

Abstract
Thymine DNA glycosylase (TDG) excises thymine from G·T mispairs, and removes a variety of damaged bases (X), with a preference for lesions in a CpG·X context. We recently reported that human TDG rapidly excises 5-halogenated uracils, exhibiting much greater activity for CpG·FU, CpG·ClU, and CpG·BrU than for CpG·T. Here, we examine the effects of altering the CpG context on the excision activity for U, T, FU, ClU, and BrU. We show that the maximal activity (kmax) for G·X substrates depends significantly on the 5′ base pair. For example, kmax decreases by 6-, 11-, and 82-fold for TpG·ClU, GpG·ClU, and ApG·ClU, respectively, as compared to CpG·ClU. For the other G·X substrates, the 5′-neighbor effects have a similar trend but vary in magnitude. The activity for G·FU, G·ClU, and G·BrU, with any 5′-flanking pair, meets and in most cases significantly exceeds the CpG·T activity. Strikingly, hTDG activity is reduced 102.3- to 104.3-fold for A·X relative to G·X pairs, and reduced further for A·X pairs with a 5′ pair other than C·G. The effect of altering the 5′ pair and/or the opposing base (G·X versus A·X) is greater for substrates that are larger (BrdU, dT) or have a more stable N-glycosidic bond (such as dT). The largest CpG context effects are observed for the excision of thymine. The potential role played by hTDG in the cytotoxic effects of ClU and BrU incorporation into DNA, which can occur under inflammatory conditions, and in the cytotoxicity of FU, a widely used anticancer agent, are discussed.
The nucleobases in DNA are subject to continuous chemical modification, generating a broad range of mutagenic and cytotoxic lesions that can lead to cancer and other diseases (1,2). To counteract this inevitable damage, the cellular machinery includes systems for DNA repair (3). Damage occurring to the nucleobases is the purview of base excision repair (BER)2, a pathway that is initiated by a damage-specific DNA glycosylase. These enzymes find damaged or mismatched bases within the vast expanse of normal DNA, and catalyze the cleavage of the base-sugar (N-glycosidic) bond, producing an abasic or a (AP) site in the DNA. The repair process is continued by follow-on BER enzymes.
Human thymine DNA glycosylase (hTDG) was discovered as an enzyme that removes thymine from G·T and uracil from G·U mispairs in DNA (4,5). In vertebrates, G·T mispairs arise from replication errors, which are handled by the mismatch repair pathway, or from the deamination of 5-methylcytosine (m5C) to T (6,7). Because cytosine methylation occurs at CpG dinucleotides (8,9), G·T mispairs caused by m5C deamination are found at CpG sites. It has been shown that hTDG is most active for G·T mispairs with a 5′ C·G pair, suggesting that a predominant biological role of the enzyme is to initiate the repair of CpG·T lesions (10,11). DNA methylation at CpG plays a fundamental role in many cellular processes, including transcriptional regulation and the silencing of repetitive genetic elements (8,9). Suggesting a biological imperative to maintain the integrity of CpG sites, another human DNA glycosylase exhibits specificity for G·T mispairs at CpG sites; methyl binding domain IV (MBD4) (12-15).
In addition to its CpG·T activity, hTDG has been shown to remove a variety of damaged bases (5,16-19), most of which are shown in Figure 1. We recently identified several new hTDG substrates (20), including 5-chlorouracil (ClU), 5-iodouracil (IU), 5-flourocytosine (FC), and 5-bromocytosine (BrC) (the activity is weak for IU, FC, and BrC, and is probably not biologically relevant). The ability of hTDG to remove a broad range of damaged nucleobases is consistent its relatively large and non-specific active site (21,22). Yet, despite its substrate promiscuity, hTDG exhibits exceedingly weak activity for the excision of cytosine and 5-methylcytosine (11,16,20). We recently showed that for a broad range of C5-substituted uracil and cytosine bases, hTDG specificity depends on substrate reactivity (i.e., the stability of the scissile C-N bond) rather than the selective recognition of substrates in the active site (20). Moreover, we showed that specificity against the excision of cytosine from the huge excess of normal G·C pairs in DNA is largely explained by the very low reactivity of dC, rather than the inability of hTDG to flip cytosine into its active site (20). Consistent with this catalytic mechanism and the enhanced reactivity of 5-halogenated dU substrates, we found that hTDG rapidly excises FU, ClU, and BrU from CpG sites (20). Indeed, compared to CpG·T, the activity is 920-fold greater for CpG·FU, 550-fold greater for CpG·ClU, and 53-fold greater for CpG·BrU (20).
FIGURE 1. hTDG removes many damaged bases and has specificity for CpG sites.
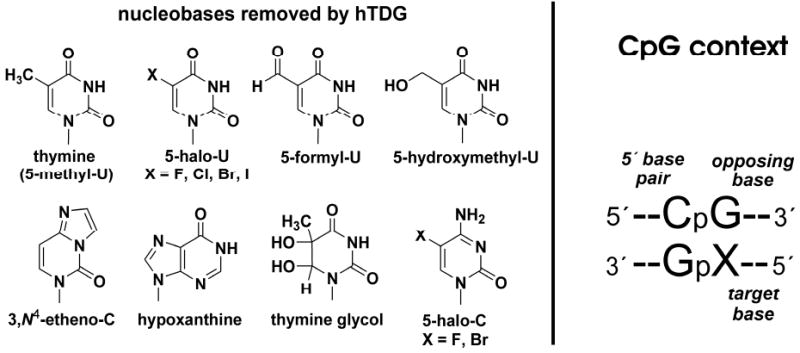
(A) Most of the known hTDG substrates are shown. Cytosine and 5-methylcytosine (m5C) are not significant substrates. (B) The CpG context is shown. Previous work has shown that hTDG is specific for removing T from G·T mispairs with a 5′-flanking C·G pair.
The robust activity observed for CpG·ClU and CpG·BrU suggests that hTDG may also have significant activity for removing ClU and BrU from DNA contexts other than CpG, raising the possibility that hTDG could play a role in the mutagenic and cytotoxic effects associated with ClU and BrU incorporation into DNA (23,24). These lesions can arise in DNA when the Cl5dUTP or Br5dUTP pools become elevated, which can be promoted by the activity of peroxidases during inflammation (25,26). The very strong hTDG activity for CpG·FU substrates is also of interest, because FU has been used for decades to treat many types of cancer (27). The mechanism of FU cytotoxicity is thought to involve multiple pathways, including a repetitive cycle of U and FU incorporation into DNA followed by the excision of these bases by a DNA glycosylase, increasing the burden abasic sites and leading to DNA strand breaks (27). Thus, hTDG could potentially be involved in the cytotoxicity of FU, as suggested by a report that inactivation of TDG in fission yeast and in mouse embryonic fibroblasts diminishes the sensitivity of these cells to FU treatment (28).
To further examine these possibilities, it is important to determine the activity of hTDG for removing FU, ClU, and BrU from DNA contexts other than CpG, because the incorporation of these bases into DNA or their presence in the template strand can be expected to give predominantly A·X pairs, but also some G·X pairs, and with no significant preference for a CpG sequence context. Although previous studies have examined the effect of altering the 5′-flanking pair on hTDG activity for G·T and G·εC (10,29), quantitative studies have not been reported for the many other hTDG substrates. Moreover, previous studies (and our findings here) indicate that the effect of the 5′-flanking pair depends strongly on the nature of the target base (29), so the results for G·T substrates do not necessarily predict the 5′-neighbor effects for other substrates. In addition, the effect of pairing the target base with adenine rather than guanine (i.e., A·X versus G·X) has not been rigorously examined for substrates other than U (10,30), and the effect of altering the 5′-flanking pair for A·X substrates is completely unexplored.
Here, we use single turnover kinetics experiments to compare the activity of hTDG (kmax) for substrates that contain a G·X lesion with various 5′-flanking base pairs, i.e., CpG·X, TpG·X, GpG·X, and ApG·X, where X represents FU, ClU, BrU, U, or T. We also examine the effect of pairing the target base with adenine rather than guanine (i.e., CpA·X versus CpG·X). Finally, we examine the combined effect of pairing the target base with adenine and altering the 5′-flanking base pair, using CpA·X, TpA·X, GpA·X, and ApA·X substrates. These studies provide the relative activity of hTDG for the excision of U, FU, ClU, and BrU from DNA contexts other than CpG, i.e., those in which they might be expected to arise in vivo. In addition, by systematically altering the CpG context for a series of target bases, our findings illuminate the catalytic role of the putative interactions that hTDG forms with the opposing guanine and with the 5′ C·G base pair.
Experimental Proceedures
DNA Synthesis and Purification
Duplex DNA substrates were hybridized in 10 mM Tris pH 8.0, 0.1 M NaCl, and 0.1 mM EDTA, by rapid heating to 80 °C and slow cooling to room temperature. Single strand DNA oligonucleotides were synthesized at the Biopolymer Genomics Core Facility, University of Maryland Baltimore, and at the Keck Foundation Biotechnology Resource Laboratory of Yale University. The 5-chlorodeoxyuridine phosphoramidite was obtained from ChemGenes Corp. (Wilmington, MA). Oligonucleotides were purified by anion exchange HPLC using a Zorbax Oligo column (Agilent Technologies), desalted by gel filtration using pre-packed Sephadex G25 columns (GE Healthcare), and stored at -20 °C. Oligonucleotide purity was verified by analytical anion-exchange HPLC under denaturing (pH 12) conditions using a DNAPac PA200 column (Dionex Corp.), as described previously (20). Oligonucleotides were quantified by absorbance (260 nm) using the pairwise extinction coefficients, calculated as described (31).
Expression and Purification of hTDG
Escherichia coli BL21(DE3) cells (Stratagene) were transformed with a pET-28-based expression plasmid for human TDG, 410 amino acids (32). Expression cells were grown in LB (typically 2 L) at 37 °C until OD600 = 0.8, the temperature was reduced to 15 °C, and hTDG expression was induced with 0.25 mM IPTG (isopropyl β-D-thiogalactoside) and continued for about 15 hrs. Cells were harvested by centrifugation, suspended in ∼25 ml lysis buffer (0.05 M sodium phosphate pH 8.0, 0.3 M NaCl, 0.02 M imidazole, 0.01 M beta-mercaptoethanol) with 1 mg/ml lysozyme and a protease inhibitor cocktail (Roche). The cell suspension was frozen on dry ice, thawed, incubated on ice for 30 min with stirring, followed by an additional 30 min with DNase (Novagen). The lysate was cleared by centrifugation, and incubated with 4 ml of Ni-NTA metal affinity resin (Qiagen) for 1 h at 4 °C. The lysate-resin mix was placed in a gravity-flow column, washed with 30 ml lysis buffer containing 1M NaCl and 20 mM imidazole, followed by 30 ml lysis buffer containing 20 mM imidazole, and hTDG was eluted with lysis buffer containing 150 mM imidazole. hTDG was purified further using an SP Sepharose HP column (GE Healthcare) with buffers IE-A (25 mM Tris pH 7.5, 75 mM NaCl, 1 mM DTT, 0.2 mM EDTA, 1% glycerol) and IE-B (IE-A with 1 M NaCl) and a gradient of 5-20% IE-B over 60 min at 2.5 ml/min. hTDG was further purified using a Q Sepharose HP column (GE Healthcare) with the same IE-A and IE-B buffers and a gradient of 0-100% IE-B over 60 min at 2.5 ml/min. The purity of hTDG was >99% as judged by SDS-PAGE stained with Coomassie. Purified hTDG was dialyzed overnight versus storage buffer (20 mM HEPES 7.5, 0.1 M NaCl, 1 mM DTT, 0.5 mM EDTA, 1% glycerol), concentrated to about 0.1 mM, flash frozen in small aliquots, and stored at -80 °C. The concentration of hTDG was determined by absorbance using ε280 = 31.5 mM-1cm-1 (33).
Single Turnover Kinetics
Because hTDG is strongly inhibited by its abasic DNA product (10), the rate constant obtained from steady-state kinetics experiments (kcat) is dominated by product release and is not useful for comparing hTDG activity for various substrates. Here, we use single turnover kinetics under saturating enzyme conditions to obtain rate constants (kmax) that are not impacted by product release or the association of enzyme and substrate, and thereby reflect the maximal enzymatic activity for a given substrate. To ensure that the observed rate constants represent the maximal value (i.e., kobs ≈ kmax), single turnover experiments were collected using a large excess of enzyme over substrate, and with an enzyme concentration that is more than 100-fold higher the KD = 41 nM reported as the apparent binding affinity of hTDG for DNA containing a CpG·T mispair (29). To confirm saturating enzyme conditions, experiments were in some cases conducted with two or more hTDG concentrations, typically 5 μM and 10 μM, providing rate constants that were equivalent within experimental uncertainty. Substrate DNA concentrations were 500 nM, unless noted otherwise. Single turnover reactions were performed either manually or using a rapid chemical quenched-flow instrument (RQF-3, Kintek Corp.). The reactions were conducted at 22°C in HEMN.1 buffer (20 mM HEPES pH 7.50, 0.2 mM EDTA, 2.5 mM MgCl2, 0.1 M NaCl) with 0.1 mg/ml bovine serum albumin, quenched with 50% (v:v) 0.3M NaOH, 0.03M EDTA, and heated for 15 min at 85 °C to induce cleavage of the DNA backbone at AP sites. The extent of product formation was analyzed by HPLC, as described below. Rate constants were determined by fitting the single turnover data to a single exponential equation using non-linear regression with Grafit 5 (34). In most cases, the reactions proceeded to full completion, except those that are very slow (i.e., kmax < ∼1 × 10-4 min‐1).
HPLC Assay for Monitoring hTDG Activity
We recently developed a HPLC assay for monitoring hTDG activity (20). Samples taken during a kinetics experiment contain a mixture of substrate and products that is comprised of four oligonucleotides: the full-length target strand and its complement, and two shorter strands resulting from alkaline cleavage of the nascent abasic strand. These strands are resolved by anion exchange HPLC using denaturing (pH 12.0) conditions with a DNAPac PA200 column (Dionex Corp.). The alkaline conditions serve to suppress hybridization of ssDNA during chromatography, and have the added benefit of resolving strands that are of the same length but differ in the number of thymine and guanine bases, which are negatively charged at pH 12. The elution buffer is 0.02 M sodium phosphate pH 12.0 containing either 0.03 M NaClO4 (buffer A) or 0.50 M NaClO4 (buffer B). The oligonucleotides are detected by absorbance (260 nm), and the fraction product (F) is determined from the integrated peak areas for the target strand (AS) and product strands (AP1 and AP2) using eq 1:
| (1) |
The determination of fraction product using this assay is reproducible to within 1%, as determined from multiple analyses of identical samples.
Results
In a recent study we determined the activity of hTDG (kmax) for a series of 5-substituted uracil and cytosine substrates in which the target base was placed in a CpG context (20). We found that kmax is much higher for CpG·FU, CpG·ClU, and CpG·BrU than for CpG·T, suggesting that hTDG may have significant activity for FU, ClU, and BrU in DNA contexts other than CpG. Here, we examine the effect of altering the CpG context on the activity of hTDG for the excision of five different target bases (X = T, U, FU, ClU, and BrU) using a series of substrates as shown in Figure 2. Thus, one set of substrates examines the effect of altering the 5′-flanking pair on G·X activity (i.e., YpG·X). Another set examines the effect of pairing the target base with adenine rather than guanine while preserving the 5′ C·G pair (CpA·X versus CpG·X). A final set examines the effect of both; pairing the target base with adenine and altering the 5′-flanking pair (YpA·X).
FIGURE 2. DNA substrates used in this work.

Substrates are designed to examine the effect of altering the CpG site context on hTDG activity for the excision of T, U, FU, ClU, and BrU. For the YpG·X substrates, the 5′-flanking pair is varied for G·X substrates. For example, TpG·X indicates that the G·X target pair has a 5′-flanking T·A pair. For the CpA·X substrates, the target base is paired with A rather than G, while the 5′ C·G pair is retained. For the YpA·X substrates, the target base is paired with A and the 5′ flanking base pair is varied.
Single Turnover Kinetics
Like many DNA glycosylases, hTDG is strongly inhibited by one of its reaction products, abasic DNA (10,14,35-37). Indeed, previous studies show that under limiting enzyme conditions, the turnover of hTDG is exceedingly slow after it converts one molar equivalent of G·T (or G·U) substrate to G·AP product (10,38,39). Thus, the rate constant obtained from steady-state kinetics, kcat, is dominated by product release and cannot provide a meaningful comparison of activity for different substrates (Figure 3). In contrast, single turnover kinetics conducted under saturating enzyme conditions provide a rate constant (kmax) that is not impacted by product release or the association of enzyme and substrate, and therefore reflects the maximal activity for a given substrate (Figure 3). For the hTDG reaction, kmax reflects the rate constant for the chemical step (kchem), and is also influenced by the equilibrium constant for base flipping (Kflip). In the base flipping step, the target nucleotide flips out of the DNA duplex and into the active site, a process that likely involves a conformational change in hTDG, as observed for uracil DNA glycosylase (40). Thus, differences in kmax that result from alterations to the CpG context reflect a change in kchem and/or Kflip.
FIGURE 3. Minimal kinetic mechanism for hTDG.
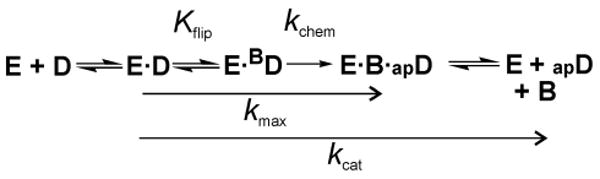
Shown is a minimal kinetic mechanism for hTDG, including the steps that comprise kcat, which is obtained from steady state kinetics, and kmax, as obtained from single turnover kinetics with a saturating enzyme concentration (used here). Association of hTDG (E) and DNA substrate (D) forms the initial collision complex, E·D, and base-flipping (Kflip) gives the reactive enzyme-substrate complex, E·BD (where BD is base-flipped DNA). Base flipping likely involves a conformational change for hTDG, which is not explicitly shown. The chemical step (kchem) involves cleavage of the base-sugar (N-glycosidic) bond and addition of the water nucleophile, producing the ternary product complex, E·B·apD (B is the nucleobase, apD is abasic DNA). Release of the excised base likely precedes the dissociation of abasic DNA, which is known to be very slow. As shown, kmax is influenced by Kflip and kchem, where kmax = kchem(Kflip/1 + Kflip).
Effect of the 5′ Base Pair on G·X Activity
We determined the effect of varying the 5′ neighboring base pair on hTDG activity (kmax) for G·FU, G·ClU, G·BrU, G·U, and G·T using the YpG·X series of substrates (Figure 2). The results are given in Table 1 and Figure 4. Previous studies showed that hTDG activity for G·T substrates depends strongly on the 5′-flanking pair, with relative activity of: CpG·T ≫ TpG·T > GpG·T > ApG·T (10,11). We find a similar trend here; compared to CpG·T, kmax is reduced by 37-, 96-, and 582-fold for TpG·T, GpG·T, and ApG·T, respectively. The influence of the 5′ neighbor is much smaller for G·U activity; compared to CpG·U, kmax is decreased by 3.3-, 2.9‐, and 22-fold for TpG·U, GpG·U, and ApG·U, respectively. The 5′-neighbor effect is also small for G·FU activity; compared to CpG·FU, kmax decreases by merely 1.8-, 1.6-, and 11-fold for TpG·FU, GpG·FU, and ApG·FU, respectively (Figure 4B). The 5′-neighbor effects are much larger for G·ClU activity; compared to CpG·ClU, kmax is decreased by 6-, 11-, and 82-fold for TpG·ClU, GpG·ClU, and ApG·ClU, respectively (Figure 4C). The results are similar for G·BrU; compared to CpG·BrU, kmax is decreased by 9-, 26‐, and 75-fold for TpG·BrU, GpG·BrU, and ApG·BrU, respectively.
TABLE 1.
Kinetic Parameters for hTDG
| substrate | kmax (min-1) a | Fold change relative to CpG·X | Fold change relative to CpG·T | Fold change relative to CpA·X | Fold change relative to YpG·X c |
|---|---|---|---|---|---|
| G·X | |||||
| CpG·T | 0.22 ± 0.04 | 1 | 1 | ||
| TpG· | 0.0060 ± 0.0001 | 0.027 | 0.027 | ||
| GpG· | 0.0023 ± 0.0002 | 0.010 | 0.010 | ||
| ApG· | 0.00038 ± 0.00005 | 0.0017 | 0.0017 | ||
| CpG·U | 2.6 ± 0.3 | 1 | 12 | ||
| TpG· | 0.79 ± 0.04 | 0.303 | 3.6 | ||
| GpG· | 0.88 ± 0.11 | 0.340 | 4.0 | ||
| ApG· | 0.117 ± 0.003 | 0.045 | 0.5 | ||
| CpG·FU | 202 ± 16 | 1 | 918 | ||
| TpG· | 113 ± 1 | 0.558 | 513 | ||
| GpG· | 125 ± 11 | 0.618 | 568 | ||
| ApG· | 18 ± 1 | 0.089 | 75 | ||
| CpG·ClU | 120 ± 6 | 1 | 546 | ||
| TpG· | 20.9 ± 0.5 | 0.174 | 95 | ||
| GpG· | 11.1 ± 0.3 | 0.093 | 51 | ||
| ApG· | 1.46 ± 0.15 | 0.012 | 6.7 | ||
| CpG·BrU | 11.6 ± 1.0 | 1 | 53 | ||
| TpG· | 1.2 ± 0.1 | 0.106 | 5.6 | ||
| GpG· | 0.44 ± 0.06 | 0.038 | 2.0 | ||
| ApG· | 0.15 ± 0.02 | 0.013 | 0.7 | ||
| A·X | |||||
| CpA·T | 1.3 × 10-5 ± 0.3 × 10-5 | 10−4.3 | 10−4.3 | 1 | |
| TpA· | N.D. b | ||||
| GpA· | N.D. b | ||||
| ApA· | N.D. b | ||||
| CpA·U | 0.0033 ± 0.0001 | 10−2.9 | 0.015 | 1 | 10−2.9 |
| TpA· | 83 × 10-5 ± 4 × 10-5 | 10−3.5 | 10−2.4 | 0.253 | 10−3.0 |
| GpA· | 5.3 × 10-5 ± 0.1 × 10-5 | 10−4.7 | 10−3.6 | 0.016 | 10−4.2 |
| ApA· | 3.2 × 10-5 ± 0.6 × 10-5 | 10−4.9 | 10−3.8 | 0.0097 | 10−3.6 |
| CpA·FU | 1.08 ± 0.08 | 10−2.3 | 4.9 | 1 | 10−2.3 |
| TpA· | 0.24 ± 0.06 | 10−2.9 | 1.08 | 0.220 | 10−2.7 |
| GpA· | 0.0138 ± 0.0004 | 10−4.2 | 0.063 | 0.013 | 10−4.0 |
| ApA· | 0.0063 ± 0.0002 | 10−4.5 | 0.029 | 0.0058 | 10−3.5 |
| CpA·ClU | 0.022 ± 0.003 | 10−3.7 | 0.100 | 1 | 10−3.7 |
| TpA· | 0.0045 ± 0.0001 | 10−4.4 | 0.021 | 0.205 | 10−3.7 |
| GpA· | 8.6 × 10-5 ± 0.2 × 10-5 | 10−6.2 | 10−3.4 | 0.0039 | 10−5.1 |
| ApA· | 5.1 × 10-5 ± 0.3 × 10-5 | 10−6.4 | 10−3.6 | 0.0023 | 10−4.5 |
| CpA·BrU | 78 × 10-5 ± 10 × 10-5 | 10−4.2 | 10−2.5 | 1 | 10−4.2 |
| TpA· | 17 × 10-5 ± 1 × 10-5 | 10−4.8 | 10−3.1 | 0.223 | 10−3.9 |
| GpA· | 1.2 × 10-5 ± 0.1 × 10-5 | 10−6.0 | 10−4.3 | 0.015 | 10−4.5 |
| ApA· | 0.8 × 10-5 ± 0.2 × 10-5 | 10−6.2 | 10−4.5 | 0.0097 | 10−4.3 |
The rate constants (kmax) reflect the maximal enzymatic activity of hTDG for a given substrate, as determined using single turnover kinetics experiments with saturating enzyme conditions.
Not determined.
Fold change relative to YpG·X gives the effect of pairing the target base (X) with adenine rather than guanine for a given 5′ base pair (Y), i.e. the rate of TpA·U relative to TpG·U.
FIGURE 4. Effect of the 5′-flanking base pair on hTDG activity for G·X substrates.
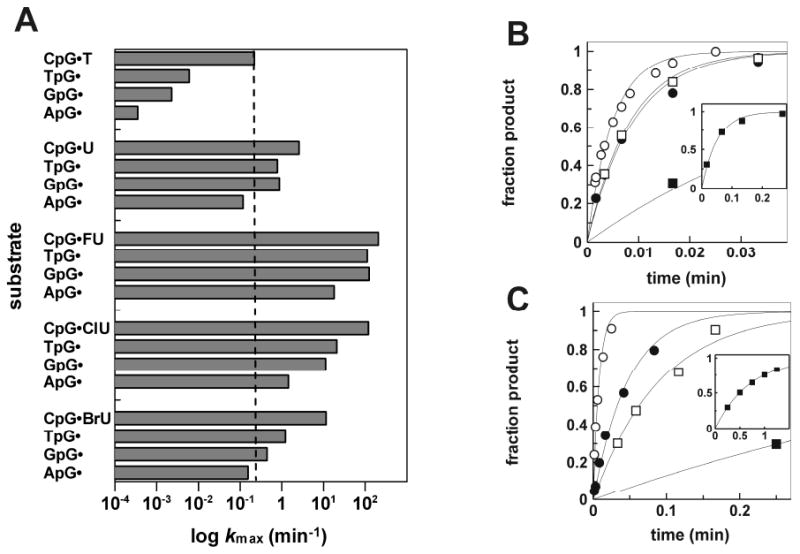
(A) Bar graph shows the maximal activity of hTDG (log kmax) for the various YpG·X substrates. For comparison, the activity for CpG·T (kmax = 0.22 min-1) is indicated by the dotted line. (B) Representative data from single turnover kinetics experiments for FU excision from CpG·FU (○), TpG·FU (●), GpG·FU (□), and ApG·FU (■) substrates. The inset shows the complete time course for ApG·FU (■). (C) Representative data from single turnover kinetics for ClU excision from CpG·ClU (○), TpG·ClU (●), GpG·ClU (□), and ApG·ClU (■) substrates. The inset shows the complete data for ApG·ClU (■).
It is important to note that the activity is significantly greater for all of the G·FU and G·ClU substrates than for CpG·T, which is generally considered the most biologically relevant substrate for hTDG (28,29). Indeed, kmax is 75- to 920-fold greater for the G·FU substrates, and 7- to 550-fold greater for the G·ClU substrates, relative to CpG·T. In addition, the activity for all of the G·U and G·BrU substrates exceeds the CpG·T activity except for ApG·U and ApG·BrU, which are merely 2-fold and 1.4-fold slower, respectively.
Activity is Greatly Diminished for A·X Relative to G·X Substrates
It has been reported that hTDG does not remove thymine from A·T pairs (10,11,41), in keeping with the biological imperative to minimize the activity against undamaged DNA. We therefore wondered to what extent the robust hTDG activity observed for G·FU, G·ClU, and G·BrU is diminished when the 5-halouracil bases are paired with adenine rather than guanine. This is relevant because 5-halogenated dU is incorporated opposite template dA, and vice versa, during replication (24). We examined the effect of pairing the target base with adenine rather than guanine, while preserving the 5′ C·G pair, using the CpA·X substrates (Figure 2). The data are listed in Table 1 and displayed in Figure 5. Under the experimental conditions used here, i.e. high concentrations of substrate (500 nM) and enzyme (5 μM), we were able to measure the exceedingly weak activity for thymine excision from the CpA·T substrate, kmax = 1.3 × 10-5 min-1. This is a striking 17,600-fold lower than the activity for CpG·T. In contrast, a much smaller 795-fold decrease is observed for CpA·U compared to CpG·U, consistent with a previously reported 600-fold difference (30). Similarly, a small 187-fold decrease is observed for CpA·FU relative to CpG·FU. For the excision of ClU, which is similar to T in terms of its steric and electrostatic properties (20), we find a much larger 5,460-fold decrease for CpA·ClU versus CpG·ClU. A significantly larger 14,870-fold decrease is observed for CpA·BrU relative to CpG·BrU, approaching the difference in activity for CpA·T versus CpG·T. Consistent with our results is a previous report that hTDG excision of 5-hydroxymethyluracil (hmU) is 104.0-fold slower for CpA·hmU compared to CpG·hmU (30). Our results show that for the substrate bases examined here, the effect on kmax of pairing the target base with A rather than G is much larger than the effect altering the 5′-flanking pair for the G·X substrates.
FIGURE 5. hTDG activity is greatly reduced for A·X relative to G·X substrates.
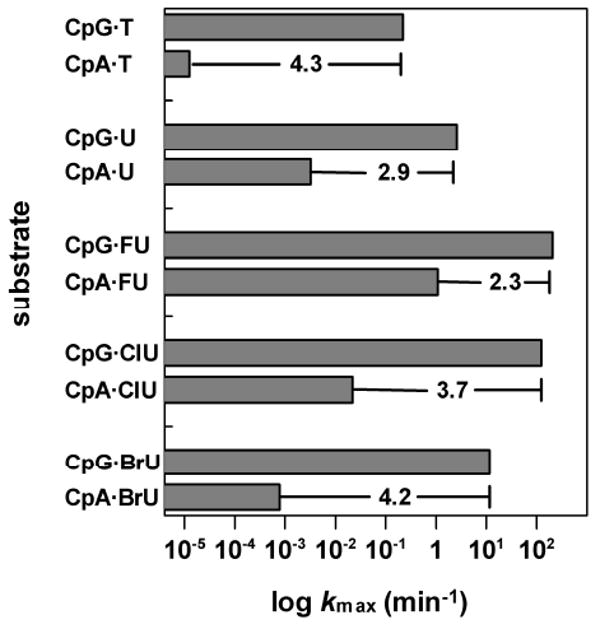
The hTDG activity (log kmax) for CpG·X versus CpA·X substrates is shown. The change in log kmax is given.
Effect of the 5′ Base Pair on A·X activity
We also examined the effect of altering the 5′-flanking pair on hTDG activity for A·X pairs, using the YpA·X substrates (Figure 2). The data are listed in Table 1 and shown in Figure 6. Generally, these effects follow the same trend observed for the 5′-neighbor effect on G·X activity, where the relative activity is: CpA·X > TpA·X > GpA·X > ApA·X. We find a significant 5′-neighbor effect for A·U activity; kmax decreases by 4-, 62-, and 104-fold for TpA·U, GpA·U, and ApA·U, respectively, as compared to the CpA·U activity. The 5′-neighbor effects are larger for A·FU pairs; kmax decreases by 5-, 78-, and 171-fold for TpA·U, GpA·U, and ApA·U, respectively, relative to CpA·FU. The effects are larger still for A·ClU; kmax decreases by 5-, 257-, and 436-fold for TpA·ClU, GpA·ClU, and ApA·ClU, respectively, compared to CpA·ClU. Somewhat smaller effects are observed for A·BrU; kmax decreases by 5-, 65-, and 103-fold for TpA·BrU, GpA·BrU, and ApA·BrU, relative to CpA·BrU. We note that of the A·X substrates examined, only CpA·FU and TpA·FU exhibit greater activity than observed for CpG·T (Figure 6).
FIGURE 6. Effects of altering the 5′-flanking pair on hTDG activity for A·X pairs.
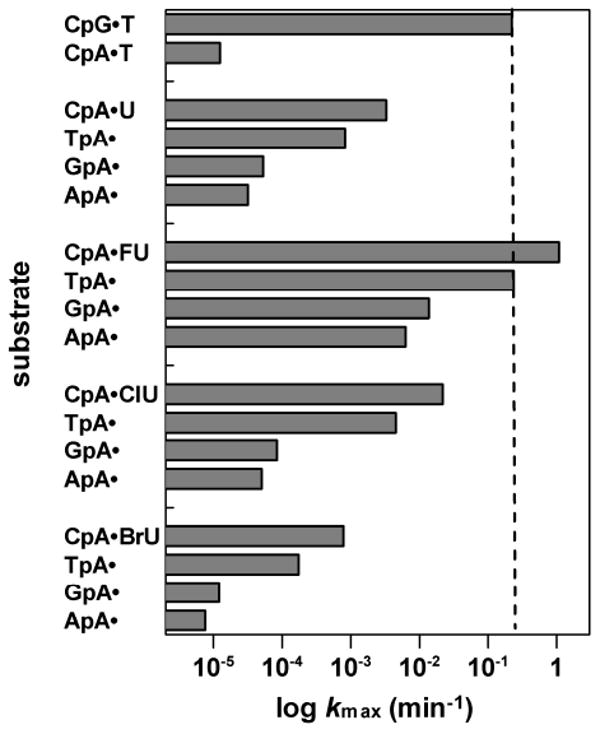
Shown is the hTDG activity (log kmax) for the YpA·X substrates, which contain an A·X pair with one of four possible 5′-flanking pairs. For comparison, the activity for CpG·T (kmax = 0.22 min-1) is indicated by the dotted line.
Discussion
Role of the 5′ C·G Pair on G·X Activity
The effect of altering the 5′ C·G pair on G·X activity was examined using the YpG·X series of substrates (Figure 2), and the results are given in Table 1 and Figure 4A. Our findings indicate that the magnitude of the 5′-neighbor effect depends on the size of the nucleobase and on substrate reactivity, i.e., the stability of the C-N bond that is cleaved by hTDG. For the uracil analogues examined here, size depends on the C5 substituent, with nucleobase volume varying as: U < FU ≪ ClU ≈ T < BrU (20). The substrate reactivity (N-glycosidic bond stability) also depends on the C5 substituent. Thus, for the hTDG-catalyzed and non-catalyzed reactions, the rate depends on the leaving ability of the departing nucleobase (20,42-44). The electron withdrawing halogens enhance the leaving ability of uracil, whereas the electron donating methyl group suppresses it. Accordingly, substrate reactivity varies as: CldU ≥ BrdU ≥ FdU ≫ dU > dT (20). As we consider the effects of altering the CpG context, we note that differences in kmax reflect a change in the equilibria for the base flipping step (Kflip) and/or the rate of the chemical step (kchem), as shown in Figure 3.
The dependence of the 5′-neighbor effect on substrate reactivity is illustrated by the comparing the effects for G·ClU, ranging from 6- to 82-fold, with the effects for G·T, which range from 37- to 582-fold. As we have noted previously, the size and electrostatic properties of T and ClU are quite similar (20), suggesting that T and ClU are not substantially discriminated by the relatively large and non-specific hTDG active site (21,22). In addition, melting studies showed that G·T and G·ClU base pairs have essentially the same stability in DNA, indicating that ClU and T have a similar propensity for flipping spontaneously out of the duplex (20). This suggests that the steeper dependence of kmax on the 5′ flanking pair for G·T relative to G·ClU is not due to substantial effects on the base flipping step. The most significant difference is that CldU is much more reactive than dT, which suggests that altering the 5′ C·G pair elicits a greater effect on kchem for G·T than for G·ClU pairs. Thus, interactions with the 5′ C·G pair may induce a conformational change in hTDG and/or the DNA substrate that stabilizes the transition state, and this effect appears to be greater for less reactive substrates.
The dependence of the 5′-neighbor effect on substrate size is illustrated by comparing the effects for G·FU, 2- to 11-fold, with G·BrU, 9- to 75-fold. The reactivity of FdU and BrdU is about the same, as is the stability of G·FU and G·BrU base pairs in DNA, however BrU is substantially larger than FU (20). Although the hTDG active site is accommodating, we found previously that kmax decreases for substrates with large C5 substituents, including BrU, IU, and BrC (20). Thus, our results suggest that interactions with the 5′ C·G pair serve to increase Kflip or stabilize the transition-state of the reaction, and that the effect is greater for substrates with a larger C5-group (BrdU and dT). A previous study found that the apparent KD is nearly the same for CpG·T and TpG·T substrates, suggesting that recognition of the 5′ C·G pair contributes largely to stabilizing the transition-state (increasing kchem) rather than promoting base flipping (29). However, additional studies are required to establish which steps of the hTDG reaction are affected by recognition of the 5′ C·G pair.
Effect of Pairing the Substrate Base with A Rather than G
Because hTDG excises a normal base, thymine, from G·T mispairs, it has an effective mechanism for avoiding the excision of T from the huge excess of A·T pairs in DNA (10,11). The specificity against A·T may involve H bond interactions that select for guanine, as observed in a crystal structure of the related mismatch uracil DNA glycosylase from Escherichia coli (eMUG) (45). Here, we have quantitatively established the specificity of hTDG for excising bases from G·X versus A·X pairs and we find that it is strikingly large. The difference in kmax ranges from 187-fold for CpA·FU versus CpG·FU, to 17,600-fold for CpA·T versus CpG·T (Table 1, Figure 5). Our findings indicate that these large differences depend on the size and reactivity (C-N bond stability) of the nucleotide substrate. The much larger effect for BrdU (104.2-fold) relative to FdU (102.3-fold) is likely due to the larger size of BrdU, because these substrates have similar reactivity (20). The larger effect for dU (102.9-fold) compared to FdU is probably attributable to the significantly greater reactivity of dFU, since dU and dFU have relatively similar steric and electrostatic properties (20). Thus, our results suggest that the putative interactions formed with the mismatched guanine serve to promote base-flipping (increase Kflip) and/or stabilize the transition state (increase kchem), and that disrupting these interactions has a greater effect for larger and less reactive substrates (i.e., dT). Additional mechanistic and/or structural studies are needed to determine how hTDG selects for G·X over A·X pairs and which step(s) of the reaction are involved.
It also of interest to consider the difference in activity for A·X versus G·X for substrates in which the 5′-flanking pair is not C·G. These effects are listed in Table 1 under “Fold change relative to YpG·X”. For example, considering U excision, the decrease in kmax is 103.0-fold for TpA·U versus TpG·U, 104.2-fold for GpA·U versus GpG·U, and 103.6-fold for ApA·U versus ApG·U, as compared to 102.9-fold for CpA·U versus CpG·U. The effects for the FU substrates have the same trend but are smaller in magnitude, whereas the effects for the ClU and BrU substrates have a similar trend and are larger in magnitude. Overall, the results indicate that the difference in activity for A·X versus G·X substrates is similar when the 5′-flanking pair is C·G or T·A, becomes larger for a 5′ A·T pair, and is maximal for a 5′ G·C pair.
Effect of the 5′ Base Pair on A·X Activity
The effects of altering the 5′ C·G pair for A·X substrates are substantial, and are larger than those observed for the G·X substrates (Figure 6, Table 1). For example, the effects are 4- to 104-fold for A·U activity as compared to 3- to 22-fold for G·U pairs. Likewise, the 5′-neighbor effects are greater for A·FU versus G·FU and for A·ClU versus G·ClU activity, whereas the effects are nearly equivalent for A·BrU versus G·BrU activity. Thus, our results do not support a previous suggestion that specificity for CpG·T involves cooperativity between the interactions formed with the mismatched guanine and the 5′-flanking C·G pair (10). If the interactions were cooperative, one would expect that the effects of altering the 5′ C·G pair would be smaller for A·X relative to G·X substrates, because the interactions formed with the mismatched guanine would be disrupted for the A·X substrates.
Combined Effect of Pairing the Target Base with Adenine and Altering the 5′ C·G Pair
It is illuminating to consider the effect on kmax of replacing the opposing guanine with adenine and altering the 5′ C·G pair. The combined effects are very large, as shown in the lower region of Table 1 under the heading “Fold change relative to CpG·X”. For example, compared to the activity for CpG·U, kmax is reduced by 103.5-, 104.7-, and 104.7-fold for TpA·U, GpA·U, and ApA·U, respectively. Similar reductions in activity of 102.9- to 104.5-fold are seen for the FU substrates. The effects are even more striking for the larger bases; ranging from 104.4- to 106.4-fold for ClU, and 104.8- to 106.2-fold for BrU. The huge effects observed for ClU and BrU suggest that the specificity of hTDG for CpG·T lesions over normal A·T pairs ranges in magnitude from 104.3-fold to 106.4-fold or more, corresponding to 6 to 8.7 kcal/mol.
Implications for CpG·T Specificity
Taken together, our findings indicate that disrupting the interactions that hTDG forms with the 5′ C·G pair and the mismatched guanine have a greater effect for substrates with a large C5-substituent and/or a more stable N-glycosidic bond. Thus, the relatively large size and low reactivity of dT likely explains the large (37- to 625-fold) 5′-neighbor effect on kmax and the huge (104.3-fold) difference in activity for CpA·T versus CpG·T. Indeed, the smaller (3- to 22-fold) 5′-neighbor effect for G·U activity and the much smaller difference between CpA·U and CpG·U (102.9-fold) is likely explained by the substantially smaller size and enhanced reactivity of dU relative to dT. These differences in activity would seem to be consistent with the requirement of the cell to restrict thymine excision to thymines that arise by 5-methylcytosine deamination at CpG sites, whereas uracil can be removed wherever it arises in DNA.
Biomedical Relevance of FU Activity
Our finding of very strong hTDG activity for G·FU lesions with any 5′ base pair, some 75- to 920-fold greater than CpG·T activity, may be relevant to the cytotoxicity of FU, which has been used for decades to treat cancer (27). The anticancer effect of FU is thought to involve multiple pathways, including the incorporation of U and FU into DNA followed by their excision by a DNA glycosylase, leading to a cycle of incorporation and excision, an increased level of abasic sites, DNA fragmentation and cell death (24,27). Consistent with a potential role for TDG in this process, it was reported that TDG inactivation diminishes the sensitivity of fission yeast and mouse embryo fibroblasts to FU treatment, and leads to a decrease in FU-induced DNA strand breaks (28). This likely reflects TDG activity against G·FU lesions, because its A·FU activity is relatively weak. Other DNA glycosylases that could potentially elicit a similar effect by removing misincorporated U and/or FU include uracil DNA glycosylase (UNG2), SMUG1, and methyl binding domain IV (MBD4). The removal of U probably involves UNG2 and SMUG1 (46), because TDG and MBD4 are much less efficient against U (Table 1)(14). Although UNG2 has the highest activity for U removal, its activity is much lower for FU (47-49), and UNG2 does not remove FU from DNA in mouse embryo fibroblasts (49). Although hMBD4 has significant activity for CpG·FU lesions, the effect of the 5′ base pair on G·FU activity is unknown for this CpG specific enzyme, and it is inactive against A·FU pairs (50). Recent studies report that MBD4 inactivation decreases the sensitivity of mice to FU treatment, although this may reflect a loss of an MBD4-mediated apoptotic response to DNA damage rather than FU excision (51).
The toxicity of FU may also depend on its presence in DNA, due to mutagenesis or perturbations of protein-DNA interactions, as was suggested by a report that FU excision by SMUG1 protects mouse embryo fibroblasts against FU-induced toxicity (49). The potential for mutagenesis is significant; FU incorporation yields mostly A·FU pairs, but also some G·FU pairs which can cause G·C → A·T mutations (52,53). Thus, repair of G·FU lesions for which G is in the parental strand would be protective. However, because G can be incorporated opposite template FU, replication of A·FU pairs can give G·FU lesions, leading to A·T → G·C mutations (52,53). It is important to note that BER processing of these G·FU lesions would facilitate the A·T → G·C transition rather than protect against it. Perhaps the protective role observed for SMUG1 (49) reflects the repair of initial A·FU lesions (i.e., FU in the daughter strand). The reported effect of hTDG in enhancing the sensitivity of mouse embryo fibroblasts to FU treatment may reflect hTDG activity against G·FU lesions in which FU is in the parental strand, or the production of abasic sites, as discussed above.
Biological Relevance of BrU and ClU Activity
We find that hTDG exhibits robust activity for G·ClU and G·BrU pairs, which meets and in most cases exceeds the activity for CpG·T (Figure 4). Our findings may have important biological implications, because it has been shown that ClU and BrU arise in DNA due to oxidative processes associated with inflammation (25,26), leading to mutagenic, genotoxic, and cytotoxic effects (23,24). As an element of host defense, peroxidases produce hypochlorous and hypobromous acid, which promote the halogenation of pyrimidines (at C5), leading to Cl5dUTP and Br5dUTP (25,26,54-58). These dTTP analogues are incorporated into DNA, giving predominantly A·ClU and A·BrU pairs, and some G·ClU and G·BrU lesions (53,59), which can cause G·C → A·T mutations. The initial A·ClU and A·BrU pairs are also mutagenic, because incorporation of G opposite template ClU or BrU gives G·ClU or G·BrU, and eventually A·T → G·C transitions (53,59,60). Until recently, it did not appear that any human enzymes could remove ClU or BrU from DNA. It had been shown that UNG2 and SMUG1 do not remove ClU or BrU, due likely due to steric hindrance in the active site (25,61-63). Our findings raise the possibility that hTDG is active against G·ClU or G·BrU lesions that arise in vivo, and a recent study suggests that hMBD4 may also be active against these lesions (50). However, A·ClU and A·BrU lesions are poor substrates for hTDG (Table 1) and hMBD4 (50), and these lesions may persist in DNA. The effect of excising ClU or BrU may depend on whether the halogenated base is misincorporated or resides in the template strand. The excision of ClU or BrU that was misincorporated opposite template G could protect against G·C → A·T mutations. On the other hand, repair of G·ClU and G·BrU lesions that have arisen from replication of A·ClU or A·BrU (i.e., template ClU or BrU) will facilitate an A·T → G·C transition rather than protect against it. In addition to these potential effects on ClU- and BrU-induced mutagenesis, hTDG could potentially contribute to a repetitive cycle of ClU and BrU misincorporation and excision, leading to abasic sites and DNA strand breaks, similar to the potential role played by hTDG in FU toxicity, as discussed above.
It has been shown that sister-chromatid exchange (SCE) increases with the amount of CldU and BrdU present in replicated DNA (64,65). Although the mechanism of ClU- and BrU-induced SCE has not been established, evidence suggests that one mechanism involves the formation of single-strand breaks (SSBs) that originate from abasic sites (23,24,66). The generally accepted model is that these abasic sites arise from dehalogenation of ClU or BrU followed by excision of the resulting uracil by UNG2 (66). Our findings suggest an alternative mechanism; that the abasic sites originate from the direct excision of ClU and BrU by TDG (and potentially MBD4). It has also been observed that CldU induces 3-5 times more SCE than BrdU when these dT analogs are incorporated at equivalent level in DNA (24,64). Our findings offer a potential explanation; the ∼10-fold higher activity of hTDG for G·ClU over G·BrU could generate more abasic sites, given a similar level of ClU and BrU incorporation into DNA. Thus, our findings and recent observations for MBD4 raise the possibility that these enzymes might contribute to the mechanism of CldU- and BrdU-induced SCE.
Footnotes
This work was supported by the National Institutes of Health, R01-GM72711 (A.C.D.) and the University of Maryland Marlene and Stewart Greenebaum Cancer Center. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: AP, apurinic/apyrimidinic; BER, base excision repair; BrU, 5-bromouracil;Br5 dUTP; 5-bromo-dUTP; dU, 2′-deoxyuridine; ClU, 5-chlorouracil; Cl5 dUTP, 5-chloro-dUTP; FU, 5-fluorouracil; HPLC, high pressure liquid chromatography; hTDG, human thymine DNA glycosylase; kmax, rate constant determined from single turnover kinetics; LB, Luria Broth; m5C, 5-methylcytosine; MBD4,methyl binding domain IV; SMUG1, single-strand selective monofunctional uracil DNA glycosylase; UNG2, uracil DNA glycosylase.
References
- 1.Lindahl T. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 2.Loeb LA, Christians FC. Mutat Res. 1996;350:279–286. doi: 10.1016/0027-5107(95)00117-4. [DOI] [PubMed] [Google Scholar]
- 3.Lindahl T, Wood RD. Science. 1999;286:1897–1905. doi: 10.1126/science.286.5446.1897. [DOI] [PubMed] [Google Scholar]
- 4.Wiebauer K, Jiricny J. Nature. 1989;339:234–236. doi: 10.1038/339234a0. [DOI] [PubMed] [Google Scholar]
- 5.Neddermann P, Jiricny J. Proc Natl Acad Sci USA. 1994;91:1642–1646. doi: 10.1073/pnas.91.5.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coulondre C, Miller JH, Farabaugh PJ, Gilbert W. Nature. 1978;274:775–780. doi: 10.1038/274775a0. [DOI] [PubMed] [Google Scholar]
- 7.Rideout WM, 3rd, Coetzee GA, Olumi AF, Jones PA. Science. 1990;249:1288–1290. doi: 10.1126/science.1697983. [DOI] [PubMed] [Google Scholar]
- 8.Jones PA, Takai D. Science. 2001;293:1068–1070. doi: 10.1126/science.1063852. [DOI] [PubMed] [Google Scholar]
- 9.Feinberg AP, Tycko B. Nat Rev Cancer. 2004;4:143–153. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- 10.Waters TR, Swann PF. J Biol Chem. 1998;273:20007–20014. doi: 10.1074/jbc.273.32.20007. [DOI] [PubMed] [Google Scholar]
- 11.Sibghat U, Gallinari P, Xu YZ, Goodman MF, Bloom LB, Jiricny J, Day RS., 3rd Biochemistry. 1996;35:12926–12932. doi: 10.1021/bi961022u. [DOI] [PubMed] [Google Scholar]
- 12.Hendrich B, Hardeland U, Ng HH, Jiricny J, Bird A. Nature. 1999;401:301–304. doi: 10.1038/45843. [DOI] [PubMed] [Google Scholar]
- 13.Bellacosa A, Cicchillitti L, Schepis F, Riccio A, Yeung AT, Matsumoto Y, Golemis EA, Genuardi M, Neri G. Proc Natl Acad Sci USA. 1999;96:3969–3974. doi: 10.1073/pnas.96.7.3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Petronzelli F, Riccio A, Markham GD, Seeholzer SH, Stoerker J, Genuardi M, Yeung AT, Matsumoto Y, Bellacosa A. J Biol Chem. 2000;275:32422–32429. doi: 10.1074/jbc.M004535200. [DOI] [PubMed] [Google Scholar]
- 15.Millar CB, Guy J, Sansom OJ, Selfridge J, MacDougall E, Hendrich B, Keightley PD, Bishop SM, Clarke AR, Bird A. Science. 2002;297:403–405. doi: 10.1126/science.1073354. [DOI] [PubMed] [Google Scholar]
- 16.Hardeland U, Bentele M, Jiricny J, Schar P. Nucleic Acids Res. 2003;31:2261–2271. doi: 10.1093/nar/gkg344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu P, Burdzy A, Sowers LC. DNA Repair (Amst) 2003;2:199–210. doi: 10.1016/s1568-7864(02)00198-2. [DOI] [PubMed] [Google Scholar]
- 18.Saparbaev M, Laval J. Proc Natl Acad Sci USA. 1998;95:8508–8513. doi: 10.1073/pnas.95.15.8508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoon JH, Iwai S, O'Connor TR, Pfeifer GP. Nucleic Acids Res. 2003;31:5399–5404. doi: 10.1093/nar/gkg730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bennett MT, Rodgers MT, Hebert AS, Ruslander LE, Eisele L, Drohat AC. J Am Chem Soc. 2006;128:12510–12519. doi: 10.1021/ja0634829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barrett TE, Scharer OD, Savva R, Brown T, Jiricny J, Verdine GL, Pearl LH. EMBO J. 1999;18:6599–6609. doi: 10.1093/emboj/18.23.6599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baba D, Maita N, Jee JG, Uchimura Y, Saitoh H, Sugasawa K, Hanaoka F, Tochio H, Hiroaki H, Shirakawa M. Nature. 2005;435:979–982. doi: 10.1038/nature03634. [DOI] [PubMed] [Google Scholar]
- 23.Morris SM. Mutat Res. 1991;258:161–188. doi: 10.1016/0165-1110(91)90007-i. [DOI] [PubMed] [Google Scholar]
- 24.Morris SM. Mutat Res. 1993;297:39–51. doi: 10.1016/0165-1110(93)90006-9. [DOI] [PubMed] [Google Scholar]
- 25.Jiang Q, Blount BC, Ames BN. J Biol Chem. 2003;278:32834–32840. doi: 10.1074/jbc.M304021200. [DOI] [PubMed] [Google Scholar]
- 26.Henderson JP, Byun J, Takeshita J, Heinecke JW. J Biol Chem. 2003;278:23522–23528. doi: 10.1074/jbc.M303928200. [DOI] [PubMed] [Google Scholar]
- 27.Longley DB, Harkin DP, Johnston PG. Nat Rev Cancer. 2003;3:330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 28.Cortazar D, Kunz C, Saito Y, Steinacher R, Schar P. DNA Repair (Amst) 2007;6:489–504. doi: 10.1016/j.dnarep.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 29.Abu M, Waters TR. J Biol Chem. 2003;278:8739–8744. doi: 10.1074/jbc.M211084200. [DOI] [PubMed] [Google Scholar]
- 30.Wibley JE, Waters TR, Haushalter K, Verdine GL, Pearl LH. Mol Cell. 2003;11:1647–1659. doi: 10.1016/s1097-2765(03)00235-1. [DOI] [PubMed] [Google Scholar]
- 31.Fasman G. CRC Handbook of Biochemistry and Molecular Biology. 3rd. CRC Press; Boca Ratton, FL: 1975. [Google Scholar]
- 32.Hardeland U, Steinacher R, Jiricny J, Schar P. EMBO J. 2002;21:1456–1464. doi: 10.1093/emboj/21.6.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gill SC, von Hippel PH. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 34.Leatherbarrow, R. J. (1998), Erithacus Software Ltd., Staines, U.K.
- 35.O'Neill RJ, Vorob'eva OV, Shahbakhti H, Zmuda E, Bhagwat AS, Baldwin GS. J Biol Chem. 2003 doi: 10.1074/jbc.M210860200. [DOI] [PubMed] [Google Scholar]
- 36.Porello SL, Leyes AE, David SS. Biochemistry. 1998;37:14756–14764. doi: 10.1021/bi981594+. [DOI] [PubMed] [Google Scholar]
- 37.McCann JA, Berti PJ. J Biol Chem. 2003;278:29587–29592. doi: 10.1074/jbc.M212474200. [DOI] [PubMed] [Google Scholar]
- 38.Waters TR, Gallinari P, Jiricny J, Swann PF. J Biol Chem. 1999;274:67–74. doi: 10.1074/jbc.274.1.67. [DOI] [PubMed] [Google Scholar]
- 39.Steinacher R, Schar P. Curr Biol. 2005;15:616–623. doi: 10.1016/j.cub.2005.02.054. [DOI] [PubMed] [Google Scholar]
- 40.Jiang YL, Stivers JT. Biochemistry. 2002;41:11236–11247. doi: 10.1021/bi026226r. [DOI] [PubMed] [Google Scholar]
- 41.Neddermann P, Jiricny J. J Biol Chem. 1993;268:21218–21224. [PubMed] [Google Scholar]
- 42.Shapiro R, Danzig M. Biochemistry. 1972;11:23–29. doi: 10.1021/bi00751a005. [DOI] [PubMed] [Google Scholar]
- 43.Shapiro R, Kang S. Biochemistry. 1969;8:1806–1810. doi: 10.1021/bi00833a004. [DOI] [PubMed] [Google Scholar]
- 44.Vanschepdael A, Macken E, Busson R, Janssen G, Herdewijn P, Roets E, Hoogmartens J. J Chromatog A. 1993;657:208–212. [Google Scholar]
- 45.Barrett TE, Savva R, Panayotou G, Barlow T, Brown T, Jiricny J, Pearl LH. Cell. 1998;92:117–129. doi: 10.1016/s0092-8674(00)80904-6. [DOI] [PubMed] [Google Scholar]
- 46.Krokan HE, Drablos F, Slupphaug G. Oncogene. 2002;21:8935–8948. doi: 10.1038/sj.onc.1205996. [DOI] [PubMed] [Google Scholar]
- 47.Mauro D, De Riel J, Tallarida R, Sirover M. Mol Pharmacol. 1993;43:854–857. [PubMed] [Google Scholar]
- 48.Kavli B, Sundheim O, Akbari M, Otterlei M, Nilsen H, Skorpen F, Aas PA, Hagen L, Krokan HE, Slupphaug G. J Biol Chem. 2002;277:39926–39936. doi: 10.1074/jbc.M207107200. [DOI] [PubMed] [Google Scholar]
- 49.An Q, Robins P, Lindahl T, Barnes DE. Cancer Res. 2007;67:940–945. doi: 10.1158/0008-5472.CAN-06-2960. [DOI] [PubMed] [Google Scholar]
- 50.Turner DP, Cortellino S, Schupp JE, Caretti E, Loh T, Kinsella TJ, Bellacosa A. Cancer Res. 2006;66:7686–7693. doi: 10.1158/0008-5472.CAN-05-4488. [DOI] [PubMed] [Google Scholar]
- 51.Sansom OJ, Zabkiewicz J, Bishop SM, Guy J, Bird A, Clarke AR. Oncogene. 2003;22:7130–7136. doi: 10.1038/sj.onc.1206850. [DOI] [PubMed] [Google Scholar]
- 52.Sowers L, Eritja R, Kaplan B, Goodman M, Fazakerly G. J Biol Chem. 1988;263:14794–14801. [PubMed] [Google Scholar]
- 53.Yu H, Eritja R, Bloom L, Goodman M. J Biol Chem. 1993;268:15935–15943. [PubMed] [Google Scholar]
- 54.Henderson JP, Byun J, Mueller DM, Heinecke JW. Biochemistry. 2001;40:2052–2059. doi: 10.1021/bi002015f. [DOI] [PubMed] [Google Scholar]
- 55.Chen HJ, Row SW, Hong CL. Chem Res Toxicol. 2002;15:262–268. doi: 10.1021/tx015578g. [DOI] [PubMed] [Google Scholar]
- 56.Whiteman M, Jenner A, Halliwell B. Chem Res Toxicol. 1997;10:1240–1246. doi: 10.1021/tx970086i. [DOI] [PubMed] [Google Scholar]
- 57.Henderson JP, Byun J, Williams MV, McCormick ML, Parks WC, Ridnour LA, Heinecke JW. Proc Natl Acad Sci USA. 2001;98:1631–1636. doi: 10.1073/pnas.041146998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Henderson JP, Byun J, Williams MV, Mueller DM, McCormick ML, Heinecke JW. J Biol Chem. 2001;276:7867–7875. doi: 10.1074/jbc.M005379200. [DOI] [PubMed] [Google Scholar]
- 59.Lasken RS, Goodman MF. J Biol Chem. 1984;259:11491–11495. [PubMed] [Google Scholar]
- 60.Trautner TA, Swartz MN, Kornberg A. Proc Natl Acad Sci USA. 1962;48:449–455. doi: 10.1073/pnas.48.3.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brandon ML, Mi LJ, Chaung W, Teebor G, Boorstein RJ. Mutat Res. 2000;459:161–169. doi: 10.1016/s0921-8777(99)00061-0. [DOI] [PubMed] [Google Scholar]
- 62.Kubareva EA, Volkov EM, Vinogradova NL, Kanevsky IA, Oretskaya TS, Kuznetsova SA, Brevnov MG, Gromova ES, Nevinsky GA, Shabarova ZA. Gene. 1995;157:167–171. doi: 10.1016/0378-1119(94)00771-j. [DOI] [PubMed] [Google Scholar]
- 63.Baker D, Liu PF, Burdzy A, Sowers LC. Chem Res Toxicol. 2002;15:33–39. doi: 10.1021/tx010113b. [DOI] [PubMed] [Google Scholar]
- 64.Heartlein MW, O'Neill JP, Preston RJ. Mutat Res. 1983;107:103–109. doi: 10.1016/0027-5107(83)90081-7. [DOI] [PubMed] [Google Scholar]
- 65.O'Neill JP, Heartlein MW, Preston RJ. Mutat Res. 1983;109:259–270. doi: 10.1016/0027-5107(83)90051-9. [DOI] [PubMed] [Google Scholar]
- 66.Wilson DM, 3rd, Thompson LH. Mutat Res. 2007;616:11–23. doi: 10.1016/j.mrfmmm.2006.11.017. [DOI] [PubMed] [Google Scholar]