H+] (calcd for C15H24N2O2H+ 265.1916).
Using Method D, triethylamine (1.7 mL, 12.0 mmol) and acetyl chloride (856 μL, 12.0 mmol) gave 1.70 g (55%) of (R)-14 as a white solid after recrystallization with EtOAc: Rf = 0.73 (EtOAc); mp 125– 126 °C; [α]26.8 D −26.0° (c 1.0, CHCl3); IR (nujol) 3280, 2920, 2860, 1636, 1544, 1456, 1374, 1301, 1247, 1197, 1119, 966, 815, 725 cm−1; 1H NMR (CDCl3) δ 1.30 (s, C(CH3)3), 1.99 (s, CH3C(O)), 3.37 (s, OCH3), 3.46 (dd, J = 7.2, 9.0 Hz, CHH′O), 3.77 (dd, J = 4.2, 9.0 Hz, CHH′O), 4.36–4.44 (m, CH2NH), 4.56–4.62 (m, CH), 6.63 (br d, J = 6.6 Hz, NHC(O)CH3), 6.89–6.98 (br t, CH2NH), 7.18 (d, J = 8.1 Hz, 2 ArH), 7.35 (d, J = 8.1 Hz, 2 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-14 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 23.1 (CH3C(O)), 31.3 (C(CH3)3), 34.4 (C(CH3)3), 43.2 (CH2NH), 52.4 (CHCH2), 59.0 (OCH3), 71.8 (CH2OCH3), 125.5, 127.2, 134.7, 150.4 (4 ArC), 169.9, 170.3 (2 C(O)); HRMS (M + H+)(ESI+) 307.2022 [M + H+] (calcd for C17H26N2O3H+ 307.2021). Anal. (C17H26N2O3): C, H, N.
Preparation of (R)-N-(4′-Aminomethyl)benzyl 2-Acetamido-3-methoxypropionamide Hydrochloride ((R)-15)
A saturated HCl solution in dioxane (11.25 mL, 45.0 mL) was added to (R)-16 (1.70 g, 4.5 mmol) at 0 °C and the solution was stirred at room temperature (4 h). The reaction solution was concentrated in vacuo and dried (30 min). The residue was triturated with Et2O and the white solid was filtered to obtain (R)-15 (1.20 g, quant.): Rf = 0.00 (EtOAc); mp > 210 °C; [α]26.2 D −1.6° (c 1.0, DMSO); IR (nujol) 3124, 2919, 2860, 1635, 1639, 1457, 1374, 1281, 1195, 1121, 974, 728 cm−1; 1H NMR (DMSO-d6) δ 1.87 (s, CH3C(O)), 3.25 (s, OCH3), 3.44–3.56 (m, CH2OCH3), 3.97 (q, J = 5.7 Hz, CH2NH3Cl), 4.28 (d, J = 6.0 Hz, CH2NH), 4.36–4.50 (m, CH), 7.26 (d, J = 7.9 Hz, 2 ArH), 7.43 (d, J = 7.9 Hz, 2 ArH), 8.15 (br d, J = 7.8 Hz, NHC(O)CH3), 8.38–8.55 (br m, NH3Cl), 8.58 (br t, J = 6.0 Hz, CH2NH); 13C NMR (DMSO-d6) δ 22.5 (CH3C(O)), 41.6, 41.8 (2 CH2NH), 52.6 (CHCH2), 58.1 (OCH3), 72.0 (CH2OCH3), 127.0, 128.7, 132.2, 139.6 (4 ArC), 169.3, 169.7 (2 C(O)); HRMS (M + H+)(ESI+) 280.1661 [M + H+] (calcd for C14H21N3O3H+ 280.1661). Anal. (C14H22ClN3O3·0.49 HCl): C, H, N.
Preparation of (R)-N-(4′-(tert-Butoxycarbonyl)aminomethyl)benzyl 2-Acetamido-3- methoxypropionamide ((R)-16)
An EtOH solution (400 mL) of (R)-N-(4′-(tertbutoxycarbonyl) aminomethyl)benzyl 2-N-(benzyloxycarbonyl)amino-3-methoxypropionamide (5.30 g, 11.2 mmol) was treated with H2 (1 atm) in presence of 10% Pd/C (530 mg) at room temperature (24 h) and then an additional 470 mg of Pd/C was added and then the mixture was allowed to stir at room temperature (12 h). The mixture was carefully filtered through a bed of Celite® and the filtrate was evaporated in vacuo to obtain a brown oil.
Using Method D, triethylamine (1.9 mL, 13.5 mmol) and acetyl chloride (0.96 mL, 13.5 mmol) gave 2.50 g (60%) of (R)-16 as a white solid after recrystallization with EtOAc: Rf = 0.47 (EtOAc); mp 153– 154 °C; [α]24.9 D −15.9° (c 1.0, CHCl3); IR (nujol) 3318, 2919, 2861, 1675, 1639, 1530, 1458, 1374, 1260, 1167, 1127, 1057, 835, 724 cm−1; 1H NMR (CDCl3) δ 1.45 (s, C(CH3)3), 2.01 (s, CH3C(O)), 3.37 (s, OCH3), 3.44 (dd, J = 7.2, 9.0 Hz, CHH′O), 3.79 (dd, J = 4.2, 9.0 Hz, CHH′O), 4.28 (d, J = 5.7 Hz, CH2NH), 4.43 (d, J = 5.7 Hz, CH2NH), 4.51–4.57 (m, CH), 4.86–4.95 (br s, t-BocNH), 6.49–6.57 (br d, NHC(O)CH3), 6.83–6.93 (br m, CH2NH), 7.17–7.26 (m, 4 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-16 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 23.1 (CH3C(O)), 28.4 (C(CH3)3), 43.2, 44.3 (2 CH2NH), 52.4 (CHCH2), 59.0 (OCH3), 71.7 (CH2OCH3), 79.5 (C(CH3)3), 127.7, 136.9, 138.3 (3 ArC), 155.9 (NC(O)O), 170.0, 170.4 (2 C(O)), one signal was not detected and is believed to overlap with nearby peaks; HRMS (M + H+)(ESI+) 380.2186 [M + H+] (calcd for C19H29N3O5H+ 380.2185). Anal. (C19H29N3O5): C, H, N.
Preparation of (R)-N-(4′-(Methoxymethyl))benzyl 2-Acetamido-3-methoxypropionamide ((R)- 17)
A MeOH solution (400 mL) of (R)-N-(4′-(methoxymethyl))benzyl 2-N-(benzyloxycarbonyl)amino- 3-methoxypropionamide (3.50 g, 9.1 mmol) was treated with H2 (1 atm) in presence of 10% Pd/C (350 mg) at room temperature (16 h). The mixture was carefully filtered through a bed of Celite® and the filtrate was evaporated in vacuo to obtain a colorless oil.
Using Method D, triethylamine (1.5 mL, 10.9 mmol) and acetyl chloride (772 μL, 10.9 mmol) gave 1.50 g (56%) of (R)-17 as a white solid after trituration with EtOAc: Rf = 0.35 (EtOAc); mp 119–120 °C; [α]25 D −25.4° (c 0.5, CHCl3); IR (nujol) 3266, 3069, 2935, 2863, 1635, 1550, 1458, 1457, 1382, 1282, 1226, 1194, 1125, 948, 836, 792, 726 cm−1; 1H NMR (CDCl3) δ 2.03 (s, CH3C(O)), 3.37, 3.38 (2 s, 2 OCH3), 3.43 (dd, J = 7.5, 9.1, CHH′O), 3.80 (dd, J = 4.2, 9.1 Hz, CHH′O), 4.41–4.49 (m, CH2OCH3, CH2NH), 4.51–4.58 (m, CH), 6.42–6.52 (br d, NHC(O)CH3), 6.75–6.84 (br t, CH2NH), 7.24 (d, J = 7.9 Hz, 2 ArH), 7.30 (d, J = 7.9 Hz, 2 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-17 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 23.2 (CH3C(O)), 43.3 (CH2NH), 52.5 (CHCH2), 58.1 (OCH3), 59.1 (OCH3), 71.8 (CH2OCH3), 74.3 (CH2OMe), 127.5, 128.1, 137.3, 137.5 (4 ArC), 170.0, 170.3 (2 C(O)); HRMS (M + H+)(ESI+) 295.1658 [M + H+] (calcd for C15H22N2O4H+ 295.1658). Anal. (C15H22N2O4): C, H, N.
Preparation of (R)-N-(4′-Trifluoromethyl)benzyl 2-Acetamido-3-methoxypropionamide ((R)-18)
An EtOH solution (250 mL) of (R)-N-(4′-trifluoromethyl)benzyl 2-N-(benzyloxycarbonyl)amino-3- methoxypropionamide (1.20 g, 3.0 mmol) was treated with H2 (1 atm) in presence of 10% Pd/C (120 mg) at room temperature (16 h). The mixture was carefully filtered through a bed of Celite®. The pad was washed with MeOH and CH2Cl2, and the washings were collected and evaporated in vacuo to obtain a yellow solid: 1H NMR (CDCl3) δ 1.62–1.67 (br d, NH2), 3.39 (s, OCH3), 3.58–3.72 (m, CH2, CH), 4.52 (d, J = 6.0 Hz, HNCH2), 7.39 (d, J = 8.2 Hz, 2 ArH), 7.58 (d, J = 8.2 Hz, 2 ArH), 7.88–7.89 (br s, NHC(O)).
Using Method D, triethylamine (0.5 mL, 3.5 mmol) and acetyl chloride (250 μL, 3.5 mmol) gave 495 mg (55%) of (R)-18 as a white solid after recrystallization with EtOAc: Rf = 0.45 (EtOAc); mp 160–161 °C; [α]26.7 D +2.6° (c 0.5, DMSO); IR (nujol) 3393, 3278, 3145, 2923, 2834, 2723, 2673, 1638, 1552, 1456, 1374, 1157, 1111, 965, 840, 726 cm−1; 1H NMR (CDCl3) δ 2.05 (s, CH3C(O)), 3.40 (s, OCH3), 3.45 (dd, J = 7.8, 9.3 Hz, CHH′O), 3.83 (dd, J = 4.2, 9.3 Hz, CHH′O), 4.50–4.61 (m, CH2NH, CH), 6.37–6.44 (br d, NHC(O)CH3), 6.85–6.94 (br t, CH2NH), 7.38 (d, J = 8.2 Hz, 2 ArH), 7.58 (d, J = 8.2 Hz, 2 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-18 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 23.1 (CH3C(O)), 42.9 (CH2NH), 52.5 (CHCH2), 59.1 (OCH3), 71.6 (CH2OCH3), 124.0 (q, J = 270.4 Hz, CF3), 125.6 (q, J = 3.4 Hz, C3), 127.5 (C2), 129.7 (q, J = 31.9 Hz, C4), 142.0 (C1), 170.3, 170.5 (2 C(O)); HRMS (M + H+)(ESI+) 319.1270 [M + H+] (calcd for C14H17F3N2O3H+ 307.1269). Anal. (C14H17F3N2O3): C, H, F, N.
Preparation of (R)-N-(4′-(3-Hydroxypropyl))benzyl 2-Acetamido-3-methoxypropionamide ((R)- 19)
4-(3-Hydroxypropyl)benzylamine (600 mg, 3.6 mmol) was added to a THF (33 mL) solution of the (R)-2-acetamido-3-methoxypropionoic acid ((R)-58)25 (532 mg, 3.3 mmol) and the mixture was stirred at room temperature (5 min). DMTMM28b (1.10 g, 4.0 mmol) was added, and the reaction was stirred at room temperature (16 h). The white precipitate was filtered and the filtrate was concentrated in vacuo. The residue was purified by flash column chromatography on silica gel with EtOAc to EtOAc/acetone (5/5) as the eluant to obtain after recrystallization with EtOAc a white solid (560 mg, 55%): Rf = 0.26 (8/2 EtOAc/acetone); mp 118 °C; [α]26.9 D −25.0° (c 0.5, CHCl3); IR (nujol mull) 3339, 3279, 2951, 2862, 1630, 1552, 1456, 1376, 1304, 1195, 1140, 1097, 1038, 909, 820, 724 cm−1; 1H NMR (CDCl3) δ 1.34–1.45 (br m, OH), 1.83–1.93 (br m, CH2CH2CH2), 2.03 (s, CH3C(O)), 2.70 (t, J = 7.8 Hz, CH2Ar), 3.38 (s, OCH3), 3.43 (dd, J = 7.5, 9.0 Hz, CHH′O), 3.63–3.72 (br m, CH2OH), 3.80 (dd, J = 3.9, 9.0 Hz, CHH′O), 4.44 (d, J = 6.0 Hz, CH2NH), 6.41–6.50 (br d, CH3C(O)NH), 6.69–6.79 (m, NH), 7.12– 7.23 (m, 4 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-19 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 23.0 (CH3C(O)), 31.7, 34.2 (2 CH2), 43.2 (NCH2), 52.6 (CHCH2), 59.0 (OCH3), 61.9 (CH2OH), 72.1 (CH2O), 127.5, 128.7, 135.3, 141.2 (4 ArC), 170.1, 170.6 (2 C(O)); HRMS (M + H+)(ESI+) 309.1815 [M + H+] (calcd for C16H24N2O4H+ 319.1814). Anal. (C16H24N2O4): C, H, N.
Preparation of (R)-N-(4′-(3-Methoxypropyl))benzyl 2-Acetamido-3-methoxypropionamide ((R)- 20)
An EtOH solution (30 mL) of (R)-27 (1.00 g, 3.1 mmol) was treated with H2 (1 atm) in the presence of 10% PtO2 (50 mg) at room temperature (16 h). The mixture was carefully filtered through a bed of Celite®. The filtrate was concentrated in vacuo and the residue was purified by flash chromatography on silica gel with EtOAc as the eluant to obtain (R)-20 (510 mg, 51%) as a white solid: Rf = 0.27 (EtOAc); mp 105–107 °C; [α]25 D +3.0° (c 0.5, DMSO); IR (nujol) 3283, 3085, 1638, 1550, 1457, 1379, 1299, 1122, 979, 725, 605 cm−1; 1H NMR (CDCl3) δ 1.81–1.92 (m, CH2), 2.03 (s, CH3C(O)), 2.67 (t, J = 7.8 Hz, CH2Ph), 3.33–3.46 (m, CHH′O, CH2O, 2 OCH3), 3.80 (dd, J = 4.0, 9.1 Hz, CHH′O), 4.44 (d, J = 5.7 Hz, CH2NH), 4.50–4.57 (m, CH), 6.45 (br d, J = 6.6 Hz, NHC(O)CH3), 6.70–6.75 (br t, CH2NH), 7.15–7.20 (m, 4 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-20 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 23.2 (CH3C(O)), 31.2, 31.9 (2 CH2), 43.3 (CH2NH), 52.4 (CHCH2), 58.6, 59.1 (2 OCH3), 71.7, 71.9 (2 CH2OMe), 127.5, 128.8, 135.3, 141.4 (ArC), 169.9, 170.2 (2 C(O)); HRMS (M + Na+)(ESI+) 345.1784 [M + Na+] (calcd for C17H26N2O4Na+ 345.1790). Anal. (C17H26N2O4): C, H, N.
Preparation of (R)-N-(4′-Vinyl)benzyl 2-Acetamido-3-methoxypropionamide ((R)-21)
pTSA (769 mg, 4.04 mmol) was added to a CH2Cl2 (6 mL) solution of (R)-N-(4′-vinyl)benzyl 2-N-(tert-butoxycarbonyl) amino-3-methoxypropionamide (900 mg, 2.7 mmol). The reaction was stirred at room temperature (24 h), and then triethylamine (2.3 mL, 16.2 mmol) followed by acetyl chloride (574 μL, 8.1 mmol) were added at 0 °C. The solution was stirred at room temperature (30 min). Aqueous 10% citric acid was added and then the organic layer was separated. The aqueous layer was extracted with CH2Cl2 (2 × 30 mL). The organic layers were combined, washed with aqueous saturated NaHCO3 and H2O, dried (MgSO4), and concentrated in vacuo. The residue was purified by column chromatography (SiO2; EtOAc) to obtain 700 mg (77%) of white solid. Rf = 0.49 (5/5 EtOAc/acetone); mp = 148–149 °C; [α]26 D = +3.5° (c 1.0, DMSO); IR (nujol) 3281, 3093, 1638, 1552, 1456, 1381, 1298, 1246, 1125, 1043, 986, 917, 825, 722, 604 cm−1; 1H NMR (CDCl3) δ 2.03 (s, CH3C(O)), 3.38 (s, OCH3), 3.43 (dd, J = 7.2, 9.0 Hz, CHH′O), 3.80 (dd, J = 4.2, 9.0 Hz, CHH′O), 4.43–4.52 (m, CH2NH), 4.53–4.58 (m, CH), 5.24 (d, Jcis = 10.8 Hz, CH=CHH′), 5.75 (d, Jtrans = 17.7 Hz, CH=CHH′), 6.46 (br d, J = 6.6 Hz, NHC(O)CH3), 6.70 (dd, Jcis = 10.8 Hz, Jtrans = 17.7 Hz, CH=CH2), 6.75–6.82 (br m, CH2NH), 7.24 (d, J = 8.1 Hz, 2 ArH), 7.35 (d, J = 8.1 Hz, 2 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-21 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 23.3 (CH3C(O)), 43.4 (CH2NH), 52.6 (CHCH2), 59.2 (OCH3), 72.0 (CH2OCH3), 114.1 (CH=CH2), 126.7, 127.8, 136.6, 137.0, 137.6 (4 ArC, CH=CH2), 170.2, 170.6 (2 C(O)). HRMS (M + K+)(ESI+) 315.1115 [M + K+] (calcd for C15H20N2O3K+ 315.1111). Anal. (C15H20N2O3): C, H, N.
Preparation of (S)-N-(4′-Vinyl)benzyl 2-Acetamido-3-methoxypropionamide ((S)-21)
Employing the same procedure utilized for (R)-N-(4′-vinyl)benzyl 2-acetamido-3-methoxypropionamide and using (S)-N-(4′-vinyl)benzyl 2-N-(tert-butoxycarbonyl)amino-3-methoxypropionamide (900 mg, 2.7 mmol), triethylamine (2.3 mL, 16.2 mmol) and acetyl chloride (574 μL, 8.1 mmol) gave 690 mg (76%) of (S)-21 after silica gel column chromatography: Rf = 0.45 (1/9 MeOH/EtOAc); mp 140–142 °C; [α]26 D = −3.1° (c 1.0, DMSO); IR (nujol) 3284, 3087, 1640, 1548, 1457, 1378, 1298, 1244, 1198, 1127, 1046, 985, 912, 826, 721 cm−1; 1H NMR (CDCl3) δ 2.02 (s, CH3C(O)), 3.38 (s, OCH3), 3.44 (dd, J = 7.5, 9.0, Hz, CHH′O), 3.80 (dd, J = 4.2, 9.0 Hz, CHH′O), 4.39–4.48 (m, CH2NH), 4.53–4.59 (m, CH), 5.24 (d, Jcis = 11.1 Hz, CH=CHH′), 5.75 (d, Jtrans = 17.4 Hz, CH=CHH′), 6.48 (br d, J = 6.6 Hz, NHC(O)CH3), 6.70 (dd, Jcis = 11.1 Hz, Jtrans = 17.4 Hz, CH=CH2), 6.82–6.85 (br m, CH2NH), 7.22 (d, J = 8.1 Hz, 2 ArH), 7.34 (d, J = 8.1 Hz, 2 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (S)- 21 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 23.4 (CH3C(O)), 43.5 (CH2NH), 52.6 (CHCH2), 59.3 (OCH3), 71.9 (CH2OCH3), 114.2 (CH=CH2), 126.7, 127.8, 136.5, 137.1, 137.6 (4 ArC, CH=CH2), 170.2, 170.5 (2 C(O)); HRMS (M + K+)(ESI+) [M + K+] 315.1114 (calcd for C15H20N2O3K+ 315.1111). Anal. (C15H20N2O3·0.10H2O): C, H, N.
Preparation of (R)-N-(Biphenyl-4-yl)methyl 2-Acetamido-3-methoxypropionamide ((R)-22)
Using Method D, triethylamine (0.79 mL, 5.7 mmol), acetyl chloride (561 μL, 2.8 mmol) and (R)-N- (biphenyl-4-yl)methyl 2-amino 3-methoxypropionamide hydrochloride (600 mg, 1.9 mmol) gave 380 mg (55%) of (R)-N-(biphenyl-4-yl)methy) 2-acetamido-3-methoxypropionamide as a white solid after purification by flash column chromatography on silica gel with EtOAc/MeOH (100/0 to 80/20) as the eluant and recrystallization with EtOAc: Rf = 0.20 (EtOAc); mp 178–180 °C; [α]26.9 D −8.8° (c 0.5, CHCl3); IR (nujol) 3293, 3087, 2870, 1642, 1547, 1457, 1376, 1298, 1127, 727 cm−1; 1H NMR (CDCl3) δ 2.04 (s, CH3C(O)), 3.40 (s, OCH3), 3.45 (dd, J = 7.5, 9.2 Hz, CHH′O), 3.83 (dd, J = 3.9, 9.2 Hz, CHH′O), 4.50–4.61 (m, CH2NH, CH), 6.45 (br d, J = 5.7 Hz, NHC(O)CH3), 6.76–6.84 (br t, CH2NH), 7.31–7.38 (m, 3 ArH), 7.41–7.48 (m, 2 ArH), 7.55–7.60 (m, 4 ArH), addition of excess (R)-(−)- mandelic acid to a CDCl3 solution of (R)-22 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 23.2 (CH3C(O)), 43.2 (CH2NH), 52.4 (CHCH2), 59.1 (OCH3), 71.6 (CH2OCH3), 127.0, 127.3, 127.4, 127.8, 128.8, 136.9, 140.5 (7 ArC), 170.0, 170.3 (2 C(O)), 1 signal was not detected and is believed to overlap with nearby peaks; HRMS (M + H+)(ESI+) 327.1709 [M + H+] (calcd for C19H22N2O3H+ 327.1708). Anal. (C19H22N2O3·0.1H2O): C, H, N.
Preparation of (R)-N-(4′-Ethynyl)benzyl 2-Acetamido-3-methoxypropionamide ((R)-23)
A 1 M THF solution of TBAF (8.7 mL, 8.66 mmol) was added to a THF (60 mL) solution (R)-26 (1.50 g, 4.33 mmol) and then the solution was stirred at room temperature (4 h). CH2Cl2 and an aqueous 10% citric acid solution were added and the organic layer was separated. The aqueous layer was extracted with CH2Cl2 (2 × 30 mL). The organic layers were combined, dried (MgSO4), and concentrated in vacuo. The residue was purified by flash chromatography on silica gel with EtOAc as the eluant to obtain (R)-23 (0.81 g, 68%) as a white solid: Rf = 0.41 (EtOAc); mp 161–162 °C; [α]24 D = +4.2° (c 0.5, DMSO); IR (nujol) 3290, 1634, 1544, 1458, 1375, 1311, 1240, 1197, 1104, 1041, 714 cm−1; 1H NMR (CDCl3) δ 2.02 (s, CH3C(O)), 3.07 (s, C≡CH), 3.37 (s, OCH3), 3.45 (dd, J = 7.2, 9.3 Hz, CHH′O), 3.77 (dd, J = 4.5, 9.3 Hz, CHH′O), 4.36–4.49 (m, CH2NH), 4.56–4.63 (m, CH), 6.60 (br d, J = 6.9 Hz, NHC(O)CH3), 7.01–7.10 (br t, CH2NH), 7.20 (d, J = 8.2 Hz, 2 ArH), 7.44 (d, J = 8.2 Hz, 2 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-23 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 23.1 (CH3C(O)), 43.1 (CH2NH), 52.5 (CHCH2), 59.0 (OCH3), 71.7 (CH2OCH3), 77.3 (C≡C), 82.2(C≡C), 121.2, 127.3, 132.4, 138.7 (4 ArC), 170.1, 170.4 (2 C(O)); HRMS (M + Na+)(ESI+) 297.1210 [M + Na+] (calcd for C15H18N2O3Na+ 297.1215). Anal. (C15H18N2O3): C, H, N.
Preparation of (S)-N-(4′-Ethynyl)benzyl 2-Acetamido-3-methoxypropionamide ((S)-23)
Employing the preceding procedure and using (S)-26 (50 mg, 0.145 mmol), and TBAF (290 μL, 0.290 mmol) gave 753 mg (91%) of (S)-23 as a white solid: Rf = 0.41 (EtOAc); mp 159–160 °C; [α]24 D = − 4.4° (c 0.5, DMSO); IR (nujol) 3289, 2728, 1635, 1544, 1458, 1375, 1304, 1234, 975, 724 cm−1; 1H NMR (CDCl3) δ 2.03 (s, CH3C(O)), 3.07 (s, C≡CH), 3.38 (s, OCH3), 3.44 (dd, J = 7.5, 9.0 Hz, CHH′O), 3.80 (dd, J = 4.2, 9.0 Hz, CHH′O), 4.41–4.51 (m, CH2NH), 4.52–4.57 (m, CH), 6.46 (br d, J = 5.4 Hz, NHC(O)CH3), 6.80–6.92 (br t, CH2NH), 7.21 (d, J = 8.4 Hz, 2 ArH), 7.45 (d, J = 8.4 Hz, 2 ArH); 1H NMR (DMSO-d6) δ 1.87 (s, CH3C(O)), 3.25 (s, OCH3), 3.44–3.55 (m, CHH′, CHH′), 4.14 (s, C≡CH), 4.29 (d, J = 6.0 Hz, CH2NH), 4.43–4.48 (m, CH), 7.25 (d, J = 8.4 Hz, 2 ArH), 7.42 (d, J = 8.4 Hz, 2 ArH), 8.11 (br d, J = 7.8 Hz, NHC(O)CH3), 8.52 (br t, J = 6.0 Hz CH2NH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (S)-23 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 23.2 (CH3C(O)), 43.2 (CH2NH), 52.5 (CHCH2), 59.1 (OCH3), 71.7 (CH2OCH3), 77.3 (C≡C), 83.3 (C≡C), 121.2, 127.3, 132.4, 138.8 (4 ArC), 170.1, 170.4 (2 C(O)), HMQC experiment showed a correlation between the δ 3.07 signal in the 1H NMR and the δ 77.3 peak in the 13C NMR; 13C NMR (DMSO-d6) δ 22.3 (CH3C(O)), 41.6 (CH2NH), 52.4 (CHCH2), 58.0 (OCH3), 71.8 (CH2OCH3), 80.2 (C≡C), 83.2 (C≡C), 119.8, 126.9, 131.3, 140.2 (4 ArC), 169.2, 169.6 (2 C(O)), HMQC experiment showed a correlation between the δ 4.14 signal in the 1H NMR and the δ 80.2 peak in the 13C NMR; HRMS (M + Na+)(ESI+) 297.1212 [M + Na+] (calcd for C15H18N2O3Na+ 297.1215). Anal. (C15H18N2O3·0.25H2O): C, H, N.
Preparation of (R)-N-(4′-(Prop-1-ynyl))benzyl 2-Acetamido-3-methoxypropionamide ((R)- 24)
To an anhydrous triethylamine solution (0.1 M, 2.66 mL) of (R)-38 (100 mg, 0.266 mmol), dichlorobis(triphenylphosphine)palladium (II) (19 mg, 0.026 mmol), and CuI (2.5 mg, 0.013 mmol) were sequentially added to a flame-dried Schlenk tube under Ar. The mixture was cooled down to −78 °C, and then the reaction vessel was evacuated and propyne was bubbled into the triethylamine solution until the solution reached ~ 1 atm. The mixture was stirred at room temperature (16 h). The mixture was cooled to −78 °C and re-evacuated. A balloon of propyne was bubbled into the mixture and the reaction was stirred at room temperature (24 h). The reaction mixture was concentrated in vacuo and the residue was purified by flash chromatography on silica gel with EtOAc/hexanes (5/5 to 10/0) as the eluant to obtain (R)-24 (70 mg, 92%) as a white solid. The desired product (60 mg) was purified with 340 mg of resin scavenger (PhosPhonics, cat# SPM32) to remove the traces of palladium to obtain 50 mg (66%) of (R)-N-(4′-(prop-1-ynyl))benzyl 2-acetamido-3-methoxypropionamide: Rf = 0.37 (EtOAc); mp 178–180 °C; [α]24.3 D = −18.0° (c 0.5, CHCl3); IR (nujol) 3474, 3273, 2960, 2856, 1683, 1550, 1457, 1375, 1299, 1125, 978, 811, 724 cm−1; 1H NMR (CDCl3) δ 2.04 (s, CH3C(O)), 2.05 (s, CH3), 3.38–3.45 (m, CHH′O, OCH3), 3.81 (dd, J = 4.2, 9.3 Hz, CHH′O), 4.39–4.50 (m, CH2NH), 4.51–4.57 (m, CH), 6.39–6.45 (br d, NHC(O)CH3), 6.71–6.79 (br t, CH2NH), 7.17 (d, J = 8.1 Hz, 2 ArH), 7.35 (d, J = 8.1 Hz, 2 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-24 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 4.33 (CH3), 23.2 (CH3C(O)), 43.3 (CH2NH), 52.4 (CHCH2), 59.1 (OCH3), 71.7 (CH2OCH3), 79.4 (C≡C), 86.1 (C≡C), 123.2, 127.3, 131.8, 137.3 (4 ArC), 170.0, 170.3 (2 C(O)); HRMS (M+H+)(ESI+) 311.1372 [M + H+] (calcd for C16H20N2O3H+ 311.1372). Anal. (C16H20N2O3·0.2H2O): C, H, N.
Preparation of (R)-N-(4′-(3,3-Dimethylbut-1-ynyl))benzyl 2-Acetamido-3-methoxypropionamide ((R)-25)
To an anhydrous THF (10 mL) solution of (R)-38 (376 mg, 1.0 mmol), triethylamine (280 μL, 2.0 mmol), 3,3-dimethylbut-1-yne (182 μl, 1.5 mmol), dichlorobis(triphenylphosphine)palladium (II) (35 mg, 0.05 mmol), and CuI (19 mg, 0.1 mmol) were sequentially added under Ar. The mixture was stirred at room temperature (4 h), and then Et2O (10 mL) was added and the precipitate filtered through a Celite® pad. The filtrate was concentrated in vacuo and the residue was purified by flash chromatography on silica gel with EtOAc/MeOH (9/1) as the eluant to obtain (R)-25 (220 mg, 66%) as a brown solid: Rf = 0.22 (EtOAc); mp 120–121 °C; [α]25 D = +4.8° (c 1.0, DMSO); IR (nujol) 3287, 2727, 2364, 1641, 1547, 1458, 1375, 1297, 1132, 972, 816, 724 cm−1; 1H NMR (CDCl3) δ 1.31 (s, (CH3)3C), 2.00 (s, CH3C(O)), 3.35 (s, OCH3), 3.42 (dd, J = 7.5, 9.0 Hz, CHH′O), 3.76 (dd, J = 4.2, 9.0 Hz, CHH′O), 4.33–4.50 (m, CH2NH), 4.50–4.61 (m, CH), 6.60 (d, J = 6.3 Hz, NHC(O)CH3), 6.91–6.99 (br t, CH2NH), 7.14 (d, J = 8.1 Hz, 2 ArH), 7.33 (d, J = 8.1 Hz, 2 ArH), addition of excess (R)-(−)- mandelic acid to a CDCl3 solution of (R)-25 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 23.2 (CH3C(O)), 27.9 (C(CH3)3), 31.0 (C(CH3)3), 43.3 (CH2NH), 52.5 (CHCH2), 59.1 (OCH3), 71.8 (CH2OCH3), 78.6 (C≡C), 98.8 (C≡C), 123.3, 127.2, 131.8, 137.1 (4 ArC), 170.2, 170.6 (2 C(O)); HRMS (M + H+)(ESI+) 331.2019 [M + H+] (calcd for C19H26N2O3H+ 331.2029). Anal. (C19H26N2O3·0.2 H2O): C, H, N.
Preparation of (R)-N-(4′-(Trimethylsilyl)ethynylbenzyl 2-Acetamido-3-methoxypropionamide ((R)-26)
To an anhydrous THF (70 mL) solution of (R)-38 (2.40 g, 6.38 mmol), triethylamine (1.8 mL, 12.76 mmol), trimethylsilylacetylene (1.35 ml, 9.57 mmol), dichlorobis(triphenylphosphine)palladium (II) (224 mg, 0.319 mmol), and CuI (121 mg, 0.638 mmol) were sequentially added under Ar. The mixture was stirred at room temperature (4 h), and then Et2O was added and the precipitate filtered through a Celite® pad. The filtrate was concentrated in vacuo and the residue was purified by flash chromatography on silica gel with EtOAc/MeOH (9/1) as the eluant to obtain (R)-26 (1.50 g, 68%). The desired product (1.50 g) was purified with 7.50 g of resin scavenger (PhosPhonics, cat# SPM32) to remove the traces of palladium to obtain 1.20 g (55%) of (R)-26 as a brown solid: Rf = 0.41 (EtOAc); mp 126–127 °C; [α]24 D = +6.1° (c 1.0, DMSO); IR (nujol) 3285, 2157, 1641, 1546, 1457, 1375, 1302, 1248, 1130, 975, 862, 723 cm−1; 1H NMR (CDCl3) δ 0.24 (s, (CH3)3Si), 1.99 (s, CH3C(O)), 3.35 (s, OCH3), 3.45 (dd, J = 7.2, 9.0 Hz, CHH′O), 3.75 (dd, J = 4.2, 9.0 Hz, CHH′O), 4.33–4.47 (m, CH2NH), 4.57–4.62 (m, CH), 6.66 (br d, J = 6.9 Hz, NHC(O)CH3), 7.07–7.13 (br t, CH2NH), 7.17 (d, J = 7.9 Hz, 2 ArH), 7.40 (d, J = 7.9 Hz, 2 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-26 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (CDCl3) δ −0.1 (CH3)3Si), 23.2 (CH3C(O)), 43.2 (CH2NH), 52.4 (CHCH2), 59.1 (OCH3), 71.6 (CH2OCH3), 94.4 (C≡C), 104.7 (C≡C), 122.4, 127.2, 132.3, 138.3 (4 ArC), 170.0, 170.3 (2 C(O)); HRMS (M + Na+)(ESI+) 369.1605 [M + Na+] (calcd for C18H26N2O3SiNa+ 369.1610). Anal. (C18H26N2O3Si): C, H, N.
Preparation of (S)-N-(4′-(Trimethylsilyl)ethynylbenzyl 2-Acetamido-3-methoxypropionamide ((S)-26)
Employing the preceding procedure and using (S)-38 (2.40 g, 6.38 mmol), triethylamine (1.8 mL, 12.76 mmol), CuI (121 mg, 0.638 mmol), dichlorobis(triphenylphosphine)palladium (II) (224 mg, 0.319 mmol), and trimethylsilylacetylene (1.35 mL, 9.57 mmol) gave 1.97 g (91%) of (S)-26 as a brown solid: Rf = 0.41 (EtOAc); mp 126–127 °C; [α]24 D = −6.2° (c 1.0, DMSO); IR (nujol) 3285, 2727, 2157, 1641, 1546, 1457, 1374, 1304, 1250, 1137, 862, 725 cm−1; 1H NMR (DMSO-d6) δ 0.22 (s, (CH3)3Si), 1.87 (s, CH3C(O)), 3.25 (s, OCH3), 3.44–3.55 (m, CHH′O, CHH′O), 4.29 (d, J = 5.7 Hz, CH2NH), 4.43–4.51 (m, CH), 7.24 (d, J = 8.2 Hz, 2 ArH), 7.40 (d, J = 8.2 Hz, 2 ArH), 8.10 (br d, J = 8.1 Hz, NHC(O)CH3), 8.53 (br t, J = 6.0 Hz, CH2NH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (S)-26 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (DMSO-d6) δ −0.2 (CH3)3Si), 22.4 (CH3C(O)), 41.7 (CH2NH), 52.6 (CHCH2), 58.1 (OCH3), 71.9 (CH2OCH3), 93.6 (C≡C), 105.1(C≡C), 120.0, 127.1, 131.4, 140.4 (4 ArC), 169.3, 169.8 (2 C(O)); HRMS (M + Na+)(ESI+) 369.1603 [M + Na+] (calcd for C18H26N2O3SiNa+ 369.01610). Anal. (C18H26N2O3Si): C, H, N.
Preparation of (R)-N-(4′-(3-Methoxyprop-1-ynyl))benzyl 2-Acetamido-3-methoxypropionamide ((R)-27)
To an anhydrous THF (10 mL) solution (R)-38 (376 mg, 1.0 mmol), triethylamine (280 μL, 2.0 mmol), 3-methoxyprop-1-yne (125 μL, 1.5 mmol), dichlorobis(triphenylphosphine)palladium (II) (70 mg, 0.1 mmol), and CuI (38 mg, 0.2 mmol) were sequentially added under Ar. The mixture was stirred at room temperature (4 h), and then Et2O (10 mL) was added and the precipitate filtered through a Celite pad. The filtrate was concentrated in vacuo and the residue was purified by flash chromatography on silica gel with EtOAc/MeOH (9/1) as the eluant to obtain (R)-27 (260 mg, 82%) as a beige solid: Rf = 0.27 (EtOAc); mp 141–142 °C; [α]27 D = +4.4° (c 1.0, DMSO); IR (nujol) 3278, 3096, 1640, 1554, 1458, 1370, 1304, 1257, 1192, 1099, 966, 903, 810, 732 cm−1; 1H NMR (CDCl3) δ 2.02 (s, CH3C(O)), 3.37 (s, OCH3), 3.45–3.47 (m, CHH′O, OCH3), 3.78 (dd, J = 4.2, 9.0 Hz, CHH′O), 4.32 (s, C≡CCH2OCH3), 4.38–4.52 (m, CH2NH), 4.54–4.61 (m, CH), 6.52 (d, J = 6.6 Hz, NHC(O)CH3), 6.91–6.99 (br t, CH2NH), 7.19 (d, J = 7.9 Hz, 2 ArH), 7.41 (d, J = 7.9 Hz, 2 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-27 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 23.2 (CH3C(O)), 43.2 (CH2NH), 52.5 (CHCH2), 57.7 (C≡CCH2OCH3), 59.1 (OCH3), 60.4 (C≡CCH2OCH3), 71.7 (CH2OCH3), 85.2 (C≡C), 86.0 (C≡C), 121.8, 127.3, 132.1, 138.4 (4 ArC), 170.4 (br, 2 C(O)); HRMS (M + H+)(ESI+) 319.1652 [M + H+] (calcd for C17H22N2O4H+ 319.1658). Anal. (C17H22N2O4·0.33 H2O): C, H, N.
Preparation of (R)-N-(4′-Cyano)benzyl 2-Acetamido-3-methoxypropionamide ((R)-28)
A MeOH solution (150 mL) of (R)-N-(4′-cyano)benzyl 2-N-(benzyloxycarbonyl)amino-3-methoxypropionamide (1.80 g, 4.8 mmol) was treated with H2 (1 atm) in presence of 10% Pd/C (250 mg) at room temperature (36 h). The mixture was carefully filtered through a bed of Celite® and the filtrate was evaporated in vacuo to obtain a colorless oil.
Using Method D, triethylamine (810 μL, 5.8 mmol) and acetyl chloride (410 μL, 5.8 mmol) gave 320 mg (42%) of (R)-28 as a white solid after trituration with EtOAc: Rf = 0.54 (9/1 EtOAc/MeOH); mp 168–169 °C; [α]24 D +4.9° (c 1.0, DMSO); IR (nujol) 3273, 2725, 2226, 1635, 1547, 1458, 1374, 1309, 1191, 1093, 907, 727 cm−1; 1H NMR (DMSO-d6) δ 1.87 (s, CH3C(O)), 3.26 (s, OCH3), 3.45–3.57 (m, CH2OCH3), 4.36 (d, J = 6.0 Hz, CH2NH), 4.43–4.49 (m, CH), 7.42 (d, J = 8.6 Hz, 2 ArH), 7.34 (d, J = 8.6 Hz, 2 ArH), 8.14 (d, J = 7.8 Hz, NHC(O)CH3), 8.61 (t, J = 6.0 Hz, CH2NH), addition of excess (R)- (−)-mandelic acid to a CDCl3 solution of (R)-28 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (DMSO-d6) δ 22.4 (CH3C(O)), 41.7 (CH2NH), 52.6 (CHCH2), 58.1 (OCH3), 71.9 (CH2OMe), 109.3 (CCN), 118.8 (CN), 127.6, 132.1, 145.3 (3 ArC), 169.4, 170.0 (2 C(O)); HRMS (M + Na+)(ESI+) 298.1163 [M + Na+] (calcd for C14H17N3O3Na+ 298.1168). Anal. (C14H17N3O3·0.25 H2O): C, H, N.
Preparation of (S)-N-(4′-Cyano)benzyl 2-Acetamido-3-methoxypropionamide ((S)-28)
Employing the preceding procedure and using (S)-N-(4′-cyano)benzyl 2-N-(benzyloxycarbonyl)amino- 3-methoxypropionamide (1.20 g, 3.2 mmol), 10% Pd/C (250 mg), triethylamine (540 μL, 3.8 mmol), and acetyl chloride (273 μL, 3.8 mmol) gave 620 mg (70%) of (S)-28 as a white solid after trituration with EtOAc: Rf = 0.54 (9/1 EtOAc/MeOH); mp 168–169 °C; [α]25 D −4.9° (c 1.0, DMSO); IR (nujol) 3271, 2726, 2228, 1630, 1552, 1458, 1374, 1312, 1194, 1095, 908, 729 cm−1; 1H NMR (CDCl3) δ 2.04 (s, CH3C(O)), 3.40 (s, OCH3), 3.45 (dd, J = 7.2, 9.6 Hz, CHH′O), 3.81 (dd, J = 3.9, 9.6 Hz, CHH′O), 4.49–4.61 (m, CH2NH, CH), 6.48 (br d, J = 5.7 Hz, NHC(O)CH3), 7.06–7.08 (br m, CH2NH), 7.36 (d, J = 8.4 Hz, 2 ArH), 7.34 (d, J = 8.4 Hz, 2 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (S)-28 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 23.1 (CH3C(O)), 43.0 (CH2NH), 52.5 (CHCH2), 59.1 (OCH3), 71.5 (CH2OCH3), 111.2 (CCN), 118.6 (CN), 127.8, 132.4, 143.5 (3 ArC), 170.3, 170.4 (2 C(O)); HRMS (M + Na+)(ESI+) 298.1163 [M + Na+] (calcd for C14H17N3O3Na+ 298.1168). Anal. (C14H17N3O3): C, H, N.
Preparation of (R)-N-(4′-Formyl)benzyl 2-Acetamido-3-methoxypropionamide ((R)-29)
(R)-N- (4′-(1,3-dioxolan-2-yl))benzyl 2-acetamido-3-methoxypropionamide (1.62 g, 5.0 mmol) was dissolved in a 2:1 THF:H2O mixture (30 mL) and 2 M HCl (10 drops) was added. The reaction was stirred at room temperature overnight and diluted with H2O (20 mL). The solution was neutralized with the dropwise addition of a saturated aqueous NaHCO3 solution at 0 °C. The THF was removed in vacuo and the remaining aqueous layer was extracted with CHCl3 (5 × 25 mL). The organic layers were combined, dried (Na2SO4), concentrated in vacuo, and the residue recrystallized from EtOAc to give 930 mg (66%) of (R)-29 as a white solid. The mother liquor was concentrated and purified by flash chromatography (5/95 MeOH/CHCl3) to yield of product 336 mg (24%) (total yield: 1.27 g (90%)): Rf = 0.40 (5/95 MeOH/CHCl3); mp 132–133°C; [α]25 D +10.4° (c 1.0, CHCl3); IR (nujol) 3288, 1687, 1642, 1549, 1458, 1375 cm−1; 1H NMR (CDCl3) δ 1.96 (s, CH3C(O)), 3.36 (s, OCH3), 3.53 (dd, J = 6.0, 9.3 Hz, CHH′O), 3.75 (dd J = 4.8, 9.3 Hz, CHH′O), 4.38–4.58 (m, NHCH2), 4.71 (app. dt, J = 5.4, 6.0 Hz, CH), 7.03 (d, J = 7.8 Hz, NHCH2), 7.38 (d, J = 8.4 Hz, 2 ArH), 7.68 (t, J = 5.4 Hz, NHC(O)CH3), 7.77 (d, J = 8.4 Hz, 2 ArH), 9.93 (s, C(O)H), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-29 gave only one signal for the methoxy protons and the acetyl peak protons, addition of excess (R)-(−)- mandelic acid to a CDCl3 solution of (S)-29 and (R)-29 (1:2 ratio) gave two signals for the acetyl methyl protons (δ 2.037 (S) and 2.023 (R) (Δppm = 0.014)) and two signals for the methoxy protons (δ 3.346 (S) and 3.377 (R) (Δppm = 0.031)); 13C NMR (CDCl3) δ 23.0 (CH3C(O)), 43.1 (NHCH2), 52.8 (CHCH2), 59.1 (OCH3), 72.0 (CH2OCH3), 127.7, 130.0, 135.5, 145.3 (4 ArC), 170.5, 170.7 (2 C(O)), 192.0 (C(O)H); HRMS [M + Na+] (ESI+) 301.1158 [M + Na+] (calcd for C14H18N2O4Na+ 301.1164). Anal. (C14H18N2O4): C, H, N.
Preparation of (S)-N-(4′-Formyl)benzyl 2-Acetamido-3-methoxypropionamide ((S)-29)
Using the preceding procedure, (S)-N-(4′-(1,3-dioxolan-2-yl))benzyl 2-acetamido-3-methoxypropionamide (1.65 g, 5.1 mmol) gave 900 mg (63%) of (S)-29 after recrystallization from EtOAc and another 268 mg (19%) after flash chromatography (total yield: 1.17 g, 82%): Rf = 0.40 (5/95 MeOH/CHCl3); mp 132– 133°C; [α]25 D −10.4° (c 1.0, CHCl3); IR (nujol) 3288, 3073, 1687, 1637, 1551, 1458, 1375 cm−1; 1H NMR (CDCl3) δ 2.04 (s, CH3C(O)), 3.40 (s, OCH3), 3.46 (dd, J = 7.5, 9.6 Hz, CHH′O), 3.83 (dd J = 4.2, 9.6 Hz, CHH′O), 4.48–4.64 (m, NHCH2, CH), 6.45 (d, J = 6.6 Hz, NHC(O)CH3), 6.99–7.15 (m, NHCH2), 7.42 (d, J = 8.1 Hz, 2 ArH), 7.85 (d, J = 8.1 Hz, 2 ArH), 9.99 (s, C(O)H), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (S)-29 gave only one signal for the acetyl peak protons and the methoxy protons, addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (S)-29 and (R)-29 (1:2 ratio) gave two signals for the acetyl methyl protons (δ 2.037 (S) and 2.023 (R) (Δppm = 0.014)) and two signals for the methoxy protons (δ 3.317 (S) and 3.351 (R) (Δppm = 0.034)); 13C NMR (CDCl3) δ 23.4 (CH3C(O)), 43.4 (NHCH2), 52.7 (CH), 59.4 (OCH3), 71.7 (CH2OCH3), 128.0, 130.3, 135.9, 145.2 (4 ArC), 170.5, 170.6 (2 C(O)), 192.0 (C(O)H); HRMS [M + Na+] (ESI+) 301.1161 [M + Na+] (calcd for C14H18N2O4Na+ 301.1164). Anal. (C14H18N2O4): C, H, N.
Preparation of (R)-N-(4′-Carboxy)benzyl 2-Acetamido-3-methoxypropionamide ((R)-30)
To a solution of (R)-31 (0.75 g, 2.6 mmol,) in THF/H2O (1/1, 50 mL) at 0 °C, was added LiOH·H2O (66 mg, 2.8 mmol,). The resulting solution was stirred (36 h). The solvent was removed in vacuo, washed with Et2O (4 × 100 mL), acidified to a pH of ~2 (1 N HCl), extracted with EtOAc (8 × 50 mL), dried (MgSO4), filtered, and concentrated in vacuo to yield 472 mg of a white powder (66%): Rf = 0.51 (9/1 CHCl3/MeOH); mp 197–198 °C; [α]25 D +6.0° (c 0.8, MeOH); IR (nujol) 3163, 2923, 2856, 1692, 1637, 1533, 1457, 1376, 1291, 1122, 939 cm−1; 1H NMR (CD3OD) δ 2.03 (s, CH3C(O)), 3.37 (s, OCH3), 3.60 (dd, J = 5.1, 9.7 Hz, CHH′O), 3.72 (dd, J = 5.1, 9.7 Hz, CHH′O), 4.48 (d, J = 6.0 Hz, CH2NH), 4.53 (t, J = 9.7 Hz, CH), 7.39 (d, J = 8.3 Hz, 2 ArH), 7.97 (d, J = 8.3 Hz, 2 ArH), 8.57–8.66 (br t, NHCH2), one amide proton and the carboxylic acid proton were not observed; 13C NMR (75 MHz, CD3OD) δ 21.4 (CH3C(O)), 42.7 (CH2NH), 54.2 (CH), 58.2 (CH3O), 72.0 (CH2OCH3), 127.1, 129.6, 129.8, 144.3 (4 ArC), 168.8 (HOC(O)), 171.6, 172.6 (2 C(O)); LRMS (ESI) 295.1 [M + H+] (calcd. for C14H18N2O5H+: 295.1). Anal. (C14H18N2O5): C, H, N.
Preparation of (R)-N-(4′-(Methyloxycarbonyl))benzyl 2-Acetamido-3-methoxypropionamide ((R)-31)
Pd/C (10%, 400 mg) was added to a solution of (R)-N-(4′-(methyloxycarbonyl))benzyl 2-N- (benzyloxycarbonyl)amino-3-methoxypropionamide (1.28 g, 3.2 mmol) in anhydrous MeOH (35 mL). The mixture was hydrogenated (45 psi, 24 h), and then filtered through Celite®. The filtrate was evaporated in vacuo leaving a mixture (0.80 g) of a white solid and an oil of the desired amine (TLC analysis using ninhydrin indicated the presence of a primary amine).
Using Method D, triethylamine (1.25 mL, 9 mmol,) and acetyl chloride (0.32 mL, 4.5 mmol,) gave 0.43 g of a white solid (47%) after recrystallization with EtOAc: Rf = 0.62 (9/1 CHCl3/MeOH); mp 167– 168 °C; [α]25 D −11.4° (c 2.2, CHCl3); IR (nujol) 3279, 2927, 1712, 1639, 1550, 1457 cm−1; 1H NMR (CDCl3) δ 2.04 (s, CH3C(O)), 3.39 (s, CH3O), 3.45 (dd, J = 7.5, 9.0 Hz, CHH′O), 3.82 (dd, J = 4.2, 9.0 Hz, CHH′O), 3.91 (s, CH3OC(O)), 4.51–4.55 (m, CH, CH2NH), 6.45 (d, J = 6.6 Hz, NHC(O)CH3), 6.85–6.95 (br m, NHCH2), 7.32 (d, J = 8.4 Hz, 2 ArH), 8.00 (d, J = 8.4 Hz, 2 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-31 gave only one signal for the acetyl methyl protons and one signal for the ether methyl protons; 13C NMR (75 MHz, CDCl3) δ 23.3 (CH3C(O)), 43.3 (CH2NH), 52.3 (CH3OC(O)), 52.7 (CH), 59.2 (CH3OCH2), 72.0 (CH2OCH3), 127.3, 129.4, 130.1, 143.4 (4 ArC), 167.0 (OC(O)), 170.4, 170.6 (2 C(O)); HRMS (ESI) 309.1451 [M + H+] (calcd. for C15H20N2O5H+ 309.1450). Anal. (C15H20N2O5): C, H, N.
Preparation of (R)-N-(4′-Amino)benzyl 2-Acetamido-3-methoxypropionamide ((R)-32). 7
A MeOH solution (150 mL) of (R)-N-(4′-nitro)benzyl 2-acetamido-3-methoxypropionamide (2.00 g, 6.8 mmol) was treated with H2 (1 atm) in presence of PtO2 (160 mg) at room temperature (16 h). The mixture was carefully filtered through a bed of Celite® and the filtrate was evaporated in vacuo to obtain a colorless oil that was triturated with EtOAc to give 1.67 g of (R)-32 as a white solid (90%): Rf = 0.39 (9/1 CHCl3/MeOH); mp 151–152 °C (lit.7 mp 183–184 °C); 1H NMR (CDCl3) δ 2.02 (s, CH3C(O)), 3.36 (s, OCH3), 3.42 (dd, J = 7.8, 9.3 Hz, CHH′O), 3.78 (dd, J = 4.5, 9.3 Hz, CHH′O), 4.34 (d, J = 5.4 Hz, CH2NH), 4.48–4.55 (m, CH), 6.48 (br d, J = 6.0 Hz, NHC(O)CH3), 6.60–6.67 (m, CH2NH, 2 ArH), 7.05 (d, J = 8.1 Hz, 2 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)- 32 gave only one signal for the acetyl methyl and one signal for the ether methyl protons.
Preparation of (R)-N-(4′-(2,2,2-Trifluoroacetamido))benzyl 2-Acetamido-3- methoxypropionamide ((R)-33)
(S)-Ethyltrifluorothioacetate (2.99 g, 18.9 mmol) was added to a MeOH (10 mL) solution of (R)-32 (1.00 g, 3.8 mmol) at room temperature and then the reaction was maintained at this temperature (3 h). Addition of EtOAc (10 mL) led to the precipitation of (R)-33 as a white solid (600 mg) after filtration. The filtrate was concentrated in vacuum and the residue purified by flash chromatography on silica gel with EtOAc as the eluant to obtain an additional 350 mg of (R)-33 as a white solid. The solids were combined to obtain 950 mg (70%) of (R)-33: Rf = 0.24 (EtOAc); mp 202– 204 °C; [α]26 D −1.2° (c 0.5, DMSO); IR (nujol) 3389, 3282, 1721, 1653, 1536, 1459, 1375, 1294, 1249, 1206, 1150, 1066, 974, 896, 838, 727, 653 cm−1; 1H NMR (DMSO-d6) δ 1.87 (s, CH3C(O)), 3.25 (s, OCH3), 3.45–3.55 (m, CHH′O), 4.27 (d, J = 5.8 Hz, CH2NH), 4.44–4.50 (m, CH), 7.27 (d, J = 8.7 Hz, 2 ArH), 7.58 (d, J = 8.7 Hz, 2 ArH), 8.11 (d, J = 7.8 Hz, CH3C(O)NH), 8.51 (t, J = 5.8 Hz, CH2NH), 11.23 (s, CF3C(O)NH); 13C NMR (DMSO-d6) δ 22.4 (CH3C(O)), 41.5 (CH2NH), 52.6 (CH), 58.1 (OCH3), 71.9 (CH2OCH3), 115.7 (q, J = 284.6 Hz, CF3), 120.1, 127.4, 134.7, 136.7 (4 ArC), 154.3 (q, J = 37.5 Hz, HNC(O)CF3), 169.3, 169.7 (2 C(O)); HRMS (M + H+)(ESI+) 362.1321 [M + H+] (calcd for C15H18F3N3O4H+ 362.1328). Anal. (C15H18F3N3O4): C, H, F, N.
Preparation of (R)-N-(4′-Azido)benzyl 2-Acetamido-3-methoxypropionamide ((R)-34)
Ag2O (4.85 g, 20.9 mmol) was added to a CH3CN solution (100 mL) of (R)-N-(4′-azido)benzyl 2-acetamido- 3-hydroxypropionamide (1.16 g, 4.2 mmol) and CH3I (2.61 mL, 41.9 mmol) at room temperature under Ar. The reaction mixture was stirred at room temperature in the dark (5 d), filtered, and the filtrate concentrated in vacuo. The solid was purified by flash column chromatography on silica gel (1/9 MeOH/CHCl3) to obtain 0.98 g (80%) of (R)-34 as a white solid: Rf = 0.5 (1/9 MeOH/CHCl3); mp 149– 150 °C; [α]26 D −15.2° (c 1.0, MeOH); IR (nujol) 3285, 2931, 2113, 1635, 1560, 1456 cm−1; 1H NMR (CDCl3) δ 2.03 (s, CH3C(O)), 3.38 (s, OCH3), 3.44 (dd, J = 7.5, 9.3 Hz, CHH′O), 3.80 (dd, J = 4.2, 9.3 Hz, CHH′O), 4.40 (1/2ABq, J = 6.2, 15.0 Hz, CHH′Ar), 4.46 (1/2ABq, J = 6.2, 15.0 Hz, CHH′Ar), 4.52–4.58 (m, CH), 6.48 (br d, J = 6.3 Hz, NHC(O)CH3), 6.82–6.85 (br m, CH2NH), 6.96–7.01 (m, 2 ArH), 7.23–7.28 (m, 2 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-34 gave only a single signal for the acetyl methyl protons and the ether methyl protons, addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-34 and (S)-34 (1:2 ratio) gave two signals for the acetyl methyl protons (δ 1.995 (R) and 2.010 (S) (Δppm = 0.015)), and two signals for the ether methyl protons (δ 3.302 (S) and 3.342 (R) (Δppm = 0.040)); 13C NMR (CDCl3) δ 23.4 (CH3C(O)), 43.1 (CH2NH), 52.7 (CH), 59.3 (OCH3), 71.8 (CH2OCH3), 119.5, 129.1, 134.9, 139.5 (4 ArC), 170.2, 170.5 (2 C(O)); HRMS (M + H+)(ESI+) 292.1406 [M + H+] (calcd. for C13H17N5O3H+ 292.1410). Anal. (C13H17N5O3): C, H, N.
Preparation of (S)-N-(4′-Azido)benzyl 2-Acetamido-3-methoxypropionamide ((S)-34)
Utilizing the preceding procedure, (S)-N-(4′-azido)benzyl 2-acetamido-3-hydroxypropionamide (2.40 g, 8.7 mmol), Ag2O (10.04 g, 43.3 mmol), and MeI (5.40 mL, 86.6 mmol) gave crude (S)-N-(4′-azido)benzyl 2-acetamido-3-methoxypropionamide after 4 d. The product was purified by column chromatography (SiO2; 1/9 MeOH/CHCl3) to obtain 2.05 g (81%) of (S)-34 as a white solid: Rf = 0.50 (1/9 MeOH/CHCl3); mp 149–150 °C; [α]26 D +15.4° (c 1.0, MeOH); IR (nujol) 3285, 2927, 2112, 1635, 1565, 1457 cm−1; 1H NMR (CDCl3) δ 2.03 (s, CH3C(O)), 3.38 (s, OCH3), 3.43 (dd, J = 7.5, 9.0 Hz, CHH′O), 3.81 (dd, J = 4.2, 9.0 Hz, CHH′O), 4.40 (1/2ABq, J = 6.0, 15.0 Hz, CHH′Ar), 4.46 (1/2ABq, J = 6.0, 15.0 Hz, CHH′Ar), 4.51–4.57 (m, CH), 6.43 (br d, J = 6.3 Hz, NHC(O)CH3), 6.78–6.83 (br m, NHCH2), 6.96–7.01 (m, 2 ArH), 7.23–7.27 (m, 2 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (S)-34 gave only a single signal for the acetyl methyl protons and the ether methyl protons, addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-34 and (S)-34 (1:2 ratio) gave two signals for the acetyl methyl protons (δ 1.995 (R) and 2.010 (S) (Δppm = 0.015)), and the ether methyl protons (δ 3.302 (S) and 3.342 (R) (Δppm = 0.040)); 13C NMR (CDCl3) δ 23.3 (CH3C(O)), 43.1 (CH2NH), 52.7 (CH), 59.2 (OCH3), 72.0 (CH2OCH3), 119.4, 129.1, 135.0, 139.4 (4 ArC), 170.3, 170.6 (2 C(O)); HRMS (M + H+)(ESI+) 292.1405 [M + H+] (calcd. for C13H17N5O3H+ 292.1410). Anal. (C13H17N5O3): C, H, N.
Preparation of (R)-N-(4′-Methoxy)benzyl 2-Acetamido-3-methoxypropionamide ((R)-35)
A MeOH solution (300 mL) of (R)-N-(4′-methoxy)benzyl 2-N-(benzyloxycarbonyl)amino-3- methoxypropionamide (2.40 g, 6.4 mmol) was treated with H2 (1 atm) in presence of 10% Pd/C (480 mg) at room temperature (16 h). The mixture was carefully filtered through a bed of Celite® and the filtrate was evaporated in vacuo to obtain a colorless oil.
Using Method D, triethylamine (1.1 mL, 7.74 mmol) and acetyl chloride (550 μL, 7.74 mmol) gave 900 mg (70%) of (R)-35 as a white solid after trituration in EtOAc: Rf = 0.82 (EtOAc); mp 146–147 °C; [α]25 D −26.0° (c 0.5, CHCl3); IR (nujol) 3283, 2861, 1642, 1520, 1458, 1377, 1299, 1255, 1176, 1127, 1031, 978, 719 cm−1; 1H NMR (CDCl3) δ 2.02 (s, CH3C(O)), 3.37 (s, OCH3), 3.43 (dd, J = 7.8, 9.3 Hz, CHH′O), 3.76–3.81 (m, CH3OC6H4, CHH′O), 4.39 (d, J = 6.0 Hz, CH2NH), 4.50–4.57 (m, CH), 6.49 (br d, J = 6.0 Hz, NHC(O)CH3), 6.71–6.82 (br t, CH2NH), 6.86 (d, J = 8.4 Hz, 2 ArH), 7.18 (d, J = 8.4 Hz, 2 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-35 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 23.2 (CH3C(O)), 43.0 (CH2NH), 52.4 (CH), 55.3 (C6H4OCH3), 59.0 (OCH3), 71.7 (CH2OCH3), 114.1, 128.8, 129.9, 159.0 (4 ArC), 169.8, 170.3 (2 C(O)); HRMS (M + Na+)(ESI+) 303.1320 [M + Na+] (calcd for C14H20N2O4Na+ 303.1321). Anal. (C14H20N2O4): C, H, N.
Preparation of (R)-N-(4′-Chloro)benzyl 2-Acetamido-3-methoxypropionamide ((R)-36)
A saturated HCl solution in dioxane (1 mmol/2 mL, 24.0 mL) was added to (R)-N-(4′-chloro)benzyl 2-N- (tert-butoxycarbonyl)amino-3-methoxypropionamide (4.10 g, 12.0 mmol) at 0 °C and the solution was stirred at room temperature (2 h). The reaction solution was concentrated in vacuo and dried (30 min).
Using Method D, triethylamine (5.0 mL, 36.0 mmol) and acetyl chloride (1.3 mL, 18.0 mmol) gave 3.10 g (80%) of (R)-36 as a white solid after recrystallization with EtOAc: Rf = 0.42 (EtOAc); mp 155 °C; [α]27.0 D −20.5° (c 1.0, CHCl3); IR (nujol) 3288, 3277, 3162, 2900, 1634, 1556, 1457, 1375, 1306, 1259, 1193, 1137, 1098, 1045, 964, 909, 801, 732 cm−1; 1H NMR (CDCl3) δ 2.01 (s, CH3C(O)), 3.37 (s, OCH3), 3.43 (dd, J = 7.8, 9.2 Hz, CHH′O), 3.78 (dd, J = 4.2, 9.2 Hz, CHH′O), 4.34–4.49 (m, CH2NH), 4.54–4.61 (m, CH), 6.54 (br d, J = 6.6 Hz, NHC(O)CH3), 6.94–7.03 (br t, CH2NH), 7.19 (d, J = 8.8 Hz, 2 ArH), 7.29 (d, J = 8.8 Hz, 2 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-36 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 23.1 (CH3C(O)), 42.8 (CH2NH), 52.5 (CHCH2), 59.1 (OCH3), 71.7 (CH2OCH3), 128.7, 133.2, 136.4, (3 ArC), 170.1, 170.4 (2 C(O)), one signal was not detected and is believed to overlap with nearby peaks; HRMS (M + H+)(ESI+) 285.1006 [M + H+] (calcd for C13H17ClN2O3H+ 285.1006). Anal. (C13H17ClN2O3): C, H, Cl, N.
Preparation of (R)-N-(4′-Bromo)benzyl 2-Acetamido-3-methoxy-propionamide ((R)-37)
Using Method D, (R)-N-(4′-bromo)benzyl 2-amino-3-methoxypropionamide hydrochloride (1.00 g, 3.1 mmol), triethylamine (1.3 mL, 9.3 mmol) and acetyl chloride (0.3 mL, 3.7 mmol) gave 690 mg (68%) of (R)-37 as a white solid after recrystallization with EtOAc: Rf = 0.08 (7/3, EtOAc/hexanes); mp 159–161 °C; [α]25 D −15.9° (c 1.0, CHCl3); IR (nujol mull) 3272, 3093, 2919, 2860, 1634, 1555, 1457, 1375, 1306, 1254, 1197, 1135, 1046, 964, 907, 795, 740, 606, 550, 468 cm−1; 1H NMR (CDCl3) δ 2.04 (CH3C(O)), 3.39 (s, OCH3), 3.43 (dd, J = 7.5, 9.0 Hz, CHH′O), 3.81 (dd, J = 4.2, 9.0 Hz, CHH′O), 4.37–4.48 (m, CH2NH), 4.51–4.57 (m, CH), 6.40–6.42 (br d, NHC(O)CH3), 6.76–6.84 (br t, CH2NH), 7.14 (d, J = 8.6 Hz, 2 ArH), 7.45 (d, J = 8.6 Hz, 2 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-37 gave only one signal for the acetyl methyl protons and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 23.1 (CH3C(O)), 42.8 (CH2NH), 52.4 (CH), 59.1 (OCH3), 71.6 (CH2OCH3), 121.3, 129.1, 131.7, 137.0 (4 ArC), 170.1, 170.4 (2 C(O)); LRMS (+ESI) 351.0 [M + Na]+ (100%), 353.0 [M+2 + Na]+ (100%) (calcd for C13H17BrN2O3Na+ 351.0 [M + Na]+). Anal. (C13H17BrN2O3): C, H, Br, N.
Preparation of (R)-N-(4′-Iodo)benzyl 2-Acetamido-3-methoxypropionamide ((R)-38)
A saturated HCl solution in dioxane (1 mmol/2 mL, 25.0 mL) was added to (R)-N-(4′-iodo)benzyl 2-N-(tertbutoxycarbonyl) amino-3-methoxypropionamide (5.50 g, 12.7 mmol) at 0 °C and the solution was stirred at room temperature (2 h). The reaction solution was concentrated in vacuo and dried (30 min).
Using Method D, triethylamine (10.7 mL, 76.0 mmol) and acetyl chloride (2.7 mL, 38.0 mmol) gave 3.40 g (71%) of (R)-38 as a white solid after recrystallization with EtOAc: Rf = 0.76 (5/5 acetone/EtOAc); mp 159–160 °C; [α]25 D = +3.3° (c 1.0, DMSO); IR (nujol) 3279, 1636, 1552, 1457, 1375, 1305, 1139, 725 cm−1; 1H NMR (CDCl3) δ 2.02 (s, CH3C(O)), 3.38 (s, OCH3), 3.44 (dd, J = 7.2, 9.0 Hz, CHH′O), 3.79 (dd, J = 4.2, 9.0 Hz, CHH′O), 4.38–4.41 (m, CH2NH), 4.52–4.59 (m, CH), 6.46 (br d, J = 6.6 Hz, NHC(O)CH3), 6.85–6.93 (br t, CH2NH), 7.00 (d, J = 8.4 Hz, 2 ArH), 7.64 (d, J = 8.4 Hz, 2 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-38 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 23.1 (CH3C(O)), 42.9 (CH2NH), 52.4 (CH), 59.1 (OCH3), 71.6 (CH2OCH3), 92.7 (CI), 129.3, 137.7, 139.1 (3 ArC), 170.1, 170.3 (2 C(O)); HRMS (M+Na+)(ESI+) 399.0177 [M + Na+] (calcd for C13H17IN2O3Na+ 399.0182). Anal. (C13H17IN2O3): C, H, I, N.
Preparation of (S)-N-(4′-Iodo)benzyl 2-Acetamido-3-methoxypropionamide ((S)-38)
Employing the preceding procedure and using (S)-2-N-(4′-iodo)benzyl 2-N-(tert-butoxycarbonyl)amino-3-methoxypropionamide (3.70 g, 8.5 mmol), saturated dioxane solution of HCl (1 mmol/2 mL, 17.0 mL), triethylamine (3.6 mL, 25.6 mmol) and acetyl chloride (906 μL, 12.3 mmol) gave 2.43 g (76%) of (S)-38 as a white solid after recrystallization with EtOAc: Rf = 0.76 (5/5 acetone/EtOAc); mp 159–160 °C; [α]24D = −3.2° (c 1.0, DMSO); IR (nujol) 3278, 1636, 1552, 1458, 1375, 1305, 1138, 725 cm−1; 1H NMR (CDCl3) δ 2.02 (s, CH3C(O)), 3.38 (s, OCH3), 3.43 (dd, J = 7.2, 9.0 Hz, CHH′O), 3.79 (dd, J = 4.2, 9.0 Hz, CHH′O), 4.38–4.42 (m, CH2NH), 4.53–4.59 (m, CH), 6.47 (br d, J = 6.0 Hz, NHC(O)CH3), 6.85–6.93 (br t, CH2NH), 7.00 (d, J = 8.4 Hz, 2 ArH), 7.64 (d, J = 8.4 Hz, 2 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (S)-38 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (DMSO-d6) δ 22.4 (CH3C(O)), 41.4 (CH2NH), 52.5 (CH), 58.1 (OCH3), 71.9 (CH2OCH3), 92.2 (CI), 129.3, 136.8, 139.1 (3 ArC), 169.3, 169.7 (2 C(O)); HRMS (M + Na+)(ESI+) 399.0177 [M + Na+] (calcd for C13H17IN2O3Na+ 399.0182). Anal. (C13H17IN2O3): C, H, I, N.
Preparation of (R)-N-(4′-Sulfamoyl)benzyl 2-Acetamido-3-methoxypropionamide ((R)-39)
Using Method A, (R)-2-acetamido-3-methoxypropionoic acid25 (1.00 g, 6.2 mmol), NMM (1.5 mL, 13.7 mmol), IBCF (1.0 mL, 7.9 mmol) and methyl 4-(aminomethyl) benzenesulfonamide hydrochloride (2.07 g, 9.3 mmol) gave 1.25 g (61%) of (R)-39 as a white solid after purification by column chromatography (1/7 MeOH/CHCl3) and recrystallized (MeOH/CHCl3): Rf = 0.35 (1/7 MeOH/CHCl3); mp 177–179°C; [α]25D +10.7° (c 1.0, MeOH); IR (nujol) 3293, 2935, 1623, 1557, 1459 cm−1; 1H NMR (DMSO-d6) δ 1.87 (s, CH3(O)), 3.26 (s, OCH3), 3.46–3.56 (m, CH2OCH3), 4.34 (d, J = 6.1 Hz, CH2NH), 4.43–4.50 (m, CH), 7.31 (s, NH2), 7.41 (d, J = 8.6 Hz, 2 ArH), 7.75 (d, J = 8.6 Hz, 2 ArH), 8.13 (d, J = 7.8 Hz, NHC(O)CH3), 8.59 (t, J = 6.1 Hz, NHCH2); 13C NMR (DMSO-d6) δ 22.5 (CH3(O)), 41.7 (CH2NH), 52.7 (CH), 58.2 (OCH3), 72.0 (CH2OCH3), 125.5, 127.2, 142.5, 143.5 (4 ArC), 169.5, 169.9 (2 C(O)); HRMS (M + H+)(ESI+) 330.1117 [M + H+] (calcd. for C13H19N3O5SH+ 330.1124). Anal. (C13H19N3O5S): C, H, N, S.
Preparation of (R)-N-(3′,4′-Dichloro)benzyl 2-Acetamido-3-methoxypropionamide ((R)-60)
Using Method D, (R)-N-(3′,4′-dichloro)benzyl 2-amino-3-methoxypropionamide hydrochloride (3.50 g, 11.2 mmol), triethylamine (4.6 mL, 33.5 mmol) and acetyl chloride (0.95 mL, 13.4 mmol) gave 1.60 g (45%) of (R)-60 as a white solid after recrystallization with EtOAc: Rf = 0.16 (EtOAc/hexanes, 7/3); mp = 165 °C; [α]24.9D −10.5° (c 1.0, CHCl3); IR (nujol) 3292, 3096, 2927, 2859, 1636, 1555, 1459, 1379, 1256, 1135, 1034, 815, 724, 604, 491 cm−1; 1H NMR (CDCl3) δ 2.05 (CH3C(O)), 3.41 (s, OCH3), 3.45 (dd, J = 7.5, 9.3 Hz, CHH′O), 3.82 (dd, J = 4.1, 9.3 Hz, CHH′O), 4.39–4.49 (m, CH2NH), 4.52–4.59 (m, CH), 6.39–6.41 (br d, NHC(O)CH3), 6.80–6.90 (br t, CH2NH), 7.10 (dd, J = 2.1, 8.1 Hz, 1 ArH), 7.36 (d, J = 2.1 Hz, 1 ArH), 7.40 (d, J = 8.1 Hz, 1 ArH)), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-60 gave only one signal for the acetyl methyl protons and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 23.1 (CH3C(O)), 42.2 (CH2NH), 52.6 (CH), 59.1 (OCH3), 71.7 (CH2OCH3), 126.6, 129.1, 130.5, 131.3, 132.6, 138.3 (6 ArC), 170.2, 170.4 (2 C(O)); LRMS (ESI+) 341.05 [M + Na+, 100%], 343.05 [M+2 + Na+, 64%], 345.05 [M+4 + Na+, 11%] (calcd for C13H16Cl2N2O3 Na+ 341.04). Anal. (C13H16Cl2N2O3): C, H, Cl, N.
Preparation of (R)-N-(2′,4′-Dichloro)benzyl 2-Acetamido-3-methoxypropionamide ((R)-61)
Using Method D, (R)-N-(2′,4′-dichloro)benzyl 2-amino-3-methoxypropionamide hydrochloride (2.70 g, 7.2 mmol) triethylamine (3.0 mL, 21.5 mmol) and acetyl chloride (0.6 mL, 8.6 mmol,) gave 1.45 g (63%) of (R)-61 as a white solid after recrystallization with EtOAc: Rf = 0.26 (EtOAc); mp = 149 °C; [α]23.7D −11.0° (c 1.0, CHCl3); IR (nujol) 3477, 3399, 3276, 2919, 2854, 2725, 2363, 1637, 1552, 1458, 1376, 1304, 1251, 1135, 1102, 1050, 974, 821, 724, 606, 505 cm−1; 1H NMR (CDCl3) δ 2.04 (CH3C(O)), 3.37–3.43 (m, OCH3, CHH′O), 3.80 (dd, J = 4.1, 9.2 Hz, CHH′O), 4.43–4.57 (m, CH2NH, CH), 6.36–6.40 (br d, NHC(O)CH3), 6.90–7.02 (br t, CH2NH), 7.22 (dd, J = 2.1, 8.3 Hz, 1 ArH), 7.30 (d, J = 8.3 Hz, 1 ArH), 7.39 (d, J = 2.1 Hz, 1 ArH), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-61 gave only one signal for the acetyl methyl protons and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 23.1 (CH3C(O)), 40.9 (CH2NH), 52.4 (CH), 59.0 (OCH3), 71.7 (CH2OCH3), 127.2, 129.2, 130.3, 133.8, 133.9 (5 ArC), 170.2, 170.4 (2 C(O)), one signal was not detected and is believed to overlap with nearby peaks; LRMS (ESI+) 341.08 [M + Na+, 100%], 343.08 [M+2 + Na+, 66%], 345.07 [M+4 + Na+, 13%] (calcd for C13H16Cl2N2O3Na+ 341.04). Anal. (C13H16Cl2N2O3): C, H, Cl, N.
Pharmacology
Compounds were screened under the auspices of the National Institutes of Health’s Anticonvulsant Screening Program (ASP). Experiments were performed in male rodents [albino Carworth Farms No. 1 mice (intraperitoneal route, ip), albino Spague-Dawley rats (oral route, po)]. Housing, handling, and feeding were in full accordance with recommendations contained in the ‘Guide for the Care and Use of laboratory Animals’. Anticonvulsant activity was established using the MES test,33,42 the scMet test,33 and the rapid hippocampal kindled rat model39 using previously reported methods.26
Supplementary Material
Acknowledgments
The authors thank the NINDS and the ASP at the National Institutes of Health with Drs. Tracy Chen and Jeffrey Jiang, for kindly performing the pharmacological studies via the ASP’s contract site at the University of Utah with Drs. H. Wolfe, H.S. White, and K. Wilcox. Funds for this study were generously provided by grants R01NS054112 (H.K.) and F31NS060358 (P.M.) from the National Institute of Neurological Disorders and Stroke, and by the Korean Research Foundation Grant funded by the Korean Government (MOEHRD) grant KRF-2006-352-C00042) (K.D.P.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Neurological Disorders and Stroke or the National Institutes of Health. Harold Kohn has a royalty-stake position in (R)-3.
Footnotes
Abbreviations: FAA, functionalized amino acid; MES, maximal electroshock seizure; SAR, structure activity relationship; AB, affinity bait; CR, chemical reporter; IBCF, isobutyl chloroformate; NMM, N-methyl morpholine; DMTMM, 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride; National Institute of Neurological Disorders and Stroke, NINDS; ASP, Anticonvulsant Screening Program; ip, intraperitoneally; po, orally; ED50, effective dose (50%); TD50, neurological impairment (toxicity, 50%); PI, protective index; scMet, subcutaneous Metrazol.
Supporting Information Available: Synthetic procedures for the intermediates leading to the preparation of (R)- 4, 7–31, 33–39, 60, and 61, and (S)-11, 21, 23, 26, 28, 29, 34, and 38, elemental analyses, 1H and 13C NMR spectra of compounds evaluated in this study. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Hauser WA, Annegers JF, Kurland LT. The prevalence of epilepsy in Rochester, Minnesota, 1940–80. Epilepsia. 1991;32:429–445. doi: 10.1111/j.1528-1157.1991.tb04675.x. [DOI] [PubMed] [Google Scholar]
- 2.Evans JH. Post-traumatic epilepsy. Neurology. 1962;12:665–674. doi: 10.1212/wnl.12.10.665. [DOI] [PubMed] [Google Scholar]
- 3.Lindsay JM. Genetics and epilepsy. Epilepsia. 1971;12:47–54. doi: 10.1111/j.1528-1157.1971.tb03914.x. [DOI] [PubMed] [Google Scholar]
- 4.(a) Begley CE, Annegers JF, Lairson DR, Reynolds TF, Hauser WA. Cost of epilepsy in the United States: A model based on incidence and prognosis. Epilepsia. 1994;35:1230–1243. doi: 10.1111/j.1528-1157.1994.tb01794.x. [DOI] [PubMed] [Google Scholar]; (b) Begley CE, Famulari M, Annegers JF, Lairson DR, Reynolds TF, Coan S, Dubinsky S, Newmark ME, Leibson C, So EL, Rocca WA. The cost of epilepsy in the United States: an estimate from population-based clinical and survey data. Epilepsia. 2000;41:342–351. doi: 10.1111/j.1528-1157.2000.tb00166.x. [DOI] [PubMed] [Google Scholar]; (c) Begley CE, Lairson DR, Reynolds TF, Coan S. Early treatment cost in epilepsy and how it varies with seizure type and frequency. Epilepsy Res. 2001;47:205–215. doi: 10.1016/s0920-1211(01)00310-2. [DOI] [PubMed] [Google Scholar]
- 5.(a) Rogawski MA, Porter RJ. Antiepileptic drugs: Pharmacological mechanisms and clinical efficacy with consideration of promising development stage compounds. Pharmacol Reviews. 1997;42:223–286. [PubMed] [Google Scholar]; (b) McNamara JO. In: Goodman & Gilman’s The Pharmacological Basis of Therapeutics. 10. Hardman JG, Limbird LE, editors. Ch 21. McGraw-Hill; New York: 2001. pp. 521–547. [Google Scholar]; (c) Aiken SP, Brown WM. Treatment of epilepsy: Existing therapies and future developments. Frontiers in Bioscience. 2000;5:124–152. doi: 10.2741/aiken. [DOI] [PubMed] [Google Scholar]
- 6.(a) Brodie MJ, Dichter MA. Antiepileptic drugs. New Eng J Med. 1996;334:168–175. doi: 10.1056/NEJM199601183340308. [DOI] [PubMed] [Google Scholar]; (b) Dichter MA, Brodie MJ. New antiepileptic drugs. New Eng J Med. 1996;334:1583–1590. doi: 10.1056/NEJM199606133342407. [DOI] [PubMed] [Google Scholar]
- 7.(a) McCorry D, Chadwick D, Marson A. Current drug treatment of epilepsy in adults. Lancet Neurol. 2004;3:729–735. doi: 10.1016/S1474-4422(04)00935-4. [DOI] [PubMed] [Google Scholar]; (b) Duncan JS. The promise of new antiepileptic drugs. Brit J Clin Pharmacol. 2002;53:123–131. doi: 10.1046/j.0306-5251.2001.01540.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bauer J, Reuber M. Medical treatment of epilepsy. Expert Opinion on Emerging Drugs. 2003;8:457–467. doi: 10.1517/14728214.8.2.457. [DOI] [PubMed] [Google Scholar]; (d) Mattson RH, Cramer JA, Collins JF, Smith DB. Comparison of carbamazepine, phenobarbital, phenytoin, and primidone in partial and secondarily generalized tonic-clonic seizures. New Eng J Med. 1985;313:145–151. doi: 10.1056/NEJM198507183130303. [DOI] [PubMed] [Google Scholar]
- 8.Pellock JM, Willmore LJ. A rational guide to monitoring in patients receiving anticonvulsants. Neurology. 1991;41:961–964. doi: 10.1212/wnl.41.7.961. [DOI] [PubMed] [Google Scholar]
- 9.Cortes S, Liao Z-K, Watson D, Kohn H. Effect of structural modification of the hydantoin ring on anticonvulsant activity. J Med Chem. 1985;28:601–606. doi: 10.1021/jm50001a012. [DOI] [PubMed] [Google Scholar]
- 10.Conley JD, Kohn H. Functionalized D, L-amino acid derivatives. Potent new agents for the treatment to epilepsy. J Med Chem. 1987;30:567–574. doi: 10.1021/jm00386a021. [DOI] [PubMed] [Google Scholar]
- 11.Kohn H, Conley JD. New antiepileptic agents. Chem Br. 1988;24:231–234. [Google Scholar]
- 12.Kohn H, Conley JD, Leander JD. Marked stereospecificity in a new class of anticonvulsants. Brain Res. 1988;457:371–375. doi: 10.1016/0006-8993(88)90709-3. [DOI] [PubMed] [Google Scholar]
- 13.Kohn H, Sawhney KN, LeGall P, Conley JD, Robertson DW, Leander JD. Preparation and anticonvulsant activity of a series of functionalized α-aromatic and α-heteroaromatic amino acids. J Med Chem. 1990;33:919–926. doi: 10.1021/jm00165a006. [DOI] [PubMed] [Google Scholar]
- 14.Kohn H, Sawhney KN, LeGall P, Robertson DW, Leander JD. Preparation and anticonvulsant activity of a series of functionalized α-heteroatom-substituted amino acids. J Med Chem. 1991;34:2444–2452. doi: 10.1021/jm00112a020. [DOI] [PubMed] [Google Scholar]
- 15.Kohn H, Sawhney KN, Bardel P, Robertson DW, Leander JD. Synthesis and anticonvulsant activities of α-heterocyclic α-acetamido-N-benzylacetamide derivatives. J Med Chem. 1993;36:3350–3360. doi: 10.1021/jm00074a016. [DOI] [PubMed] [Google Scholar]
- 16.Bardel P, Bolanos A, Kohn H. Synthesis and anticonvulsant activities of α-acetamido-N-benzylacetamide derivatives containing an electron-deficient α-heteroaromatic substituent. J Med Chem. 1994;37:4567–4571. doi: 10.1021/jm00052a017. [DOI] [PubMed] [Google Scholar]
- 17.Kohn H, Sawhney KN, Robertson DW, Leander JD. Anticonvulsant properties of N-substituted α,α-diamino acid derivatives. J Pharm Sci. 1994;83:689–691. doi: 10.1002/jps.2600830519. [DOI] [PubMed] [Google Scholar]
- 18.Choi D, Stables JP, Kohn H. Synthesis and anticonvulsant activities of N-benzyl-2-acetamidopropionamide derivatives. J Med Chem. 1996;39:1907–1916. doi: 10.1021/jm9508705. [DOI] [PubMed] [Google Scholar]
- 19.(a) Paruszewski R, Rostafinska-Suchar G, Strupinska M, Jaworski P, Stables JP. Synthesis and anticonvulsant activity of some amino acid derivative. Pharmazie. 1996;3:145–148. [PubMed] [Google Scholar]; (b) Paruszewski R, Rostafinska-Suchar G, Strupinska M, Jaworski P, Winiecka I, Stables JP. Synthesis and anticonvulsant activity of some amino acid derivatives. Pharmazie. 1996;51:212–215. [PubMed] [Google Scholar]
- 20.Levy RH, Mattson R, Meldrum B. Antiepileptic Drugs. 4. Chapter 6 Raven Press; New York: 1995. [Google Scholar]
- 21.Eudismic ratio = ratio of activities of the two enantiomers. Lehmann PA. Quantifying stereoselectivity or how to choose a pair of shoes when you have two left feet. Trends Pharmacol Sci. 1982;3:103–106.
- 22.Perucca E, Yasothan U, Clincke G, Kirkpatrick P. Lacosamide. Nat Rev Drug Disc. 2008;7:973–974. doi: 10.1038/nrd2764. [DOI] [PubMed] [Google Scholar]
- 23.Stoehr T, Kupferberg HJ, Stables JP, Choi D, Harris RH, Kohn H, Walton N, White HS. Lacosamide, a novel anti-convulsant drug, shows efficacy with a wide safety margin in rodent models for epilepsy. Epilepsy Res. 2007;74:147–154. doi: 10.1016/j.eplepsyres.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 24.Park KD, Morieux P, Salome C, Cotten SW, Reamtong O, Eyers C, Gaskell SJ, Stables JP, Liu R, Kohn H. Lacosamide isothiocyanate-based agents: Novel agents to target and identify lacosamide receptors. J Med Chem. doi: 10.1021/jm9012054. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morieux P, Stables JP, Kohn H. Synthesis and anticonvulsant activities of N-benzyl (2R)-2-acetamido-3-oxysubstituted propionamide derivatives. Bioorg Med Chem. 2008;16:8968–8975. doi: 10.1016/j.bmc.2008.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.LeTiran A, Stables JP, Kohn H. Design and evaluation of affinity labels of functionalized amino acid anticonvulsants. J Med Chem. 2002;45:4762–4773. doi: 10.1021/jm020225f. [DOI] [PubMed] [Google Scholar]
- 27.Kohn H, Robertson D, Leander JD. unpublished results. [Google Scholar]
- 28.(a) Anderson GW, Zimmerman JE, Callahan FM. A reinvestigation of the mixed carbonic anhydride method of peptide synthesis. J Am Chem Soc. 1967;87:5012–5017. doi: 10.1021/ja00995a032. [DOI] [PubMed] [Google Scholar]; (b) Kunishima M, Kawachi C, Monta J, Terao K, Iwasaki F, Tani S. 2-(4,6-Dimethoxy-1,3,5-triazin-2-yl)-4-methyl-morpholinium chloride: an efficient condensing agent leading to the formation of amides and esters. Tetrahedron. 1999;55:13159–13170. [Google Scholar]
- 29.Chinchilla R, Najera C. The Sonogashira reaction: a booming methodology in synthetic organic chemistry. Chem Rev. 2007;107:874–922. doi: 10.1021/cr050992x. [DOI] [PubMed] [Google Scholar]
- 30.Scherrmann MC, Boutboul A, Estramareix B, Hoffmann AS, Lubineau A. Binding properties and esterase activity of monoclonal antibodies elicited against sucrose 6-heptylphosphonate. Carb Res. 2001;334:295–307. doi: 10.1016/s0008-6215(01)00199-9. [DOI] [PubMed] [Google Scholar]
- 31.Barral K, Moorhouse AD, Moses JE. Efficient conversion of aromatic amines into azides: A one-pot synthesis of triazole linkages. Org Lett. 2007;9:1809–1811. doi: 10.1021/ol070527h. [DOI] [PubMed] [Google Scholar]
- 32.For comparable procedures for resolving stereoisomers, see the following: Weisman GR. In: Asymmetric Synthesis-Analytical Methods. Morrison JD, editor. Vol. 1. Academic Press; New York: 1983. pp. 153–171.Parker D, Taylor RJ. Direct 1H NMR assay of the enantiomeric composition of amines and β-amino alcohols using O-acetyl mandelic acid as a chiral solvating agent. Tetrahedron. 1987;43:5431–5456.
- 33.Stables JP, Kupferberg HG. In: Molecular and Cellular Targets for Antiepileptic Drugs. Avanzini G, Tanganelli P, Avoli M, editors. John Libbey; London: 1977. pp. 191–198. [Google Scholar]
- 34.Porter RJ, Cereghino JJ, Gladding GD, Hessie BJ, Kupferberg HJ, Scoville B, White BG. Antiepileptic drug development program. Cleveland Clin Q. 1984;51:293–305. doi: 10.3949/ccjm.51.2.293. [DOI] [PubMed] [Google Scholar]
- 35.Dunham NW, Miya TS. A note on a simple apparatus for detecting neurological deficit in rats and mice. J Am Pharm Assoc. 1957;46:208–209. doi: 10.1002/jps.3030460322. [DOI] [PubMed] [Google Scholar]
- 36.Almenningen A, Bastiansen O, Fernholt L, Cyvin BN, Cyvin SJ, Samdal S. Structure and barrier of internal rotation of biphenyl derivatives in the gaseous state. Part 1. The molecular structure and normal coordinate analysis of normal biphenyl and perdeuteriated biphenyl. J Mol Struct. 1985;128:59–76. [Google Scholar]
- 37.Bastiansen O, Samdal S. Structure and barrier of internal rotation of biphenyl derivatives in the gaseous state. Part 4. Barrier of internal rotation in biphenyl, perdeuteriated biphenyl and seven non-ortho-substituted halogen derivatives. J Mol Struct. 1985;128:115–125. [Google Scholar]
- 38.Topliss JG. Utilization of operational schemes for analog synthesis in drug design. J Med Chem. 1972;15:1006–1011. doi: 10.1021/jm00280a002. [DOI] [PubMed] [Google Scholar]
- 39.(a) Lothman EW, Williamson JM. Closely spaced recurrent hippocampal seizures elicit two types of heightened epileptogenesis: a rapidly developing, transient kindling and a slowly developing, enduring kindling. Brain Res. 1994;649:71–84. doi: 10.1016/0006-8993(94)91050-2. [DOI] [PubMed] [Google Scholar]; (b) Morimoto K, Fahnestock M, Racine RJ. Kindling and status epilepticus models of epilepsy: rewiring the brain. Prog Neurobiol. 2004;73:1–60. doi: 10.1016/j.pneurobio.2004.03.009. [DOI] [PubMed] [Google Scholar]; (c) Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- 40.(a) McNamara JO, Byrne MC, Dasheiff RM, Fitz JG. The kindling model of epilepsy: a review. Prog Neurobiol. 1980;15:139–159. doi: 10.1016/0301-0082(80)90006-4. [DOI] [PubMed] [Google Scholar]; (b) Loscher W, Schmidt D. Which animal models should be used in the search for new antiepileptic drugs? A proposal based on experimental and clinical considerations. Epilepsy Res. 1998;2:145–181. doi: 10.1016/0920-1211(88)90054-x. [DOI] [PubMed] [Google Scholar]; (c) McNamara JO. Development of new pharmacological agents for epilepsy: lessons learned from the kindling model. Epilepsia. 1989;30(Suppl 1):S13–8. doi: 10.1111/j.1528-1157.1989.tb05809.x. [DOI] [PubMed] [Google Scholar]
- 41.Stables JP. NINDS ASP internal control data. private communication. [Google Scholar]
- 42.Krall RL, Penry JK, Kupferberg HJ, Swinyard EA. Antiepileptic drug development: I. History and a program for progress. Epilepsia. 1978;19:393–408. doi: 10.1111/j.1528-1157.1978.tb04506.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.