

Abstract
Elevated plasma homocysteine (Hcy) is associated with cerebrovascular disease and activates matrix metalloproteinases (MMPs), which lead to vascular remodeling that could disrupt the blood-brain barrier. To determine whether Hcy administration can increase brain microvascular leakage secondary to activation of MMPs, we examined pial venules by intravital video microscopy through a craniotomy in anesthetized mice. Bovine serum albumin labeled with fluorescein isothiocyanate (BSA-FITC) was injected into a carotid artery to measure extravenular leakage. Hcy (30 μM/total blood volume) was injected 10 min after FITC-BSA injection. Four groups of mice were examined: 1) wild type (WT) given vehicle; 2) WT given Hcy (WT + Hcy); 3) MMP-9 gene knockout given Hcy (MMP-9−/− + Hcy); and 4) MMP-9−/− with topical application of histamine (10−4 M) (MMP-9−/− + histamine). In the WT + Hcy mice, leakage of FITC-BSA from pial venules was significantly (P < 0.05) greater than in the other groups. There was no significant leakage of pial microvessels in MMP-9−/− + Hcy mice. Increased cerebrovascular leakage in the MMP-9−/− + histamine group showed that microvascular permeability could still increase by a mechanism independent of MMP-9. Treatment of cultured mouse microvascular endothelial cells with 30 μM Hcy resulted in significantly greater F-actin formation than in control cells without Hcy. Treatment with a broad-range MMP inhibitor (GM-6001; 1 μM) ameliorated Hcy-induced F-actin formation. These data suggest that Hcy increases microvascular permeability, in part, through MMP-9 activation.
Keywords: blood-brain barrier, F-actin, matrix metalloproteinases, matrix metalloproteinase-9, pial microvessels
Homocysteine (Hcy) is a toxic, nonprotein sulfur-containing amino acid that is formed exclusively upon demethylation of methionine. Hcy is nutritionally regulated and metabolized through remethylation or trans-sulfuration pathways. Total plasma content of Hcy varies from 3 to 15 μM, and higher plasma levels are called hyperhomocysteinemia (HHcy). The ranges of HHcy have been referred to as moderate (16 to 30 μM), intermediate (31 to 100 μM), or severe (>100 μM) (12).
Recent data indicate that moderate HHcy is an independent risk factor for several cardiovascular and cerebrovascular disorders, including atherosclerosis (12, 13, 44), vascular thrombosis (38), diabetes (1), Alzheimer's and Parkinson's diseases (28, 44), and stroke (39, 44, 47). However, some studies point to a lesser role of Hcy in the development of atherosclerosis in human coronary arteries (23). Other work (31) suggests that essential hypertension may be associated with increased plasma Hcy levels and indicates that it is unrelated to endothelial damage (assessed by the plasma von Willebrand factor) and clinical indexes or prognosis. Discrepancies in associating Hcy with vascular damage may be due to variations in the criteria used to determine the role of Hcy in various cardiovascular and cerebrovascular disorders and suggest a lack of systemic functional studies.
Many cerebrovascular disorders are accompanied by alterations in the blood-brain barrier that result in microvascular leakage and edema formation. Therefore, the vascular endothelium has a key role in these processes. We (20) and others (7) showed that enhanced formation of filamentous actin (F-actin) in endothelial cells is associated with increased gap formation between the cells that could result in microvascular leakage. In the present study, we investigated the role of acute HHcy on cerebrovascular leakage. We tested the hypothesis that high plasma Hcy levels can mediate macromolecular leakage in mouse brain pial microvessels via activation of matrix metalloproteinases (MMPs) and increased formation of F-actin in endothelial cells. The results of this study suggest that Hcy has a significant role in development of microvascular leakage and that this process can be drastically ameliorated by inhibition of MMPs.
Materials and Methods
In accordance of with National Institutes of Health Guidelines for animal research, all animal procedures for these experiments were reviewed and approved by the Institutional Animal Care and Use Committee of the University of Louisville.
Animals and microvascular observation of pial vascular bed
Male, wild-type (WT) C57BL/6J or MMP-9 gene-knockout (MMP-9−/−) mice (28–32 g) were anesthetized with tribromoethanol (240 mg/kg ip), and a tracheal cannula was inserted to maintain a patent airway. Body temperature was kept at 37 ± 1°C with a heating pad. Mean arterial blood pressure (MABP) and pulse rate were continuously monitored through a polyethylene catheter (PE-10) inserted into a carotid artery and connected to a transducer and a blood pressure analyzer (Micro-Med, Louisville, KY).
Brain pial microcirculation was prepared for observations similarly to the methods described by others (11, 17, 32). Briefly, the scalp and connective tissues were removed over the parietal cranial bone above the left hemisphere. A craniotomy was done with a small trephine attached to a high-speed microdrill (Fine Science Tool, Foster City, CA). During the drilling, the cranium was continuously washed with room temperature physiological salt solution (PSS). The dura was reflected with the bone disk using a microrongeur with extra-fine tips (Fine Science Tool) to form a cranial window about 3 mm in diameter. From this time, the exposed brain surface and pial circulation were bathed continuously by applying oxygenated artificial cerebrospinal fluid (composition in mM: 132 NaCl, 2.95 KCl, 1.71 CaCl, 1.4 MgSO4, 24.6 NaHCO3, 3.71 glucose, and 6.7 urea) with pH 7.4 and temperature of 37°C. Others have found that responses of pial vessels observed from an opened cranial window are generally representative of the responses of the pial microcirculation (30, 31).
Microvascular leakage observation
Hcy-induced microvascular leakage was observed according to a method described previously (36). The animals were positioned on the stage of a Nikon MM-11 trinocular microscope so that the exposed pial circulation could be observed by epi-illumination. After the surgical preparation there was a 1-h equilibration period. Before each experiment, autofluorescence of the observed area was recorded over a standard range of camera gains. Fluorescein isothiocyanate (FITC, 300 μg/ml) (24) tagged to bovine serum albumin (BSA, Sigma Chemicals, St. Louis, MO) was then injected intra-arterially (0.2 ml/100 g of body wt) and allowed to circulate for about 10 min. The pial circulation was surveyed to ensure that there was no spontaneous leakage in the observed area that would indicate compromised vascular integrity.
Venules were identified by observing the topology of the pial circulation and blood flow direction (with increasing diameters in the direction of blood flow). In each experiment, a rectangular area of interest (AOI), about 2,000 μm2 in the interstitium adjacent to a venular wall, was chosen for microvascular leakage assessment. There was no spontaneous leakage or other visible vessels in the chosen area. Hcy (0.028 mg/100 g of body wt in 100 μl resulting in a final blood concentration of 30 μM) or the same volume (100 μl) of the vehicle PSS was injected via the carotid artery catheter. Interstitial fluorescence was recorded before injection (baseline) and after 5 and 40 min of Hcy or vehicle injection in WT and MMP-9−/− mice.
Microvascular leakage was measured in WT and MMP-9−/− mice before and after PSS or Hcy administration. In separate MMP-9−/− and WT mice, we measured and compared the leakage caused by topical application of histamine (10−4 M). This dose of histamine is known to cause significant microvascular leakage (2, 3, 22, 25, 27, 34, 35, 48, 49). The diameters of the venules, adjacent to the AOIs were measured at baseline and 40 min after Hcy, vehicle injection, or topical application of histamine.
An epi-illumination system, consisting of a mercury arc lamp and a ploem system with appropriate filters, was used to excite intravascular FITC. The AOI was exposed to blue light (450–490 nm) for 10–15 s with a power density of 2 J/cm2. The microscope images were acquired by a light-sensitive silicon-intensified tube camera (Hamamatsu C2400) and image acquisition system (Marvel G450-eTV, Matrox Graphics). The camera output voltage was standardized with a 50 ng/ml fluorescein diacetate standard (Estman Kodak, Rochester, NY) for each experiment. The magnification of the system was determined by a stage micrometer, and vessel diameters were measured directly on the video monitor by using a video caliper.
The separate groups of animals (3 WT mice treated with PSS, 3 WT mice treated with Hcy, and 3 MMP-9−/− mice treated with Hcy) were anesthetized with pentobarbital sodium (65 mg/kg body wt ip). One milliliter of blood was collected from the vena cava of the anesthetized WT and MMP-9−/− mice as described earlier (18, 21). Animals were then overdosed (euthanized) with pentobarbital sodium (100 mg/kg body wt ip), and brain samples were collected and frozen for subsequent MMP analyses. Blood was centrifuged at 4,000 g for 5 min. After centrifugation, the Hcy level was determined in plasma samples from WT and MMP-9−/− mice according to the method described earlier (46).
Determination of macromolecular leakage
A fluorescein diacetate standard curve was determined (25) to ensure the consistency of fluorescence excitation/emission spectra of the FITC-BSA conjugate. Before the injection, the emission intensity of the volume of FITC-BSA to be injected was measured and plotted as a function of the fluorescein diacetate curve. It was the same in all the experiments. Before injection of FITC-BSA, the autofluorescence intensity of the brain surface was measured and later subtracted from the fluorescence intensity measured in the AOI adjacent to a vessel. This analysis quantifies the increase in light intensity in the interstitium and provides an index of macromolecular leakage into the area adjacent to a venule (24, 25, 36). The fluorescent image was digitized by using image analysis software (Matrox Inspector, Matrox Electronics Systems, Dorval, Canada). The digitized image is composed of pixels of varying gray levels depending on the light intensity (gray levels range from 0 to 255, with zero representing a black image and 255 representing a white image). The average pixel gray level in the AOI was determined at each observation time. Neutral density filters were used in the light path before the camera at high levels of emission intensity to ensure that detection was in the linear range of the camera. Fluorescence intensity is represented by the recorded gray level multiplied by the appropriate neutral density filter factor and presented as fluorescence intensity units (FIU).
Evaluation of enzymatic cleavage of FITC-BSA by MMP-9 (gelatinize B)
To confirm the absence of enzymatic cleavage of albumin by MMP-9, proMMP-9 (10 μM; EMD Biosciences) was activated with l-(tosylamido-2-phenyl)-ethyl-chloromethyl ketone-treated trypsin (10 nM) by incubation at 37°C for 45 min (42). The reaction was terminated by heating at 100°C for 15 min. Two milliliters of FITC-BSA conjugate (30 μg/ml) were treated with activated MMP-9 for 16 h. The enzymatic effect of active MMP-9 on albumin was evaluated by comparing the SDS-PAGE fluorescence analysis of FITC-BSA conjugates treated with activated MMP-9 with that of FITC-BSA treated with proMMP-9. Fluorescence of the FITC-BSA was detected by exiting the bands at 495 nm and recording the fluorescence at 518 nm. Activity of MMP-9 in FITC-BSA samples was confirmed by gelatin zymography performed on the same gels after fluorescence analyses. The experiment was done in duplicate.
Endothelial cell culture
Mouse cardiac microvascular endothelial cells (MEC) were obtained from WT mice by using a modification of a method described elsewhere (15). Briefly, hearts from two mice were removed aseptically, rinsed in Hanks' balanced salt solution (HBSS; GIBCO) to remove excess blood, and minced into ∼2-mm square pieces. The heart pieces were digested in 10 ml of collagenase type B (0.2%, Boehringer-Mannheim Biochemicals) for 25 min at 37°C with occasional agitation. Further digestion was done with 1 ml of 0.25% trypsin/EDTA (GIBCO) for 5 to 10 min at 37°C. The cellular digest was filtered through sterile 40-μm nylon mesh and then centrifuged at 120 g for 5 min. After the supernatant was removed, the cell pellet was washed twice in DMEM containing 20% fetal bovine serum. MECs were resuspended in growth media consisting of DMEM, 20% fetal bovine serum, 2 mM l-glutamine, 2 mM sodium pyruvate, 20 U/ml heparin, 20 mM HEPES, and antibiotics (100 μg/ml streptomycin and 100 UI/ml penicillin) (19). The MECs were then plated on eight-well chambered coverglass plates (Fisher) that were coated with bovine fibronectin. After 2 h, the attached cells were washed with DMEM. Cultures were maintained in a humidified atmosphere of 5% CO2 at 37°C until they formed a complete monolayer on the coverglass surface.
Endothelial cell cytoskeletal F-actin formation assay
Primary cultured MECs from mice were grown for 3 days in two, chambered coverglass plates that each had eight wells (Fisher). Before experimentation, the cells were washed with HBSS to remove the medium. The cells were divided into four groups (two wells in each group). Cells in each group were treated for 40 min at 37°C with either 3, 30, or 30 μM Hcy in the presence of 0.1, 1, 10, or 100 μM GM-6001 (EMD Biosciences), which is a potent broad-spectrum hydroxamic acid inhibitor of MMPs. In a separate group of cells, 30 μM Hcy in the presence of a negative control (inactive form) for the MMP inhibitor GM-6001 (0.1, 1, 10, or 100 μM) was added. MMP inhibitor (GM-6001) or its negative control was added to the wells 5 min before the addition of 30 μM Hcy. In the control group, the cells were kept in PSS for 40 min. Another two groups of cells (two wells in each group) were treated with 1 or 100 μM of GM-6001 or its negative control (1 or 100 μM) without Hcy treatment. The well contents were then aspirated, and the cells were incubated with BODIPY-Phalloidin (10 U, Molecular Probes) and lysopalmitoylphosphatidylcholine (100 μg/ml) dissolved in 3.7% formaldehyde for 30 min at 4°C in the dark (20). After incubation, the cells were washed three times with HBSS, and digital images of the formed intercellular F-actin were recorded by an Olympus FV1000 confocal microscope (with ×60 objective). A HeNe-G laser (543 nm) was used to excite the dye, and the readings were obtained above 560 nm of the emission wavelength. The images were compared according to the following criteria: 1) presence of nonactin staining areas (gaps between the adjacent cells), 2) formation of individual stress fibers, 3) increased peripheral banding F-actin, and 4) presence of actin foci (21).
Hcy-induced formation of F-actin fibers and total fluorescence intensity were assessed for each well by analyzing three random fields with image analysis software (Image-Pro Plus, Media Cybernetics). The data (expressed as FIU) were obtained in duplicate (two wells per experimental group) and averaged for each experimental group described above.
Gelatin zymography
Activity of MMP-2 and MMP-9 was measured in brain cortical tissue homogenates and in FITC-BSA solution treated with active MMP-9. Zymography was performed on 1% gelatin SDS-PAGE. The lytic activity was scanned and normalized to total protein of samples loaded into each lane of the gel as described previously (45). Data are presented as light intensity units (LIU).
Data analysis
Data are expressed as means ± SE. To compare the groups before and after treatment, one-way repeated measures ANOVA was used. Differences in means were compared by the Tukey test and considered statistically significant if P < 0.05.
Results
Macromolecular leakage from pial venules
During baseline before injection of Hcy or vehicle (PSS), MABP and pulse rate were similar in WT (88 ± 3 mmHg, 385 ± 15 beats/min; n = 5) and MMP-9−/− (85 ± 3 mmHg, 381 ± 13 beats/min; n = 4) mice. Forty minutes after Hcy injection, MABP and pulse rate did not change significantly in these WT (79 ± 6 mmHg, 415 ± 26 beats/min) and MMP-9−/− (77 ± 5 mmHg, 418 ± 22 beats/min) mice. In separate WT (n = 5) and MMP-9−/− (n = 3) mice, baseline MABP and pulse rate (84 ± 5 mmHg, 397 ± 15 beats/min in WT, and 86 ± 6 mmHg, 381 ± 16 beats/min) also did not change significantly after injection of PSS (86 ± 4 mmHg, 380 ± 12 beats/min in WT; 83 ± 4 mmHg, 376 ± 11 beats/min in MMP-9−/−). In a different group of MMP-9−/− mice (n = 4), 40 min after topical application of histamine, MABP and pulse rate were similar to baseline (81 ± 5 vs. 84 ± 3 mmHg and 393 ± 11 vs. 387 ± 9 beats/min). Similarly, in WT mice (n = 4), topical application of histamine did not cause changes in MABP (89 ± 4 vs. 86 ± 5 mmHg at baseline) or pulse rate (382 ± 10 vs. 393 ± 11 beats/min at baseline).
Macromolecular leakage significantly increased in WT mice after Hcy administration (Fig. 1). Leakage was noticeable 5 min after Hcy injection and became greater after 40 min (Fig. 1). Injection of PSS did not cause leakage in WT mice over the same time course (Fig. 1). During these observations, in the WT mice, baseline venular diameters (36 ± 5 μm) were not different 40 min after Hcy injection (40 ± 6 μm). In a separate group of WT mice, baseline venular diameters (34 ± 3 μm) were also not different 40 min after PSS administration (32 ± 5 μm).
Fig. 1.
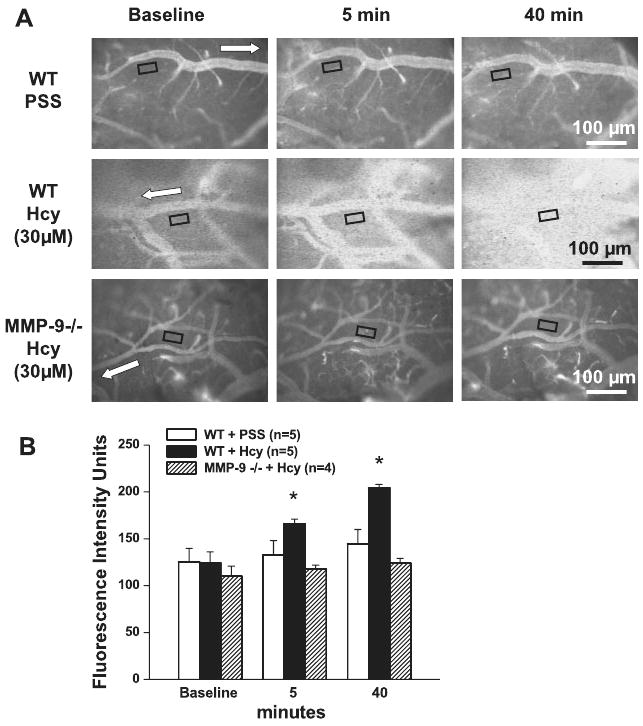
Homocysteine (Hcy)-induced macromolecular leakage of pial venules. A: examples of images recorded before (baseline) and 5 and 40 min after injection of either PSS (top row) or Hcy (final blood concentration 30 μM) in wild-type (WT; top rows) and matrix metalloproteinase (MMP)-9 gene knockout mice (bottom row). Microvascular leakage was assessed by measuring fluorescence intensity in the rectangular area of interest (AOI) shown on images. White arrows indicate flow direction in veins. B: summary of changes in fluorescence intensity after injection of PSS or Hcy measured in the AOI. Values are means ± SE. *P < 0.05 vs. control at the same time.
When Hcy was injected into MMP-9−/− mice (n = 4), it did not cause venular leakage (Fig. 1). Similarly, injection of PSS in MMP-9−/− mice did not cause leakage (110 ± 7 FIU at baseline, 118 ± 4 FIU after 5 min, and 124 ± 6 FIU after 40 min). Venular diameters were the same before and 40 min after injection of Hcy (30 ± 2 vs. 33 ± 4 μm). Injection of PSS in MMP-9−/− mice also did not change venular diameters (32 ± 4 vs. 32 ± 5 μm).
In the MMP-9−/− mice group (n = 4) treated with topical application of histamine, venular leakage was significantly increased (112 ± 4 FIU at baseline to 127 ± 3 FIU after 5 min, and to 145 ± 5 FIU after 40 min). In WT mice (n = 4) topical application of histamine increased fluorescence intensity by a similar extent (106 ± 8 FIU at baseline to 112 ± 6 FIU after 5 min, and to 141 ± 7 FIU after 40 min). Venular diameters in MMP-9−/− mice were similar before and 40 min after histamine application (34 ± 4 vs. 37 ± 5 μm). In WT mice, venular diameters increased significantly from 33 ± 7 μm (baseline) to 40 ± 6 μm after histamine application.
Effect of MMP-9 on stability of FITC-BSA conjugate
There was no detectable cleavage of albumin by activated MMP-9 (Fig. 2A). The presence of active MMP-9 in our FITC-BSA samples was confirmed by gelatin zymography (Fig. 2B).
Fig. 2.
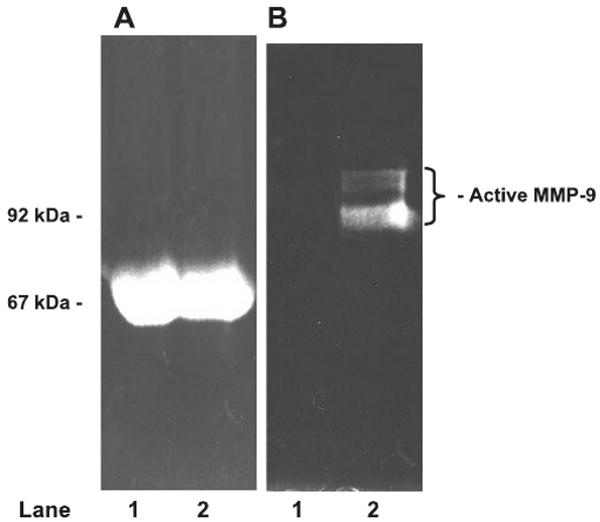
Effect of activated MMP-9 on FITC-BSA. Lane 1 contains fluorescein isothiocyanate-bovine serum albumin (FITC-BSA) treated with pro-MMP-9. Lane 2 contains FITC-BSA treated with activated MMP-9. A: SDS-PAGE analysis of samples by fluorescence. Absence of fluorescence below BSA band in lane 2 (67 kDa) confirms absence of FITC-BSA cleavage by activated MMP-9. B: presence of activated MMP-9 in FITC-BSA sample (same gel, lane 2) was confirmed by zymography.
Effect of Hcy on formation of MMP-2 and MMP-9
Activities of MMP-2 and MMP-9 were significantly increased in brain tissue samples collected from the separate group of WT mice after 40 min of Hcy injection compared with samples collected from a group of untreated mice (Fig. 3). As expected, formation of MMP-9 was not noticeable in brain tissue samples from MMP-9 gene knockout mice, and there were no significant changes in formation of MMP-2 (Fig. 3A). Furthermore, activities of MMP-2 and MMP-9 were not altered in brain tissue samples from WT mice after injection of PSS (69 ± 4 and 82 ± 6 LIU, respectively; n = 3) compared with the MMP-2 and MMP-9 activities in samples from untreated mice (65 ± 4 and 84 ± 3 LIU, respectively; n = 3).
Fig. 3.
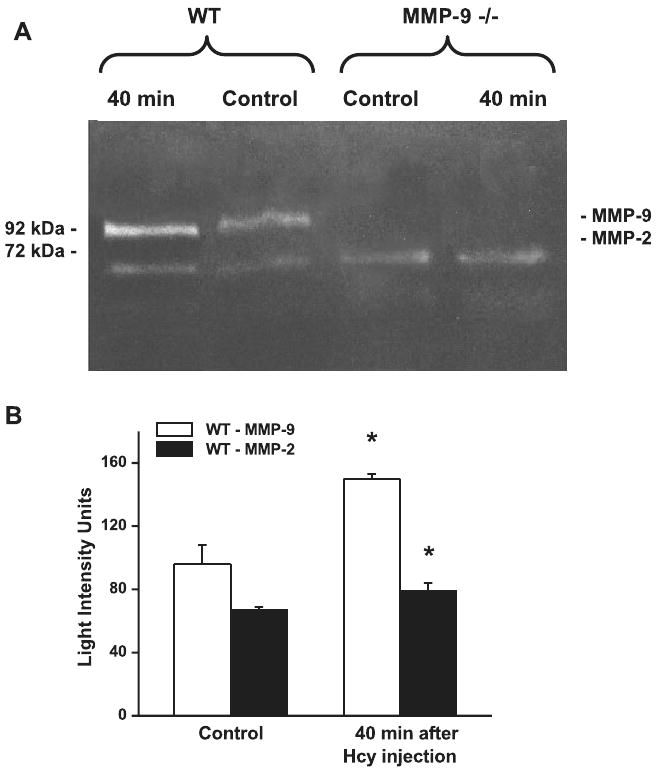
Hcy-induced formation of MMP-9 and MMP-2 in brain tissue samples. A: MMP-9 and MMP-2 activity in WT and MMP-9 gene knockout (MMP-9−/−) mice shown by zymography. B: increases in activity of MMP-9 and MMP-2 after Hcy treatment in WT mice compared with control as quantified by density of the bands. Values are means ± SE. *P < 0.05 vs. control. n = 3 for all groups.
The level of plasma Hcy in WT and MMP-9−/− mice
Plasma levels of Hcy in WT (3.2 ± 0.2 μM; n = 3) and MMP-9−/− (4.8 ±1.2 μM; n = 3) mice were in the normal range (12).
Endothelial cell F-actin formation
Alterations in formation of endothelial F-actin were assessed in mouse MECs by the binding of the BODIPY-phalloidin to actin in control (PSS-treated), Hcy-treated, and Hcy-treated in the presence of the MMPs blocker (GM-6001) groups. Figure 4A is a representative fluorescence photomicrograph, showing the effects of either PSS, 30 μM Hcy, or 30 μM Hcy + GM-6001 (1 μM) on F-actin distribution. Analysis of the fluorescent intensity showed significantly greater F-actin staining in the 30 μM Hcy-treated endothelial cells than in cells under unstimulated control conditions (Fig. 4B). The presence of 1 μM GM-6001 significantly decreased F-actin staining (90.3 ± 5.0 FIU; n = 4); however, it was still greater than in the control group (Fig. 4). Higher concentrations of GM-6001 (10 and 100 μM) completely blocked the F-actin formation caused by 30 μM Hcy (64 ± 7 FIU and 60.3 ± 2.8 FIU, respectively; n = 4 in both groups). However, a lower dose of GM-6001 (0.1 μM) failed to block the Hcy-induced F-actin formation (119.2 ± 6.4 FIU; n = 4). There were no differences in fluorescence intensities between cells treated with 30 μM Hcy (127.3 ± 4.3 FIU; n = 4) and cells treated with 30 μM Hcy in the presence of the negative control for 0.1,1,10, and 100 μM GM-6001 (125.8 ± 3.8 FIU, 121.2 ± 3.8 FIU, 118.6 ± 7.9 FIU, 134.8 ± 3.1 FIU, respectively; n = 4 for all groups). These results show dose-dependent effect of GM-6001. They also show that the optimal concentration of GM-6001 for inhibition of MMP-9 activity in our study was 1 μM.
Fig. 4.
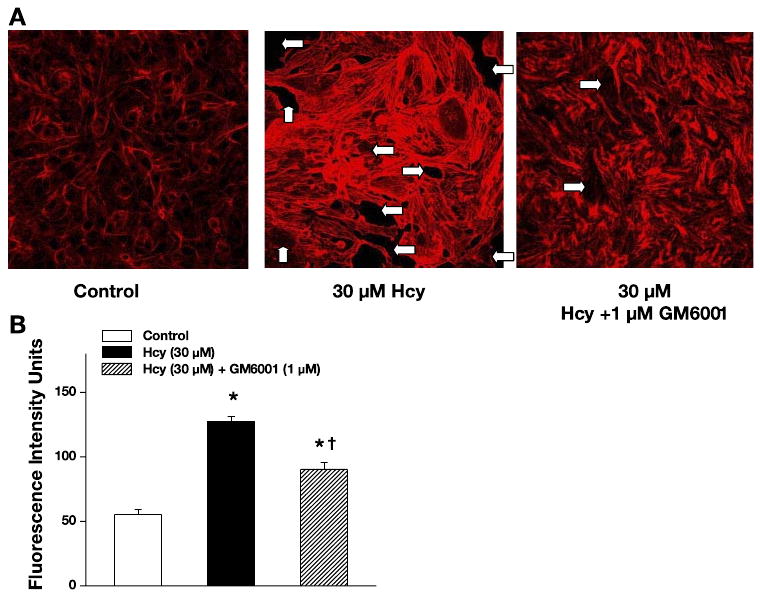
Hcy-induced formation of F-actin in mouse endothelial cells. A: examples of images recorded after treatment with PSS (control), 30 μM Hcy, or 30 μM Hcy in the presence of MMP blocker GM-6001 (1 μM). White arrows indicate gap formation in the initially confluent endothelial cell monolayer. B: increase in formation of F-actin after Hcy treatment and its inhibition by GM-6001. Values are means ± SE. *P < 0.05 vs. control; †P < 0.05 vs. Hcy treated. n = 4 for all groups.
F-actin staining in the control (55.3 ±4.1 FIU; n = 4) and 3 μM Hcy-treated groups (59.8 ± 5.2 FIU; n = 4) was similar. No difference in fluorescence intensity was found between the control cells and the cells treated with GM-6001 (1 and 100 μM) alone (53.1 ± 8.6 FIU; n = 4 and 52.5 ± 4.8 FIU; n = 3, respectively) or with the same concentrations of their negative controls (59.4 ± 3.8 FIU; n = 4 and 59.3 ± 5.9 FIU; n = 3, respectively). The 30 μM Hcy-treated group also showed decreased cell size and increased space between the cells (Fig. 4A), possibly the result of endothelial cell contraction and formation of gaps in the tight junction of the cells.
Discussion
HHcy is an established risk factor for atherosclerosis and thrombotic diseases. Endothelial cell dysfunction and injury is one of the important factors associated with increased blood Hcy levels. In the present study, we examined the acute effect of a moderate dose of Hcy on cerebrovascular permeability in mice. An increase of blood Hcy concentration to about 30 μM did not cause significant alterations of MABP in mice during the observation period (40 min). Venular diameters did not change during the experiments, but the rapid increase of Hcy content to 30 μM as a result of intra-arteriolar injection, resulted in noticeable macromolecular venular leakage starting shortly after the Hcy administration. Macromolecular leakage from pial venules was significantly increased 40 min after Hcy injection. However, the increased content of Hcy had no notable effect on macromolecular leakage of pial venules in mice lacking the MMP-9 gene and suggest that the destructive effect of Hcy can be abrogated in the absence of MMP-9. These results suggest that increased plasma level of Hcy may have a significant effect on venular wall integrity resulting in alteration of vascular endothelium leading to macromolecular leakage. However, other mechanisms may also be involved. It has also been shown that γ-aminobutyric acid (GABA) (14, 37) and N-methyl-d-aspartate (NMDA) (4, 5) receptors have a role in increasing blood-brain barrier permeability. Because Hcy competitively inhibits GABA receptors (9) and activates NMDA receptors (8), it is likely that changes in their functional activity induced by Hcy may result in increased microvascular permeability, in conjunction with activation of MMP-9, seen in the present study.
Although Hcy did not cause significant leakage in MMP-9−/− mice, leakage could still occur in response to histamine. Furthermore, histamine induced similar cerebrovascular leakage in MMP-9−/− and WT mice. This finding suggests that the decreased leakage in response to Hcy is not likely due to a nonspecific effect. Although histamine caused the same amount of leakage in WT and MMP-9−/− mice, there was a slightly greater increase in venular diameters in WT mice. Others have reported the vasodilatory effect of topical application of histamine on venules (25). Separate studies are needed to determine whether the effect of histamine on diameter results from a different mechanism than its effect on leakage.
St-Pierre et al. (43) showed that MMP-9 almost completely dissociates FITC-gelatin, but not FITC-casein conjugate from microspheres, in 16 h. Formation of active MMP-9 as a result of increased Hcy could cause cleavage of albumin in circulating blood, leading to leakage of FITC-BSA breakdown products thought microvascular endothelial gaps. The presence of nonbound FITC in FITC-BSA solution could result in leakage of the dye through the lipid portion of the endothelial cell membrane (10), even if the integrity of the endothelial cell layer is not compromised (contrary to the presented data). Our in vitro experiments (Fig. 2) confirm that activated MMP-9 did not cleave albumin even after 16 h, and no free FITC was present in our samples. Thus our results suggest that increased Hcy alters the blood-brain barrier so that large molecules such as albumin may leak through the formed endothelial gaps. The significant increase in MMP-9 activity at 40 min and lesser increase of MMP-2 activity as a result of elevation of blood Hcy in WT mice support a role for MMPs in the microvascular leakage. The lack of increase in MMP-2 activity and absence of MMP-9 activity in response to Hcy treatment in MMP-9−/− mice and the fact that both MMP-2 and particularly MMP-9 are activated in WT mice, suggest the genotype purity of MMP-9−/− mice used in the present study.
We (20) and others (6) found that increased formation of F-actin leads to gap formation in the endothelial cell layer. Gap formation is suggested as one of the reasons for macromolecular vascular leakage in response to inflammatory stimuli (7, 20). Increased expression of MMP-9 in endothelial cells has been implicated during the development of cerebral stroke, which has been significantly reduced after selective inhibition of MMP-9 (29). A significant role of MMPs in formation of F-actin has also been demonstrated by others (6, 33). These data suggest that formation of MMPs may result in enhanced formation of endothelial F-actin leading to macromolecular leakage.
Treatment of MECs with 30 μM of Hcy resulted in significantly greater formation of F-actin. This finding suggests that increased macromolecular leakage of pial venules in the present study may be directly related to Hcy-induced F-actin polymerization and the resultant increase in endothelial cell gap formation. Our data show that F-actin formation in MECs in response to Hcy treatment was significantly decreased in the presence of an MMPs blocker GM-6001 (1 μM), but it was still greater than in the control group. These results show a role of MMP in Hcy-induced formation of F-actin and a resultant formation of endothelial gaps. These results suggest that Hcy may act like other inflammatory agents (e.g., histamine, thrombin) and cause F-actin polymerization through an MMP-involved pathway, leading to gap formation in the endothelial layer and increased macromolecular leakage of pial venules in mice. Thus an increased Hcy content in endothelial cells primes the contractile mechanism associated with F-actin polymerization. Previously, we reported that HHcy leads to vascular remodeling by effects on vascular elastin (26) and collagen (40, 41), where the role of GABA and NMDA receptors were suggested (44). The results of the present study indicate that an elevated blood Hcy may first target the endothelial cell lining and then cause changes in the subendothelial matrix. They also point to an existence of some other mechanism (possibly GABA and/or NMDA receptor mediated) that may be involved in Hcy-induced endothelial gap formation.
Our study compared Hcy effects in age-matched mice that underwent similar surgical preparations. Vascular permeability was assessed by comparing fluorescence intensity values measured in a similar size AOI in the interstitum next to a venule before and after treatment. Because each animal served as its own control, small differences in baseline permeability are unlikely to account for the changes in permeability in response to the agonists (34). In MMP-9−/− and WT mice, histamine induced similar microvascular leakage compared with baseline values. However, it was significantly less than that induced by Hcy in WT mice (Fig. 1). Our data show that cerebral vessels of MMP-9−/− mice have the ability to leak in response to an inflammatory agent, and that this leakage occurs independently of an MMP-9-involved mechanism. Study of mechanisms of the histamine-induced leakage and vascular responses are beyond the scope of the present investigation. A limitation of this study is that the ability of histamine to cross an intact arachnoid is controversial and could be related to damage the meninges when the dura was removed. For example, Sarker et al. (34), who removed the arachnoid with the dura in juvenile rats, clearly showed that topical application of histamine at doses ≥ 50 μM reduced cerebrovascular permeability. However, in other studies, topical application of 10−4 M histamine (the dose used in our study) increased cerebrovascular permeability in rats (2, 3, 22, 27, 48), cats (35), and mice (49).
In conclusion, our study shows for the first time that an acute increase in blood Hcy content to the moderate level of 30 μM results in macromolecular leakage of pial venules in mice. We demonstrated that this detrimental effect of Hcy occurs through a pathway that involves MMP-9 formation, which in turn causes formation of F-actin in endothelial cells. Increased polymerization of actin filaments leads to gap formation between adjacent endothelial cells. Increased formation of F-actin in endothelial cells and macromolecular leakage of pial venules were both inhibited by blocking MMPs. Thus the present study suggests yet another destructive role of Hcy and a possible pathway of its effect on venular permeability changes in brain pial circulation.
Acknowledgments
Grants: This study was supported in part by the American Heart Association National Grant 0235317N to D. Lominadze, by the National Heart, Lung, and Blood Institute Grants HL-71010 and HL-74185 to S. C. Tyagi, and the American Lung Association, Kentucky Affiliate to A. M. Roberts.
References
- 1.Audelin MC, Genest J., Jr Homocysteine and cardiovascular disease in diabetes mellitus. Atherosclerosis. 2001;159:497–511. doi: 10.1016/s0021-9150(01)00531-7. [DOI] [PubMed] [Google Scholar]
- 2.Butt AM. Effect of inflammatory agents on electrical resistance across the blood-brain barrier in pial microvessels of anaesthetized rats. Brain Res. 1995;696:145–150. doi: 10.1016/0006-8993(95)00811-4. [DOI] [PubMed] [Google Scholar]
- 3.Butt AM, Jones HC. Effect of histamine and antagonists on electrical resistance across the blood-brain barrier in rat brain-surface microvessels. Brain Res. 1992;569:100–105. doi: 10.1016/0006-8993(92)90374-i. [DOI] [PubMed] [Google Scholar]
- 4.Chi OZ, Chang Q, Weiss HR. Effects of topical N-methyl-d-aspartate on blood-brain barrier permeability in the cerebral cortex of normotensive and hypertensive rats. Neurol Res. 1997;19:539–544. doi: 10.1080/01616412.1997.11740854. [DOI] [PubMed] [Google Scholar]
- 5.Du C, Hu R, Hsu CY, Choi DW. Dextrorphan reduces infarct volume, vascular injury, and brain edema after ischemic brain injury. J Neurotrauma. 1996;13:215–222. doi: 10.1089/neu.1996.13.215. [DOI] [PubMed] [Google Scholar]
- 6.Ehringer WD, Wang OL, Haq A, Miller FN. Bradykinin and alpha-thrombin increase human umbilical vein endothelial macromolecular permeability by different mechanisms. Inflammation. 2000;24:175–193. doi: 10.1023/a:1007037711339. [DOI] [PubMed] [Google Scholar]
- 7.Ehringer WD, Yamany S, Steier K, Farag A, Roisen FJ, Dozier A, Miller FN. Quantitative image analysis of F-actin in endothelial cells. Microcirculation. 1999;6:291–303. [PubMed] [Google Scholar]
- 8.Folbergrova J. NMDA and not non-NMDA receptor antagonists are protective against seizures induced by homocysteine in neonatal rats. Exp Neurol. 1994;130:344–350. doi: 10.1006/exnr.1994.1213. [DOI] [PubMed] [Google Scholar]
- 9.Griffiths R, Williams DC, O'Neill C, Dewhurst IC, Ekuwem CE, Sinclair CD. Synergistic inhibition of muscimol binding to calf-brain synaptic membrances in the presense of l-homocysteine and pyridoxal 5′-phosphate. Eur J Biochem. 1983;137:467–478. doi: 10.1111/j.1432-1033.1983.tb07850.x. [DOI] [PubMed] [Google Scholar]
- 10.Grimes PA, Stone RA, Laties AM, Li W. Carboxyfluorescein. A probe of the blood-ocular barriers with lower membrane permeability than fluorescein. Arch Ophthalmol. 1982;100:635–639. doi: 10.1001/archopht.1982.01030030637022. [DOI] [PubMed] [Google Scholar]
- 11.Hudetz AG, Feher G, Weigle CGM, Knuese DE, Kampine JP. Video microscopy of cerebrocortical capillary flow: response to hypotension and intracranial hypertension. Am J Physiol Heart Circ Physiol. 1995;268:H2202–H2210. doi: 10.1152/ajpheart.1995.268.6.H2202. [DOI] [PubMed] [Google Scholar]
- 12.Ji C, Kaplowitz N. Hyperhomocysteinemia, endoplasmic reticulum stress, and alcoholic liver injury. World J Gastroenterol. 2004;10:1699–1708. doi: 10.3748/wjg.v10.i12.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lawrence de Koning AB, Werstuck GH, Zhou J, Austin RC. Hyperhomocysteinemia and its role in the development of atherosclerosis. Clin Biochem. 2003;36:431–441. doi: 10.1016/s0009-9120(03)00062-6. [DOI] [PubMed] [Google Scholar]
- 14.Lee WS, Limmroth V, Ayata C, Cutrer FM, Waeber C, Yu X, Moskowitz MA. Peripheral GABA-A receptor-mediated effects of sodium valproate on dural plasma protein extravasation to substance P and trigeminal stimulation. Br J Pharmacol. 1995;116:1661–1667. doi: 10.1111/j.1476-5381.1995.tb16388.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lincoln DW, II, Larsen AM, Phillips PG, Bove K. Isolation of murine aortic endothelial cells in culture and the effects of sex steroids on their growth. In Vitro Cell Devel Biol Anim. 2003;39:140–145. doi: 10.1007/s11626-003-0008-x. [DOI] [PubMed] [Google Scholar]
- 16.Lip GY, Edmunds E, Martin SC, Jones AF, Blann AD, Beevers DG. A pilot study of homocyst(e)ine levels in essential hypertension: relationship to von Willebrand factor, an index of endothelial damage. Am J Hypertens. 2001;14:627–631. doi: 10.1016/s0895-7061(00)01321-2. [DOI] [PubMed] [Google Scholar]
- 17.Liu Y, Rusch NJ, Lombard JH. Loss of endothelium and receptor-mediated dilation in pial arterioles of rats fed a short-term high salt diet. Hypertension. 1999;33:686–688. doi: 10.1161/01.hyp.33.2.686. [DOI] [PubMed] [Google Scholar]
- 18.Lominadze D, Dean WL. Involvement of fibrinogen specific binding in erythrocyte aggregation. FEBS Lett. 2002;517:41–44. doi: 10.1016/s0014-5793(02)02575-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lominadze D, Saari JT, Miller FN, Catalfamo JL, Persival SS, Schuschke DA. In vitro platelet adhesion to endothelial cells at low shear rates during copper deficiency in rats. J Trace Ele Exper Med. 1999;12:25–36. [Google Scholar]
- 20.Lominadze D, Saari JT, Persival SS, Schuschke DA. Proinflammatory effects of copper deficiency on neutrophils and lung endothelial cells. Immunol Cell Biol. 2004;82:231–238. doi: 10.1046/j.1440-1711.2004.01231.x. [DOI] [PubMed] [Google Scholar]
- 21.Lominadze D, Schuschke DA, Joshua IG, Dean WL. Increased ability of erythrocytes to aggregate in spontaneously hypertensive rats. Clin Exp Hypertens. 2002;24:397–406. doi: 10.1081/ceh-120005376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mayhan WG. Role of nitric oxide in histamine-induced increases in permeability of the blood-brain barrier. Brain Res. 1996;743:70–76. doi: 10.1016/s0006-8993(96)01021-9. [DOI] [PubMed] [Google Scholar]
- 23.Mehrabi MR, Huber K, Serbecic N, Wild T, Wojta J, Tamaddon F, Morgan A, Ullrich R, Dietmar Glogar H. Elevated homocysteine serum level is associated with low enrichment of homocysteine in coronary arteries of patients with coronary artery disease. Thromb Res. 2002;107:189–196. doi: 10.1016/s0049-3848(02)00308-0. [DOI] [PubMed] [Google Scholar]
- 24.Miller FN, Joshua IG, Anderson GL. Quantitation of vasodilator-induced macromolecular leakage by in vivo fluorescent microscopy. Microvasc Res. 1982;24:56–67. doi: 10.1016/0026-2862(82)90042-5. [DOI] [PubMed] [Google Scholar]
- 25.Miller FN, Joshua IG, Fleming JT, Parekh N. Histamine-induced protein leakage in hypertensive rats: inhibition by verapamil. Am J Physiol Heart Circ Physiol. 1986;250:H284–H290. doi: 10.1152/ajpheart.1986.250.2.H284. [DOI] [PubMed] [Google Scholar]
- 26.Mujumdar VS, Aru GM, Tyagi SC. Induction of oxidative stress by homocyst(e)ine impairs endothelial function. J Cell Biochem. 2001;82:491–500. doi: 10.1002/jcb.1175. [DOI] [PubMed] [Google Scholar]
- 27.Olesen SP. Leakiness of rat brain microvessels to fluorescent probes following craniotomy. Acta Physiol Scand. 1987;130:63–68. doi: 10.1111/j.1748-1716.1987.tb08112.x. [DOI] [PubMed] [Google Scholar]
- 28.Polidori MC, Marvardi M, Cherubini A, Senin U, Mecocci P. Heart disease and vascular risk factors in the cognitively impaired elderly: implications for Alzheimer's dementia. Aging. 2001;13:231–239. doi: 10.1007/BF03351481. [DOI] [PubMed] [Google Scholar]
- 29.Romanic AM, White RF, Arleth AJ, Ohlstein EH, Barone FC. Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke. 1998;29:1020–1030. doi: 10.1161/01.str.29.5.1020. [DOI] [PubMed] [Google Scholar]
- 30.Rosenblum WI. Contractile response of pial arterioles to norepinephrine. Arch Neurol. 1974;31:197–199. doi: 10.1001/archneur.1974.00490390079009. [DOI] [PubMed] [Google Scholar]
- 31.Rosenblum WI, El-Sabban F. Influence of shear rate on platelet aggregation in cerebral microvessels. Microvasc Res. 1982;23:311–315. doi: 10.1016/s0026-2862(82)80003-4. [DOI] [PubMed] [Google Scholar]
- 32.Rosenblum WI, Zweifach BW. Cerebral Microcirculation in the mouse brain. Arch Neurol. 1963;9:414–423. doi: 10.1001/archneur.1963.00460100102012. [DOI] [PubMed] [Google Scholar]
- 33.Saito S, Yamaji N, Yasunaga K, Saito T, Matsumoto S, Katoh M, Kobayashi S, Masuho Y. The fibronectin extra domain A activates matrix metalloproteinase gene expression by an interleukin-1-dependent mechanism. J Biol Chem. 1999;274:30756–30763. doi: 10.1074/jbc.274.43.30756. [DOI] [PubMed] [Google Scholar]
- 34.Sarker MH, Easton AS, Fraser PA. Regulation of cerebral microvascular permeability by histamine in the anaesthetized rat. J Physiol. 1998;507:909–918. doi: 10.1111/j.1469-7793.1998.909bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schilling L, Wahl M. Opening of the blood-brain barrier during cortical superfusion with histamine. Brain Res. 1994;653:289–296. doi: 10.1016/0006-8993(94)90403-0. [DOI] [PubMed] [Google Scholar]
- 36.Schuschke DA, Miller FN, Lominadze DG, Feldhoff RC. l-arginine restores cholesterol-attenuated microvascular responses in the rat cremaster. Int J Microcirc Clin Exp. 1994;14:204–211. doi: 10.1159/000178830. [DOI] [PubMed] [Google Scholar]
- 37.Seiffert E, Dreier JP, Ivens S, Bechmann I, Tomkins O, Heinemann U, Friedman A. Lasting blood-brain barrier disruption induces epileptic focus in the rat somatosensory cortex. J Neurosci. 2004;24:7829–7836. doi: 10.1523/JNEUROSCI.1751-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Selhub J, D'Angelo A. Hyperhomocysteinemia and thrombosis: acquired conditions. Thromb Haemost. 1997;78:527–531. [PubMed] [Google Scholar]
- 39.Selhub J, Jacques PF, Bostom AG, D'Agostino RB, Wilson PW, Belanger AJ, O'Leary DH, Wolf PA, Schaefer EJ, Rosenberg IH. Association between plasma homocysteine concentrations and extracranial carotid-artery stenosis. N Engl J Med. 1995;332:286–291. doi: 10.1056/NEJM199502023320502. [DOI] [PubMed] [Google Scholar]
- 40.Shastry S, Moning L, Tyagi N, Steed M, Tyagi SC. GABA receptors and nitric oxide ameliorate constrictive collagen remodeling in hyperhomocysteinemia. J Cell Physiol. 2005;205:422–427. doi: 10.1002/jcp.20416. [DOI] [PubMed] [Google Scholar]
- 41.Shastry S, Tyagi N, Moshal KS, Lominadze D, Hayden MR, Tyagi SC. GABA receptors ameliorate Hcy-mediated integrin-shedding and constrictive collagen remodeling in microvascular endothelial cells. Cell Biochem Biophys. doi: 10.1385/CBB:45:2:157. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sorsa T, Salo T, Koivunen E, Tyynelä J, Konttinen YT, Bergmann U, Tuuttila A, Niemi E, Teronen O, Heikkilä P, Tschesche H, Leinonen J, Osman S, Stenman UH. Activation of type IV procollagenases by human tumor-associated trypsin-2. J Biol Chem. 1997;272:21067–21074. doi: 10.1074/jbc.272.34.21067. [DOI] [PubMed] [Google Scholar]
- 43.St-Pierre Y, Desrosiers M, Tremblay P, Esteve PO, Opdenakker G. Flow cytometric analysis of gelatinase B (MMP-9) activity using immobilized fluorescent substrate on microspheres. Cytometry. 1996;25:374–380. doi: 10.1002/(SICI)1097-0320(19961201)25:4<374::AID-CYTO9>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 44.Tyagi SC, Lominadze D, Roberts AM. Homocysteine in microvascular endothelial cell barrier permeability. Cell Biochem Biophys. 2005;43:1–8. doi: 10.1385/CBB:43:1:037. [DOI] [PubMed] [Google Scholar]
- 45.Tyagi SC, Matsubara L, Weber KT. Direct extraction and estimation of collagenase(s) activity by zymography in microquantities of rat myocardium and uterus. Clin Biochem. 1993;26:191–198. doi: 10.1016/0009-9120(93)90025-2. [DOI] [PubMed] [Google Scholar]
- 46.Tyagi SC, Smiley LM, Mujumdar VS, Clonts B, Parker JL. Reduction-oxidation (Redox) and vascular tissue level of homocyst(e)ine in human coronary atherosclerotic lesions and role in extracellular matrix remodeling and vascular tone. Mol Cell Biochem. 1998;181:107–116. doi: 10.1023/a:1006882014593. [DOI] [PubMed] [Google Scholar]
- 47.Van Beynum IM, Smeitink JA, den Heijer M, te Poele Pothoff MT, Blom HJ. Hyperhomocysteinemia: a risk factor for ischemic stroke in children. Circulation. 1999;99:2070–2072. doi: 10.1161/01.cir.99.16.2070. [DOI] [PubMed] [Google Scholar]
- 48.Wahl M, Unterberg A, Baethmann A, Schilling L. Mediators of blood-brain barrier dysfunction and formation of vasogenic brain edema. J Cereb Blood Flow Metab. 1988;8:621–634. doi: 10.1038/jcbfm.1988.109. [DOI] [PubMed] [Google Scholar]
- 49.Yong T, Bebo BF, Jr, Sapatino BV, Welsh CJ, Orr EL, Linthicum DS. Histamine-induced microvascular leakage in pial venules: differences between the SJL/J and BALB/c inbred strains of mice. J Neurotrauma. 1994;11:161–171. doi: 10.1089/neu.1994.11.161. [DOI] [PubMed] [Google Scholar]