

Cryptosporidium spp. are a major cause of the “vicious cycle” of diarrhea and malnutrition in the developing world and a potential bioterrorism agent 1,2. This disease is prolonged and life-threatening in immuno-compromised patients. Currently no effective therapy exists for Cryptosporidium infections. Theparasite obtains guanine nucleotides via a streamlined pathway that requires inosine 5’-monophosphate dehydrogenase (IMPDH) 3-5. Curiously, the gene encoding CpIMPDH appears to have been obtained from a bacteria via lateral gene transfer 6,7; we have exploited this unexpected divergence of parasite and host enzymes to identify CpIMPDH-specific inhibitors in a high throughput screen 8. Here we report x-ray crystal structures of CpIMPDH that explain the selectivity of one inhibitor series and use this information to design more potent and selective analogs.
Recombinant CpIMPDH was purified as described previously 9 and crystallized using the hanging drop vapor diffusion method. Protein solution (4 mg/mL IMPDH, 50 mM Tris-HCl, pH 7.5, 150 mM KCl, 5% glycerol and 2 mM DTT) was mixed with well solution (34% PEG 4000, 25 mM sodium acetate and 100 mM Tris-HCl, pH 8.5) in a 1:1 ratio. Data were collected from a single crystal at 100K at beamline 8-BM at Advanced Photon Source (Argonne National Laboratory, Argonne, IL). The crystals had the symmetry of space group P21212. The asymmetric unit contains one tetramer, which is the active form of IMPDH12. The structure was solved to 3.2 Å resolution (R = 27%, Rfree = 33%) by molecular replacement using the structure of IMPDH from Borrelia burgdorferi (PDB accession 1EEP10) as a search model11. Only 301 of 400 residues are visible in the most structured monomer; the disordered regions include catalytically important segments 214-222, 299-333 and 380-400 as well as residues 92-122, which are not required for enzymatic activity 12. Unfortunately, we were unable to improve this crystal form. This structure has been deposited in the PDB (3FFS).
To facilitate crystallization, residues 90-134 were replaced with SerGlyGly11; this modification has no effect on enzymatic activity (Figure S1). A crystallization screen was performed in the presence of IMP and various inhibitors that emerged from initial evaluation of the SAR. Compound C64 was a particularly attractive candidate for crystallization because of its improved potency relative to the parent compound C and the presence of a bromine atom which would allow the two aromatic groups to be easily distinguished (Table 1). Crystals were obtained in the presence of saturating concentrations of inhibitor C64 (20 μM), IMP (1 mM), 100 mM sodium acetate, pH 4.6, 20 mM CaCl2 and 30% MPD under oil. These crystals had the symmetry of space group P21 with two tetramers in the asymmetric unit. Data were collected at a wavelength of 0.9194 Å, enabling the simultaneous collection of bromine k-edge anomalous dispersion data. The structure was solved by molecular replacement to 2.8 Å resolution using the native CpIMPDH structure as the starting model (R = 26.6%, Rfree = 22.4%).
Table 1.
Inhibition of CpIMPDH and hIMPDH2.
 | ||||
|---|---|---|---|---|
|
Scheme 1. Synthesis of inhibitors. | ||||
| Cmpd |
R1 |
R2 |
IC50 (nM) |
|
|
CpIMPDH |
hIMPDH2 |
|||
|
C |
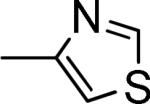 |
4-OMePh |
1200 ± 200a |
≥200,000a,b |
|
C10 |
4-ClPh |
120 ± 40 |
≥20,000c |
|
|
C14 |
4-BrPh |
60 ± 30 |
≥20,000c |
|
|
C86 |
2,3-Di-ClPh |
30 ± 10 |
≥40,000d |
|
|
C90 |
2-Naph |
7 ± 4 |
≥40,000d |
|
|
C61 |
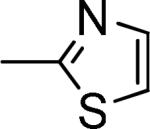 |
4-ClPh |
30 ± 10 |
≥40,000d |
|
C64 |
4-BrPh |
28 ± 9 |
≥40,000d |
|
|
C84 |
2,3-di-ClPh |
18 ± 5 |
≥8,000e |
|
| C97 | 2-Naph | 8 ± 3 | ≥40,000d | |
values from 8.
≤20% inhibition at 50 μM
≤20% inhibition at 5 μM
≤ 10% inhibition at 5 μM
≤ 20% inhibition observed at 2 μM.
While the overall structure of the E•IMP•C64 complex is similar to that of the unliganded enzyme, several additional residues are observed. Residues 214-226, which include the catalytic Cys219, are observed in most of the monomers, as are parts of the active site flap (residues 302-330) containing the characteristic ArgTyr motif 12. Lastly, the SerGlyGly linker is visible in all monomers. Electron density for IMP is observed in all eight monomers. Monomers B, D and H contained extra electron density near IMP (Figure 1). Bromine k-edge anomalous dispersion maps allowed the unambiguous assignment of the bromine atom in C64 in all three monomers. The rest of C64 was modeled into the remaining electron density; similar conformations of C64 are obtained in all three monomers. This structure has been deposited in the PDB (3KHJ).
Figure 1.

Co-crystallized structure of CpIMPDH (light gray) with IMP (salmon) and C64 (slate) shown from two different perspectives. The electron density map prior to C64 modeling with coefficients 2Fo-Fc is contoured to 1σ and shown as a slate cage. The electron density map prior to C64 modeling with coefficients Fo-Fc is contoured to 3σ and shown as a green cage. Bromine K-edge peak anomalous dispersion map is contoured to 4σ and shown as a red cage. The red and blue surfaces denote regions of negative and positive electrostatic potential, respectively.
Surprisingly, C64 binds in an unprecedented fashion. Inhibitors of human IMPDH2 such as mycophenolic acid and merimepodib bind in the nicotinamide subsite, stacking against the purine ring of IMP in a parallel fashion, and extend either into the NAD site or into a pocket adjacent the active site but within the same monomer 12-14. In contrast, the thiazole ring of C64 stacks against the purine ring of IMP perpendicularly, and the remainder of C64 extends across the subunit interface into a pocket in the adjacent monomer, where the bromoaniline moiety interacts with Tyr358’ (where ’ denotes a residue from the adjacent subunit; Figure 2). This residue forms a hydrogen bonding network involving Glu329, Ser354, Thr221 and possibly the amide nitrogen of C64 (Figure 2). Ser22’, Pro26’, Ala165, Gly357’ form the remainder of the inhibitor binding pocket. With the exception of Thr221, all of these residues are different in human IMPDHs (Figure 2). Thus these interactions account for the selectivity of C64 for CpIMPDH over human IMPDHs.
Figure 2.

The C64 binding pocket of CpIMPDH (green) superposed with human IMPDH2 (cyan). CpIMPDH residues are labeled.
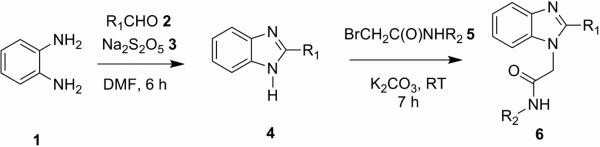
The structure also revealed the presence of a cavity adjacent to the bromoaniline moiety (Figure 1), which suggested that more potent inhibitors might be created by increasing the bulk of this substituent. Additional benzimidazole based inhibitors were prepared by condensing o-phenylenediamine 1 with thiazole carboxaldehydes 2 in the presence of the oxidizing reagent sodium metabisulfite 3 (Scheme 1)15. The resulting 2-substituted benzimidazoles 4 were then coupled with different bromoacetylamides 5 under mild basic conditions to give the new analogs 6 (Table 1).
The CpIMPDH inhibitory activity of the compounds was assessed by monitoring the production of NADH by fluorescence (Table 1)16. Replacing the p-MeO of the parent compound C with Cl or Br increased potency by 10-fold (C10) and 20-fold (C14), respectively, as has been similarly observed with another inhibitor series.16 To fill the cavity observed in the crystal structure, the para-substituted aniline group was replaced with 3,4-dichloroaniline (C86) or 2-naphthylamine (C90); the addition of a second Cl improved potency by a factor of 2, while fusing an additional aromatic ring increased potency by a factor of 8. Similar trends were observed when the thiazole ring was attached at the 2-position (C61, C64, C84 and C90). None of the compounds displayed significant inhibitory activity against human IMPDH2. The best CpIMPDH inhibitor, C90, has an IC50 = 7.4 nM with selectivity >103 for the parasite enzyme.
In conclusion, the crystal structure of CpIMPDH reveals the structural basis of inhibitor selectivity and a strategy for further optimization. This information was used to design more potent and selective inhibitors of CpIMPDH that are potential lead compounds for the treatment of cryptosporidiosis.
Supplementary Material
Acknowlegement
This work was supported by funding from NIH/NIAID (U01AI075466) to LH. GDC thanks NERCE/BEID, the Harvard NeuroDiscovery Center and the Partners Center for Drug Discovery for financial support. Initial crystal screening was performed at the Hauptman Woodward Institute. We thank Jennifer Lu for assistance in obtaining crystals of the unliganded complex, Kene Piasta for data collection and our colleague Gregory Petsko for advice and comments on the manuscript.
Footnotes
Supporting Information Available: Methods, crystallographic data table, spectra, chromatograms and complete Refs. 4 & 5 (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Fayer R. Veterinary Parasitology. 2004;126:37–56. doi: 10.1016/j.vetpar.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 2.Huang DB, White AC. Gastroenterol Clin North Am. 2006;35:291–314. viii. doi: 10.1016/j.gtc.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 3.Striepen B, Pruijssers AJ, Huang J, Li C, Gubbels MJ, Umejiego NN, Hedstrom L, Kissinger JC. Proc Natl Acad Sci U S A. 2004;101:3154–9. doi: 10.1073/pnas.0304686101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abrahamsen MS, et al. Science. 2004;304:441–445. doi: 10.1126/science.1094786. [DOI] [PubMed] [Google Scholar]
- 5.Xu P, et al. Nature. 2004;431:1107–1112. doi: 10.1038/nature02977. [DOI] [PubMed] [Google Scholar]
- 6.Striepen B, White MW, Li C, Guerini MN, Malik SB, Logsdon JM, Jr., Liu C, Abrahamsen MS. Proc Natl Acad Sci U S A. 2002;99:6304–6309. doi: 10.1073/pnas.092525699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Umejiego NN, Li C, Riera T, Hedstrom L, Striepen B. J Biol Chem. 2004;279:40320–40327. doi: 10.1074/jbc.M407121200. [DOI] [PubMed] [Google Scholar]
- 8.Umejiego NN, Gollapalli D, Sharling L, Volftsun A, Lu J, Benjamin NN, Stroupe AH, Riera TV, Striepen B, Hedstrom L. Chem Biol. 2008;15:70–77. doi: 10.1016/j.chembiol.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Riera TV, Wang W, Josephine HR, Hedstrom L. Biochemistry. 2008;47:8689–96. doi: 10.1021/bi800674a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McMillan FM, Cahoon M, White A, Hedstrom L, Petsko GA, Ringe D. Biochemistry. 2000;39:4533–4542. doi: 10.1021/bi992645l. [DOI] [PubMed] [Google Scholar]
- 11.See supplementary information for details.
- 12.Hedstrom L. Chem. Rev. 2009;109:2903–2928. doi: 10.1021/cr900021w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sintchak MD, Fleming MA, Futer O, Raybuck SA, Chambers SP, Caron PR, Murcko M, Wilson KP. Cell. 1996;85:921–930. doi: 10.1016/s0092-8674(00)81275-1. [DOI] [PubMed] [Google Scholar]
- 14.Sintchak MD, Nimmesgern E. Immunopharmacology. 2000;47:163–184. doi: 10.1016/s0162-3109(00)00193-4. [DOI] [PubMed] [Google Scholar]
- 15.Yang D, Fokas D, Li J, Yu L, Baldino CM. Synthesis. 2005:47–56. [Google Scholar]
- 16.Maurya SK, Gollapalli DR, Kirubakaran S, Zhang M, Johnson CR, Benjamin NN, Hedstrom L, Cuny GD. J Med Chem. 2009;52:4623–30. doi: 10.1021/jm900410u. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.