

Abstract
Essential to cells and their organelles, water is both shuttled to where it is needed and trapped within cellular compartments and structures. Moreover, ordered waters within protein structures often co-localize with strategically placed polar or charged groups critical for protein function. Yet it is unclear if these ordered water molecules provide structural stabilization, mediate conformational changes in signaling, neutralize charged residues, or carry out a combination of all these functions. Structures of many integral membrane proteins, including G protein-coupled receptors (GPCRs), reveal the presence of ordered water molecules that may act like prosthetic groups in a manner quite unlike bulk water. Identification of ‘ordered’ waters within a crystalline protein structure requires sufficient occupancy of water to enable its detection in the protein's X-ray diffraction pattern and thus the observed waters likely represent a subset of tightly-bound functional waters. In this review, we highlight recent studies that suggest the structures of ordered waters within GPCRs are as conserved (and thus as important) as conserved side chains. In addition, methods of radiolysis, coupled to structural mass spectrometry (protein footprinting), reveal dynamic changes in water structure that mediate transmembrane signaling. The idea of water as a prosthetic group mediating chemical reaction dynamics is not new in fields such as catalysis. However, the concept of water as a mediator of conformational dynamics in signaling is just emerging, owing to advances in both crystallographic structure determination and new methods of protein footprinting. Although oil and water do not mix, understanding the roles of water is essential to understanding the function of membrane proteins.
Keywords: Footprinting, water, integral membrane proteins, mass spectrometry, G protein-coupled receptors
1. Water and its importance in biological processes
A fundamental problem in modern structural biology is to understand the details of macromolecular structure and dynamics. Subtle structural changes induced by ligand binding or complex formation represent the quintessence of protein function in processes ranging from enzyme activity and signal transmission to nucleic acid binding. Many biological events are mediated to some extent by water. Water has been shown to mediate protein-protein interactions (1) and protein ligand interactions (2), and was suggested to facilitate protein folding (3-5). In the case of rhodopsin (6), a model for membrane bound G protein-coupled receptors (GPCRs), it has been shown that water has a significant role in forming the active site (7). Moreover, a comparison of known GPCR X-ray structures (8-13) revealed that water molecules in the hydrophobic core of these proteins interact with conserved residues implying that these waters are probably as important for GPCR function as the conserved residues themselves (14). Buried water molecules are critical for the overall function of rhodopsin (and bacteriorhodopsin as well) and these have been characterized not only by X-ray crystallography but also by Fourier transform IR spectroscopy (15). Recently radiolytic protein footprinting has emerged as a promising tool for the analysis of side chain dynamics in the vicinity of bound water molecules. In this review we present new developments and potential future directions for the study of transmembrane protein water molecules by radiolytic protein footprinting and the integration of these data with those from other biophysical techniques.
2. Radiolytic Protein Footprinting
Why hydroxyl radicals?
X-ray crystallography and nuclear magnetic resonance (NMR) are currently the most valuable methods to reveal the structural details of macromolecules at atomic resolution. However in many cases where proteins resist such analyses for certain conformational states of interest because of limitations of a specific system, structural mass spectrometry (MS) has become increasingly important as an alternative approach to address questions about structure and dynamics in relevant conformational states (16, 17). Structural changes that facilitate protein function are conveniently probed by covalent labeling methods like hydroxyl radical footprinting coupled to MS where radiolysis or other methods (see below) are used to generate radicals that can decorate solvent-accessible surfaces of macromolecules (18-25). These approaches allow detailed examination of surface residues in conformational states of interest with resolution at the side chain level.
There are several unique benefits of structural MS. For example, MS approaches are not limited by the size of a biomolecular complex. Only limited amounts (in the range of nanomoles or picomoles) of sample are required and the technique provides the ability to test a wide range of solution conditions at physiologically relevant concentrations of protein and/or ligands in the nanomolar to micromolar range (26-28). For macromolecular footprinting with hydroxyl radicals, time-resolved studies that examine dynamic processes over periods ranging from microseconds to minutes are feasible (24, 29-31). In addition, radiolytic protein footprinting generates stable modified side-chains, permitting a wide range of analytical approaches to isolate the protein and its peptides to yield a convenient readout of specific sites of modification that provide high-resolution structural information about the macromolecular species of interest (23, 32-34).
However, there are disadvantages in the application of structural MS methods as compared to more traditional procedures. For example, deuterium exchange and covalent labeling methods (including radiolytic footprinting) provide only local structural information without a tertiary or quaternary context. Cross-linking can provide distance information, but has its own difficulties with respect to experimental design and data interpretation. Thus, the field has moved towards approaches that leverage structural information obtained at atomic resolution with examination of particular species of interest by MS to explore detailed mechanisms of conformational changes intimately associated with function (27, 28, 32, 33, 35-45).
Generation of hydroxyl (OH·) radicals
Hydroxyl radicals used for covalent labeling in structural mass spectrometry may be generated by a variety of methods such as radiolysis (18, 19, 44, 45), electric discharge of an electrospray ionization source (42, 46, 47), Fenton chemistry (48) and photochemistry of peroxide (49, 50). The primary focus for the remainder of this review is on the use of radiolysis for generating OH·, specifically studies that rely on synchrotron X-ray radiation using beamline X28C of the National Synchrotron Light Source (51) because of its ability to implement dynamics studies by probing water molecules trapped inside the hydrophobic core of proteins (52).
Mechanisms of amino acid – OH· interactions
Mechanisms of amino acid modification by OH· radicals are very complex and depend largely on side chain chemistry. Extensive work in the field proposes that typical modifications of amino acid side chains include mass additions of +16 or +14 Da because of incorporation of hydroxyl or carbonyl groups. Residue-specific reaction mechanisms will produce characteristic mass changes subsequent to OH· radical exposure as follows: -43 Da for an Arg residue (53), -30 Da for Asp and Glu residues (54), +32 and +48 for Cys residues (55), and -22, -10, and + 5 for His residues (53) in addition to multiple incorporations of hydroxyl groups for aromatic residues (+16n, n=1,2,3). These modifications and the well understood attenuations of retention time they induce in reverse-phase liquid chromatographic (LC) analyses (56), can be used either to set up rules to identify modifications on specific peptides based on their LC-MS spectra as analyzed by software such as ProtMapMS (57) or to identify modifications of peptides in database search algorithms, such as Mascot (58).
Footprinting experimental setup and data analysis
The general steps involved in protein footprinting are illustrated in Fig. 1. During exposure to ionizing radiation (Fig. 1A), protein amino acid side chains react with OH· radicals generated from water molecules within the bulk solvent. Following radiation exposure, the remaining reaction products such as H2O2 or transient radical species are neutralized with methionine-amides (59). Because of inherent covalent modifications produced by radiolytic labeling, a range of experimental conditions can be employed for this procedure such as different buffers, pH and a wide range of proteases. One problem, however, is buffer components that scavenge generated radicals, such as glycerol, nucleotides, or other components used to stabilize proteins or conformations of interest. In such cases the radiation dose (or the hydroxyl radical dose in general) must be carefully adjusted to provide sufficient energy for desired peptide modifications while limiting secondary reactions resulting from overexposure. A number of studies have found that a rapidly delivered, high-flux dose is preferable to minimize unwanted overexposure and maximize the signal to noise ratio in LC-MS (21, 22, 25, 35, 51, 60-64).
Figure 1. Radiolytic protein footprinting experiments and data analysis.
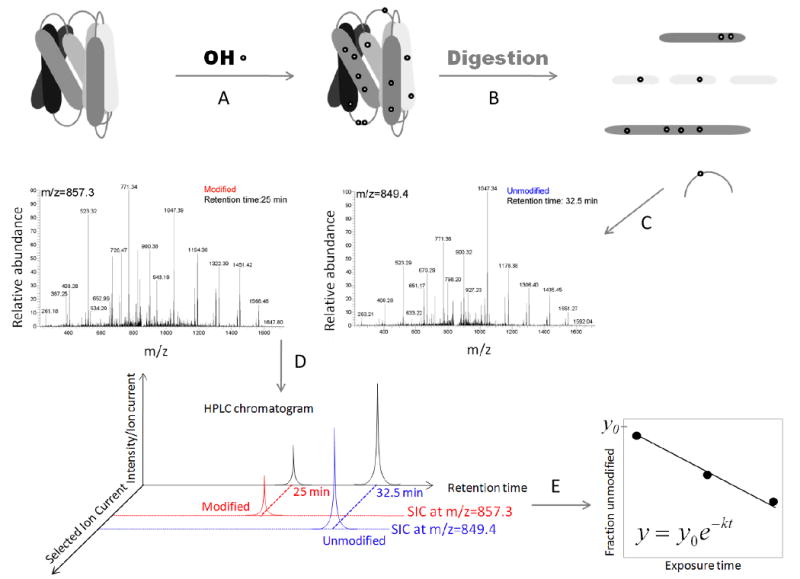
A) Proteins are exposed to X-rays for different time periods (ms). B) Following exposure the protein is digested with a protease and its fragments are separated by using reverse phase high-performance liquid chromatography coupled to a mass spectrometer. Searches for amino acid modifications such as +16 Da and other specific modifications (see Mechanisms of amino acid – OH· interactions) are performed using programs such as Mascot (Matrix Science) and ProtMapMS (57). C) Identification of the modified species based on its MS/MS spectra. D) The fraction unmodified is calculated by using the selected ion chromatogram peak area (modified in red and unmodified in blue). E) The fraction unmodified is plotted as a function of exposure time and the rate of modification is calculated by using nonlinear regression.
Following exposure to X-rays labeled proteins are digested with a protease (Fig. 1B) and the protein proteolytic fragments are separated by reverse-phase high performance chromatography (Fig. 1C) and analyzed by mass spectrometry. Unmodified peptides and their modified counterparts are identified by tandem mass spectrometry (MS/MS). To accelerate these time-consuming steps, recent efforts have concentrated on automating data analyses. These methods identify different modified species and also extract retention times (modified peptides elute before unmodified peptides) of modified and unmodified peptides (57). Following identification of the modified and unmodified species based on tandem MS, the fraction unmodified is calculated from the change in the total area of the selected ion current (Fig. 1D). Finally, the fraction unmodified is plotted as a function of dosage (exposure time) to determine the rate of modification by using a first order function (Fig. 1E).
Protein dynamics tested by footprinting
Historically, hydroxyl radical mediated protein footprinting was shown to be particularly well suited for testing solvent exposure of the modified residues. However, in recent experiments on the transmembrane protein rhodopsin, it was observed that several residues buried deep inside the hydrophobic core of the protein could exhibit modifications (52). Even more interesting, some of these modifications were not only extensive but they occurred after very short periods of exposure as compared to residues exposed to bulk solvent. Recent structural studies of bovine rhodopsin have shed some light on this peculiar reactivity of buried residues to hydroxyl radicals (14). These high-resolution studies identified water molecules in close proximity to many amino acid side chains that were efficiently modified. These findings are featured in this review to illustrate how radiolytic footprinting-mediated structural MS can be used as a unique tool to answer questions regarding the structure/function of membrane proteins and the dynamics of conformational changes involving internally bound water.
3. Footprinting of a model GPCR – rhodopsin
Rhodopsin is responsible for the initiation of signaling in visual transduction. Signaling starts upon absorption of a photon of light that causes a rapid trans isomerization of the bound 11-cis-retinal visual chromophore (65). After rhodopsin passes through several conformations corresponding to different bleached intermediates, the last one being Meta II, signal transduction is mediated by formation of the rhodopsin-transducin (G protein) complex. Contrary to its anticipated behavior, rhodopsin does not undergo large conformational changes following absorption of a photon of light – an event similar to binding of an agonist to other GPCRs (66). Superposed structures of bovine rhodopsin show little deviation (up to 5 Å for a tip of helix VI, and less or within 3 Å of normal protein dynamics for other parts) hence the signal is probably transmitted by a different mechanism (67, 68). Such a mechanism was recently proposed following studies of radiolytic footprinting of this GPCR (52). The study confirmed that only small structural variations occur in the vicinity of the bound chromophore and furthermore offered a probable mechanism by which the absorption of a photon of light results in the conversion of GDP to GTP by the rhodopsin-transducin complex.
Fig. 2 shows the residues of rhodopsin found to be modified following exposure to X-rays (52). Many modified residues are within the hydrophobic core; moreover several amino acid residues exhibited unusually high rates of modification following exposure to X-rays. Phe116 had a particularly high modification rate – about 10-fold higher than any other residue in either rhodopsin or any soluble protein previously studied by radiolytic footprinting (18, 43, 53-55). Phe116 is located in the near vicinity of the Schiff base and close to a crystallographically identified water molecule (10) (see Figure 3A and also below). The high modification rate of Phe116 is attributed to its proximity to this water molecule #2021 (10). In activated rhodopsin, by contrast, the decreased modification rate of Phe116 results from unfavorable positioning of the same internal water molecule. The residue with the largest change in relative rate constants upon rhodopsin activation was Met86. This residue is found in the vicinity of the highly conserved residue Asp83 previously determined to be critical for rhodopsin activation. A somewhat smaller change in modification rate (i.e., ∼2-fold) was observed for residue Met288 upon activation. Although located in helix VII, Met288 is near the chromophore-binding pocket and crystallographically determined water molecules. Several water molecules were found to be conserved when X-ray structures of other GPCRs were compared (Fig. 3B) (14). Moreover, these water molecules seem not to exchange with bulk solvent in any of the three rhodopsin states as demonstrated by rapid mixing with 18O labeled water (52). These results confirm previous predictions about the long-standing question of how rhodopsin transmits a visual signal in the absence of large conformational changes (69).
Figure 2. Footprinting of a model GPCR – rhodopsin.
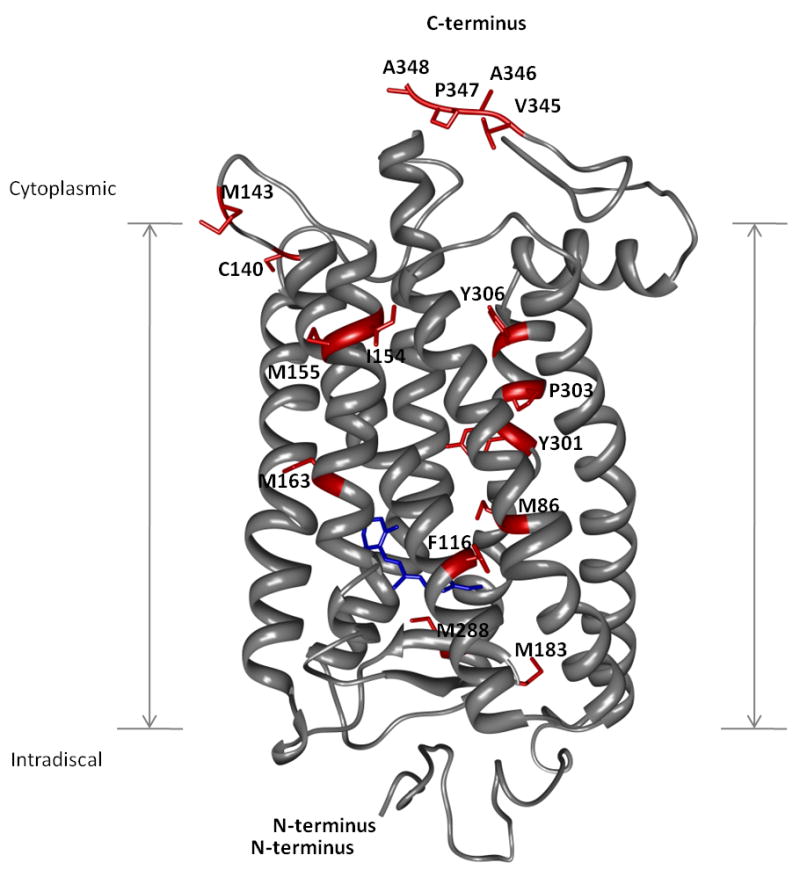
Rhodopsin is shown as gray ribbons whereas amino acid residues found to be modified following exposure to X-rays are shown as red sticks. The N- and C-termini are indicated together with the transmembrane region. The rhodopsin chromophore, 11-cis-retinal, is shown in blue near the intradiscal side of the membrane.
Figure 3. Transmembrane proteins and waters detected by crystallography.
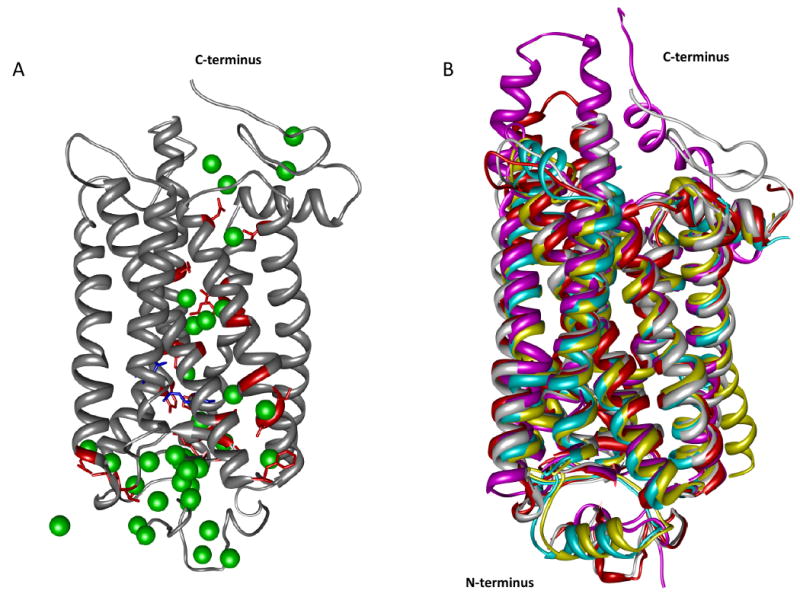
In panel A rhodopsin (pdb: 1U19) is depicted by gray ribbons and crystallographically determined water molecules are shown as green spheres. The rhodopsin chromophore, 11-cis-retinal, is shown as blue sticks whereas amino acid residues from the transmembrane region in contact with water molecules are shown as red sticks. Panel B shows superposed structures of GPCRs such as squid rhodopsin in magenta, β1-adrenergic receptor in cyan, β2-adrenergic receptor in yellow and bovine opsin in red.
4. Transmembrane waters detected by other biophysical methods
Transmembrane proteins and waters detected by crystallography
GPCRs represent one of the most attractive drug targets because a high percentage of today's drugs act on these receptors. Protein X-ray crystallography can elucidate structure of proteins with atomic resolution that can ultimately be used as the foundation of structure-based rational drug design. Obtaining well ordered three-dimensional crystals is a critical step in solving protein structure by X-ray crystallography. Although a considerable number of soluble proteins have been solved to date, protein crystallization is still regarded an art. As one can imagine, additional challenges are presented in case of membrane proteins attributable to their amphiphilic character. Unfortunately, the challenges posed by protein X-ray crystallography, the number of membrane proteins submitted to the Protein Data Bank (PDB) database is quite limited. Following the initial breakthrough of crystallization and subsequent solving of the structure of rhodopsin (6) several other GPCR structures were solved such as bovine and squid rhodpsins (10,11), bovine opsin (9), β1- and β2-adrenergic receptors (8, 13) and the A2A-adenosine receptor (12). A recent study (14) performed in light of rhodopsin footprinting experiments (52) has shown that although the sequence similarity of these class A GPCRs is not high, several water molecules identified crystallographically within the hydrophobic region were conserved. Fig. 3A shows the structure of rhodopsin, a seven helix transmembrane protein (70), together with the crystallographically determined water molecules (pdb: 1U19). A superposition of selected high-resolution GPCR structures is shown in Fig. 3B. Although the alignment based on the α-carbons of these proteins is highly similar in the transmembrane region, regions found in either side of the membrane show considerable diversity as expected (69). The structure of several regions, such as the NPXXY motif (where X can be any amino acid) representing a highly conserved region among GPCRs (71), is greatly influenced by the presence of water molecules. Conserved water molecules were found to participate in hydrogen bonding networks that link transmembrane helices 2 with 6 (12) (Trp265 ⋯ water #5772 ⋯ water # 5774 ⋯ Asp83). These two residues, Asp83 and Trp265 are 94 and 71% conserved among family A GPCRs. Another region linked by a hydrogen bonding network mediated by a water molecule is located between helices 6 and 7 in rhodopsin (72). In the case of the histamine H1 receptor (73), the hydrogen bonding network can be maintained with only one water molecule. But in the delta opioid receptor (74) the hydrogen bonding network is formed only by means of side chain interactions and the presence of water molecules seems not to be required.
Waters as integral part of proteins detected by Fourier transform IR spectroscopy (FTIR)
Fourier transform infrared spectroscopy (FTIR) was historically used to provide secondary structure information about biological molecules (frequency region <1800 cm-1). It also has been successfully used to study water molecules within transmembrane proteins. FTIR spectroscopy can detect changes in hydrogen bonding strength based on the frequency of OH stretching vibrations (75). In the case of proteins, these vibrations are shifted from 2900 and 3200 cm-1 to 3200 and 3600 cm-1, respectively, as a result of the strong absorption of bulk water. In the case of bacteriorhodopsin, water molecules were found to be involved in the mechanism of proton pumping (76, 77). Specifically, several hydrogen bonding networks were identified where amino acid residues Asp85, Asp 212, and Arg82 were implicated in the proton transfer mechanism (76). Two water molecules were found to be altered in rhodopsin – the first in Meta I and the second at the Meta I to Meta II transition stage (15). Fig. 4 shows rhodopsin with labeled amino acid residues Asp83 and Gly120 (in red) and water molecule #2030 (blue sphere, PDB 1U19) (10). Asp83, an important player in rhodopsin activation, is found near Met86 – another residue shown to be important in rhodopsin activation by recent footprinting experiments (52). An important water molecule recently identified in the sensory rhodopsin (SRII)-transducer (HtrII) complex also was found to be located near residue Tyr199 (78). Thus, FTIR, crystallography and footprinting all identify conserved and functionally important water molecules in membrane proteins and these techniques can work in concert to understand the role of water in mediating important functions such as ion transport and signaling.
Figure 4. Transmembrane water molecules determined by FTIR.
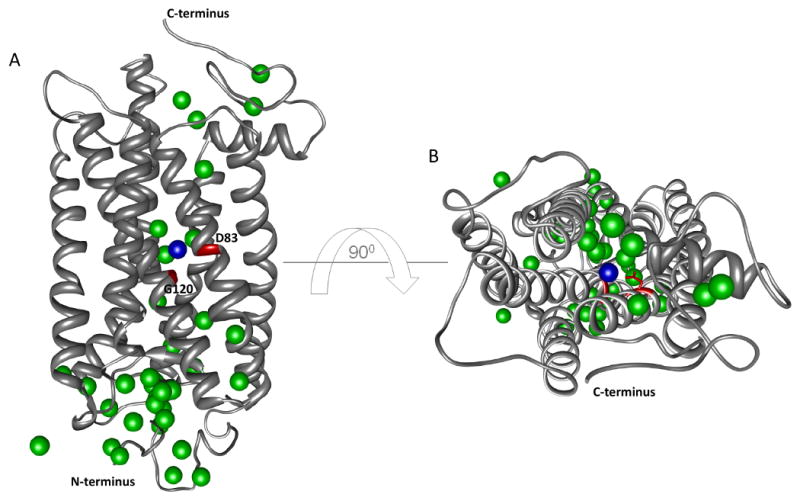
Panel A shows rhodopsin as gray ribbons, Asp83 and Gly120 are shown in red whereas the interacting water molecule is shown as a blue sphere. Water molecules determined by X-ray crystallography (pdb 1U19-chain A) are shown as green spheres. Panel B shows a top view of rhodopsin with the same coloring scheme used as in panel A.
5. Future directions
Other membrane proteins and ion channels
The possibility of using footprinting to examine the details of water structure in membrane proteins for a wide range of membrane proteins, both in vitro and in vivo is discussed in this section. For example, structures of GPCRs other than rhodopsin have recently been published (8, 12); these structures, which have great similarities to rhodopsin, provide interesting potential insights into GPCR signaling. But engineering of these proteins to enable the generation of diffraction quality crystals raises questions as to their functionality. Radiolytic protein footprinting and FTIR studies, on both native forms and the engineered species could provide novel insights into the presence and activity of bound waters in these proteins as well as their ligand dependent signaling mechanisms.
Recently radiolytic protein footprinting was used to understand the details of the conformational dynamics and the amino acids involved in K-channel gating (79). The closed state of bacterial inwardly rectifying K-channels is well understood (80), although high-resolution data for the open state are not yet available (81, 82). Footprinting data for detergent preparations of the inwardly rectifying (Kir) potassium channel of Magnetospirillum magnetotacticum (KirBac 3.1) in both open and closed states also were collected and the data were analyzed in the context of the known closed state structure. Differences in the extent of modification observed between the closed and open states were then used to reveal local conformational changes that occur during channel gating. The data suggest that specific side chain residues located in the inner helices of the trans-membrane membrane pore as well as residues towards the extra-cellular side of the transmembrane region are involved in gating. These findings provide a basis for understanding the dynamics of gating at the side chain level for a wide variety of bacterial and eukaryotic channels in the presence of a range of ligands and other effectors that tune the gating process.
Because strongly bound waters can have long residence times, radiolytic labeling by short highly intense pulses of synchrotron X-rays or electron beams can generate local spurs of hydroxyl radicals that activate bound waters that rapidly modify adjacent side chain groups to provide information on the local dynamics of functionally important amino acid residues. In addition, recent advances in radiolytic labeling coupled to H218O exchange can probe the dynamics of membrane protein signaling and gating (52). In the latter experiments, H218O mixing of proteins followed by X-ray irradiation or pulse radiolysis generates 18O-labeled covalent modifications primarily of aromatic or sulfur containing groups that are accessible to bulk solvent. Timescales of these mixing experiments can be as short as 1-2 milliseconds, thus one can delay irradiation subsequent to mixing and monitor the time-course of bulk water movement into crevices, channels, or pores for defined states of interest.
Prospects for developing new footprinting tools for in vivo probes of macromolecules are encouraging. In vivo footprinting of cells has a long history and the use of X-rays to analyze nucleic acids structure in cells has been an important tool (83). Since a predominant constituent of a cell is water, ionizing radiation can generate hydroxyl radicals transiently inside and outside cells. However, non-radiolytic techniques of in vivo footprinting require harsh treatment of cells, whereas radiolytic approaches traditionally have required long exposures to gamma rays. Recently, in vivo X-ray footprinting was used to identify the folding pattern of a 16S ribosomal RNA structure in frozen E. coli after sub-second exposures (84). The use high-flux X-rays from a synchrotron as the source of hydroxyl radicals reduced the exposure time considerably. With respect to protein footprinting, in situ generation of hydroxyl radicals by Fenton's reagents has shown different extents of oxidation for the open and close states of the porin, OmpF, in E. Coli cells (85), thus providing a proof-of-principle for in vivo hydroxyl radical footprinting analysed by MS. But this requires a prolonged exposure to Fenton's reagent so the approach is suitable only for analysis of extracellular motifs, unless cells are made porous. However, high-flux radiolysis sources coupled to MS analysis have the potential to provide very short timescales of exposure to examine structures and dynamics of living systems.
Generating cross-linked side-chains
Additional applications of radiolysis chemistry yet to be explored may represent future directions for research. There are a myriad of chemical species, including hydrated electrons, which can be produced or consumed depending on experimental conditions. The history of radiolysis research includes not only aerobic radiolysis of bulk aqueous solutions (86), but also radiolysis: 1) under anaerobic conditions, 2) in the presence of scavengers, 3) after addition of N2O to convert hydrated electrons to hydroxyl radicals, and 4) applied to gels and solids (87, 88). There are many biological structures andstates, including membranes, which could be explored with this technology. In the presence of oxygen, radicals transferred from hydroxyl radicals to protein side chains are efficiently quenched, leading to the stable products observed. However, in the absence of oxygen when the observed yield of oxidized products is reduced (23), lifetimes of protein-derived radicals are extended. This raises the prospect of potentially effective zero-length cross-linking of radicals found on side chains in close proximity. This could be explored as an efficient cross-linking strategy both in vitro and in vivo that would target multiple native residues without any engineering, compared to current methods that typically require use of native Cys or Lys residues, or their insertion in defined positions. The resulting data could provide distance constraints informative of tertiary structures as well of quaternary structures within a protein complex.
In summary, visualizing water in transmembrane proteins using mass spectrometry is an important advance, and the use of radiolysis related technologies to permit labeling of many types of macromolecules mediated by their interactions with water is developing rapidly. The coupling of hydroxyl radical labeling and MS is especially powerful for the study of membrane proteins, as femtomoles to picomoles of material are all that is required to provide very detailed examinations of structure. In addition, the study of more complex systems as well as the state of membrane proteins in vivo, is increasingly possible. With further increases in the time of molecular dynamics simulations, it will be possible to merge experimental and theoretical approaches to gain a more realistic and mechanistic view of how membrane proteins work, including how they respond to the binding of ligands and transmit signals to partner proteins.
Acknowledgments
We thank Rujita Di Mello, Mike Sullivan, John Toomey, and Don Able for assistance at the X28C beamline.
Abbreviations
- FTIR
Fourier transform infrared
- GPCRs
G protein-coupled receptors
- MS
mass spectrometry
- NMR
nuclear magnetic resonance
- PDB
protein data bank
Footnotes
This work was supported in part by U.S. Public Health Service grants EY018085, EY009339, EB09998, EB09688, and GM079191, from the National Institutes of Health, Bethesda, MD.
References
- 1.Papoian GA, Ulander J, Wolynes PG. Role of water mediated interactions in protein-protein recognition landscapes. J Am Chem Soc. 2003;125:9170–9178. doi: 10.1021/ja034729u. [DOI] [PubMed] [Google Scholar]
- 2.Tame JR, Murshudov GN, Dodson EJ, Neil TK, Dodson GG, Higgins CF, Wilkinson AJ. The structural basis of sequence-independent peptide binding by OppA protein. Science. 1994;264:1578–1581. doi: 10.1126/science.8202710. [DOI] [PubMed] [Google Scholar]
- 3.Papoian GA, Ulander J, Eastwood MP, Luthey-Schulten Z, Wolynes PG. Water in protein structure prediction. Proc Natl Acad Sci U S A. 2004;101:3352–3357. doi: 10.1073/pnas.0307851100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Head-Gordon T, Brown S. Minimalist models for protein folding and design. Curr Opin Struct Biol. 2003;13:160–167. doi: 10.1016/s0959-440x(03)00030-7. [DOI] [PubMed] [Google Scholar]
- 5.Cheung MS, Garcia AE, Onuchic JN. Protein folding mediated by solvation: Water expulsion and formation of the hydrophobic core occur after the structural collapse. Proc Natl Acad Sci USA. 2002;99:685–690. doi: 10.1073/pnas.022387699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 7.Wald G, Durell J, St George CC. The light reaction in the bleaching of rhodopsin. Science. 1950;111:179–181. doi: 10.1126/science.111.2877.179. [DOI] [PubMed] [Google Scholar]
- 8.Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, Kobilka BK. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 9.Park JH, Scheerer P, Hofmann KP, Choe HW, Ernst OP. Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature. 2008;454:183–187. doi: 10.1038/nature07063. [DOI] [PubMed] [Google Scholar]
- 10.Okada T, Sugihara M, Bondar AN, Elstner M, Entel P, Buss V. The retinal conformation and its environment in rhodopsin in light of a new 2.2 A crystal structure. J Mol Biol. 2004;342:571–583. doi: 10.1016/j.jmb.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 11.Murakami M, Kouyama T. Crystal structure of squid rhodopsin. Nature. 2008;453:363–367. doi: 10.1038/nature06925. [DOI] [PubMed] [Google Scholar]
- 12.Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, Ijzerman AP, Stevens RC. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Angel TE, Chance MR, Palczewski K. Conserved waters mediate structural and functional activation of family A (rhodopsin-like) G protein-coupled receptors. Proc Natl Acad Sci USA. 2009;106:8555–8560. doi: 10.1073/pnas.0903545106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rath P, Delange F, Degrip WJ, Rothschild KJ. Hydrogen bonding changes of internal water molecules in rhodopsin during metarhodopsin I and metarhodopsin II formation. The Biochemical journal. 1998;329(Pt 3):713–717. doi: 10.1042/bj3290713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chance MR, editor. Mass Spectrometry Analysis for Protein Interactions and Dynamics. Wiley-Blackwell; 2008. [Google Scholar]
- 17.Konermann L, Tong X, Pan Y. Protein structure and dynamics studied by mass spectrometry: H/D exchange, hydroxyl radical labeling, and related approaches. J Mass Spectrom. 2008;43:1021–1036. doi: 10.1002/jms.1435. [DOI] [PubMed] [Google Scholar]
- 18.Xu G, Chance MR. Hydroxyl radical-mediated modification of proteins as probes for structural proteomics. Chem Rev. 2007;107:3514–3543. doi: 10.1021/cr0682047. [DOI] [PubMed] [Google Scholar]
- 19.Takamoto K, Chance MR. Radiolytic protein footprinting with mass spectrometry to probe the structure of macromolecular complexes. Annu Rev Biophys Biomol Struct. 2006;35:251–276. doi: 10.1146/annurev.biophys.35.040405.102050. [DOI] [PubMed] [Google Scholar]
- 20.Brenowitz M, Chance MR, Dhavan G, Takamoto K. Probing the structural dynamics of nucleic acids by quantitative time-resolved and equilibrium hydroxyl radical “footprinting”. Curr Opin Struct Biol. 2002;12:648–653. doi: 10.1016/s0959-440x(02)00366-4. [DOI] [PubMed] [Google Scholar]
- 21.Aye TT, Low TY, Sze SK. Nanosecond laser-induced photochemical oxidation method for protein surface mapping with mass spectrometry. Analytical chemistry. 2005;77:5814–5822. doi: 10.1021/ac050353m. [DOI] [PubMed] [Google Scholar]
- 22.Hambly DM, Gross ML. Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. Journal of the American Society for Mass Spectrometry. 2005;16:2057–2063. doi: 10.1016/j.jasms.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 23.Maleknia SD, Brenowitz M, Chance MR. Millisecond radiolytic modification of peptides by synchrotron X-rays identified by mass spectrometry. Analytical chemistry. 1999;71:3965–3973. doi: 10.1021/ac990500e. [DOI] [PubMed] [Google Scholar]
- 24.Maleknia SD, Ralston CY, Brenowitz MD, Downard KM, Chance MR. Determination of macromolecular folding and structure by synchrotron x-ray radiolysis techniques. Anal Biochem. 2001;289:103–115. doi: 10.1006/abio.2000.4910. [DOI] [PubMed] [Google Scholar]
- 25.Konermann L, Stocks BB, Pan Y, Tong X. Mass spectrometry combined with oxidative labeling for exploring protein structure and folding. Mass Spectrom Rev. 2009 doi: 10.1002/mas.20256. [DOI] [PubMed] [Google Scholar]
- 26.Chance MR. Unfolding of apomyoglobin examined by synchrotron footprinting. Biochem Biophys Res Commun. 2001;287:614–621. doi: 10.1006/bbrc.2001.5628. [DOI] [PubMed] [Google Scholar]
- 27.Kiselar JG, Janmey PA, Almo SC, Chance MR. Visualizing the Ca2+-dependent activation of gelsolin by using synchrotron footprinting. Proc Natl Acad Sci U S A. 2003;100:3942–3947. doi: 10.1073/pnas.0736004100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kiselar JG, Mahaffy R, Pollard TD, Almo SC, Chance MR. Visualizing Arp2/3 complex activation mediated by binding of ATP and WASp using structural mass spectrometry. Proc Natl Acad Sci U S A. 2007;104:1552–1557. doi: 10.1073/pnas.0605380104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chance MR, Sclavi B, Woodson SA, Brenowitz M. Examining the conformational dynamics of macromolecules with time-resolved synchrotron X-ray ‘footprinting’. Structure. 1997;5:865–869. doi: 10.1016/s0969-2126(97)00241-4. [DOI] [PubMed] [Google Scholar]
- 30.Sclavi B, Sullivan M, Chance MR, Brenowitz M, Woodson SA. RNA folding at millisecond intervals by synchrotron hydroxyl radical footprinting. Science. 1998;279:1940–1943. doi: 10.1126/science.279.5358.1940. [DOI] [PubMed] [Google Scholar]
- 31.Stocks BB, Konermann L. Structural characterization of short-lived protein unfolding intermediates by laser-induced oxidative labeling and mass spectrometry. Analytical chemistry. 2009;81:20–27. doi: 10.1021/ac801888h. [DOI] [PubMed] [Google Scholar]
- 32.Guan JQ, Almo SC, Chance MR. Synchrotron radiolysis and mass spectrometry: a new approach to research on the actin cytoskeleton. Acc Chem Res. 2004;37:221–229. doi: 10.1021/ar0302235. [DOI] [PubMed] [Google Scholar]
- 33.Guan JQ, Vorobiev S, Almo SC, Chance MR. Mapping the G-actin binding surface of cofilin using synchrotron protein footprinting. Biochemistry. 2002;41:5765–5775. doi: 10.1021/bi0121104. [DOI] [PubMed] [Google Scholar]
- 34.Kiselar JG, Maleknia SD, Sullivan M, Downard KM, Chance MR. Hydroxyl radical probe of protein surfaces using synchrotron X-ray radiolysis and mass spectrometry. Int J Radiat Biol. 2002;78:101–114. doi: 10.1080/09553000110094805. [DOI] [PubMed] [Google Scholar]
- 35.Jennings LD, Bohon J, Chance MR, Licht S. The ClpP N-terminus coordinates substrate access with protease active site reactivity. Biochemistry. 2008;47:11031–11040. doi: 10.1021/bi8010169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kamal JK, Benchaar SA, Takamoto K, Reisler E, Chance MR. Three-dimensional structure of cofilin bound to monomeric actin derived by structural mass spectrometry data. Proc Natl Acad Sci U S A. 2007;104:7910–7915. doi: 10.1073/pnas.0611283104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kamal JK, Chance MR. Modeling of protein binary complexes using structural mass spectrometry data. Protein Sci. 2008;17:79–94. doi: 10.1110/ps.073071808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guan JQ, Almo SC, Reisler E, Chance MR. Structural reorganization of proteins revealed by radiolysis and mass spectrometry: G-actin solution structure is divalent cation dependent. Biochemistry. 2003;42:11992–12000. doi: 10.1021/bi034914k. [DOI] [PubMed] [Google Scholar]
- 39.Guan JQ, Takamoto K, Almo SC, Reisler E, Chance MR. Structure and dynamics of the actin filament. Biochemistry. 2005;44:3166–3175. doi: 10.1021/bi048021j. [DOI] [PubMed] [Google Scholar]
- 40.Guan JQ, Chance MR. Structural proteomics of macromolecular assemblies using oxidative footprinting and mass spectrometry. Trends Biochem Sci. 2005;30:583–592. doi: 10.1016/j.tibs.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 41.Liu R, Guan JQ, Zak O, Aisen P, Chance MR. Structural reorganization of the transferrin C-lobe and transferrin receptor upon complex formation: the C-lobe binds to the receptor helical domain. Biochemistry. 2003;42:12447–12454. doi: 10.1021/bi0352973. [DOI] [PubMed] [Google Scholar]
- 42.Wong JW, Maleknia SD, Downard KM. Hydroxyl radical probe of the calmodulin-melittin complex interface by electrospray ionization mass spectrometry. Journal of the American Society for Mass Spectrometry. 2005;16:225–233. doi: 10.1016/j.jasms.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 43.Xu G, Liu R, Zak O, Aisen P, Chance MR. Structural allostery and binding of the transferrin*receptor complex. Mol Cell Proteomics. 2005;4:1959–1967. doi: 10.1074/mcp.M500095-MCP200. [DOI] [PubMed] [Google Scholar]
- 44.Watson C, Janik I, Zhuang T, Charvatova O, Woods RJ, Sharp JS. Pulsed electron beam water radiolysis for submicrosecond hydroxyl radical protein footprinting. Analytical chemistry. 2009;81:2496–2505. doi: 10.1021/ac802252y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Charvatova O, Foley BL, Bern MW, Sharp JS, Orlando R, Woods RJ. Quantifying protein interface footprinting by hydroxyl radical oxidation and molecular dynamics simulation: application to galectin-1. Journal of the American Society for Mass Spectrometry. 2008;19:1692–1705. doi: 10.1016/j.jasms.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maleknia SD, Chance MR, Downard KM. Electrospray-assisted modification of proteins: a radical probe of protein structure. Rapid Commun Mass Spectrom. 1999;13:2352–2358. doi: 10.1002/(SICI)1097-0231(19991215)13:23<2352::AID-RCM798>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 47.Wong JW, Maleknia SD, Downard KM. Study of the ribonuclease-S-proteinpeptide complex using a radical probe and electrospray ionization mass spectrometry. Anal Chem. 2003;75:1557–1563. doi: 10.1021/ac026400h. [DOI] [PubMed] [Google Scholar]
- 48.Sharp JS, Becker JM, Hettich RL. Protein surface mapping by chemical oxidation: structural analysis by mass spectrometry. Anal Biochem. 2003;313:216–225. doi: 10.1016/s0003-2697(02)00612-7. [DOI] [PubMed] [Google Scholar]
- 49.Sharp JS, Becker JM, Hettich RL. Analysis of protein solvent accessible surfaces by photochemical oxidation and mass spectrometry. Anal Chem. 2004;76:672–683. doi: 10.1021/ac0302004. [DOI] [PubMed] [Google Scholar]
- 50.Sharp JS, Guo JT, Uchiki T, Xu Y, Dealwis C, Hettich RL. Photochemical surface mapping of C14S-Sml1p for constrained computational modeling of protein structure. Anal Biochem. 2005;340 doi: 10.1016/j.ab.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 51.Gupta S, Sullivan M, Toomey J, Kiselar J, Chance MR. The Beamline X28C of the Center for Synchrotron Biosciences: a national resource for biomolecular structure and dynamics experiments using synchrotron footprinting. J Synchrotron Radiat. 2007;14:233–243. doi: 10.1107/S0909049507013118. [DOI] [PubMed] [Google Scholar]
- 52.Angel TE, Gupta S, Jastrzebska B, Palczewski K, Chance MR. Structural waters define a functional channel mediating activation of the GPCR, rhodopsin. Proc Natl Acad Sci U S A. 2009;106:14367–14372. doi: 10.1073/pnas.0901074106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu G, Takamoto K, Chance MR. Radiolytic modification of basic amino acid residues in peptides: probes for examining protein-protein interactions. Analytical chemistry. 2003;75:6995–7007. doi: 10.1021/ac035104h. [DOI] [PubMed] [Google Scholar]
- 54.Xu G, Chance MR. Radiolytic modification of acidic amino acid residues in peptides: probes for examining protein-protein interactions. Analytical chemistry. 2004;76:1213–1221. doi: 10.1021/ac035422g. [DOI] [PubMed] [Google Scholar]
- 55.Xu G, Chance MR. Radiolytic modification of sulfur-containing amino acid residues in model peptides: fundamental studies for protein footprinting. Analytical chemistry. 2005;77:2437–2449. doi: 10.1021/ac0484629. [DOI] [PubMed] [Google Scholar]
- 56.Kiselar JG, Janmey PA, Almo SC, Chance MR. Structural analysis of gelsolin using synchrotron protein footprinting. Mol Cell Proteomics. 2003;2:1120–1132. doi: 10.1074/mcp.M300068-MCP200. [DOI] [PubMed] [Google Scholar]
- 57.Kaur P, Kiselar JG, Chance MR. Integrated algorithms for high-throughput examination of covalently labeled biomolecules by structural mass spectrometry. Anal Chem. 2009;81:8141–8149. doi: 10.1021/ac9013644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Perkins DN, Pappin DJC, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 59.Xu G, Kiselar J, He Q, Chance MR. Secondary reactions and strategies to improve quantitative protein footprinting. Analytical chemistry. 2005;77:3029–3037. doi: 10.1021/ac048282z. [DOI] [PubMed] [Google Scholar]
- 60.Bohon J, Jennings LD, Phillips CM, Licht S, Chance MR. Synchrotron Protein Footprinting Supports Substrate Translocation by ClpA via ATP-Induced Movements of the D2 Loop. Structure. 2008;16:1157–1165. doi: 10.1016/j.str.2008.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sullivan MR, Rekhi S, Bohon J, Gupta S, Abel D, Toomey J, Chance MR. Installation and testing of a focusing mirror at beamline X28C for high flux x-ray radiolysis of biological macromolecules. Rev Sci Instrum. 2008;79:025101. doi: 10.1063/1.2839027. [DOI] [PubMed] [Google Scholar]
- 62.Tong X, Wren JC, Konermann L. gamma-Ray-mediated oxidative labeling for detecting protein conformational changes by electrospray mass spectrometry. Analytical chemistry. 2008;80:2222–2231. doi: 10.1021/ac702321r. [DOI] [PubMed] [Google Scholar]
- 63.Sharp JS, Tomer KB. Analysis of the oxidative damage-induced conformational changes of apo- and holocalmodulin by dose-dependent protein oxidative surface mapping. Biophys J. 2007;92:1682–1692. doi: 10.1529/biophysj.106.099093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Venkatesh S, Tomer KB, Sharp JS. Rapid identification of oxidation-induced conformational changes by kinetic analysis. Rapid Commun Mass Spectrom. 2007;21:3927–3936. doi: 10.1002/rcm.3291. [DOI] [PubMed] [Google Scholar]
- 65.Palczewski K. G protein-coupled receptor rhodopsin. Annual review of biochemistry. 2006;75:743–767. doi: 10.1146/annurev.biochem.75.103004.142743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mustafi D, Palczewski K. Topology of class A G protein-coupled receptors: insights gained from crystal structures of rhodopsins, adrenergic and adenosine receptors. Molecular pharmacology. 2009;75:1–12. doi: 10.1124/mol.108.051938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Salom D, Lodowski DT, Stenkamp RE, Le Trong I, Golczak M, Jastrzebska B, Harris T, Ballesteros JA, Palczewski K. Crystal structure of a photoactivated deprotonated intermediate of rhodopsin. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:16123–16128. doi: 10.1073/pnas.0608022103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lodowski DT, Angel TE, Palczewski K. Comparative analysis of GPCR crystal structures. Photochemistry and photobiology. 2009;85:425–430. doi: 10.1111/j.1751-1097.2008.00516.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mirzadegan T, Benkö G, Filipek S, Palczewski K. Sequence analyses of G-protein-coupled receptors: similarities to rhodopsin. Biochemistry. 2003;42:2759–2767. doi: 10.1021/bi027224+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Okada T, Fujiyoshi Y, Silow M, Navarro J, Landau EM, Shichida Y. Functional role of internal water molecules in rhodopsin revealed by X-ray crystallography. Proc Natl Acad Sci U S A. 2002;99:5982–5987. doi: 10.1073/pnas.082666399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Probst WC, Snyder LA, Schuster DI, Brosius J, Sealfon SC. Sequence alignment of the G-protein coupled receptor superfamily. DNA and cell biology. 1992;11:1–20. doi: 10.1089/dna.1992.11.1. [DOI] [PubMed] [Google Scholar]
- 72.Pardo L, Deupi X, Dolker N, Lopez-Rodriguez ML, Campillo M. The role of internal water molecules in the structure and function of the rhodopsin family of G protein-coupled receptors. Chembiochem. 2007;8:19–24. doi: 10.1002/cbic.200600429. [DOI] [PubMed] [Google Scholar]
- 73.Jongejan A, Bruysters M, Ballesteros JA, Haaksma E, Bakker RA, Pardo L, Leurs R. Linking agonist binding to histamine H1 receptor activation. Nature chemical biology. 2005;1:98–103. doi: 10.1038/nchembio714. [DOI] [PubMed] [Google Scholar]
- 74.Xu W, Campillo M, Pardo L, Kim de Riel J, Liu-Chen LY. The seventh transmembrane domains of the delta and kappa opioid receptors have different accessibility patterns and interhelical interactions. Biochemistry. 2005;44:16014–16025. doi: 10.1021/bi050938a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kandori H. Role of internal water molecules in bacteriorhodopsin. Biochim Biophys Acta. 2000;1460:177–191. doi: 10.1016/s0005-2728(00)00138-9. [DOI] [PubMed] [Google Scholar]
- 76.Garczarek F, Gerwert K. Functional waters in intraprotein proton transfer monitored by FTIR difference spectroscopy. Nature. 2006;439:109–112. doi: 10.1038/nature04231. [DOI] [PubMed] [Google Scholar]
- 77.Mathias G, Marx D. Structures and spectral signatures of protonated water networks in bacteriorhodopsin. Proc Natl Acad Sci U S A. 2007;104:6980–6985. doi: 10.1073/pnas.0609229104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bergo VB, Spudich EN, Spudich JL, Rothschild KJ. Active water in protein-protein communication within the membrane: the case of SRII-HtrII signal relay. Biochemistry. 2009;48:811–813. doi: 10.1021/bi802180a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gupta S, Bravo V, Tucker S, Venien-Bryan C, Chance MR. Probing the structure of an ion channel by structural mass spectrometry. Biophysical J in press 2010 [Google Scholar]
- 80.Kuo A, Gulbis JM, Antcliff JF, Rahman T, Lowe ED, Zimmer J, Cuthbertson J, Ashcroft FM, Ezaki T, Doyle DA. Crystal structure of the potassium channel KirBac1.1 in the closed state. Science. 2003;300:1922–1926. doi: 10.1126/science.1085028. [DOI] [PubMed] [Google Scholar]
- 81.Kuo A, Domene C, Johnson LN, Doyle DA, Venien-Bryan C. Two different conformational states of the KirBac3.1 potassium channel revealed by electron crystallography. Structure. 2005;13:1463–1472. doi: 10.1016/j.str.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 82.Jaroslawski S, Zadek B, Ashcroft F, Venien-Bryan C, Scheuring S. Direct visualization of KirBac3.1 potassium channel gating by atomic force microscopy. J Mol Biol. 2007;374:500–505. doi: 10.1016/j.jmb.2007.09.043. [DOI] [PubMed] [Google Scholar]
- 83.Hayes JJ, Kam L, Tullius TD. Footprinting protein-DNA complexes with gamma-rays. Methods Enzymol. 1990;186:545–549. doi: 10.1016/0076-6879(90)86148-o. [DOI] [PubMed] [Google Scholar]
- 84.Adilakshmi T, Lease RA, Woodson SA. Hydroxyl radical footprinting in vivo: mapping macromolecular structures with synchrotron radiation. Nucleic Acids Res. 2006;34:e64. doi: 10.1093/nar/gkl291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhu Y, Guo T, Park JE, Li X, Meng W, Datta A, Bern M, Lim SK, Sze SK. Elucidating in vivo structural dynamics in integral membrane protein by hydroxyl radical footprinting. Mol Cell Proteomics. 2009;8:1999–2010. doi: 10.1074/mcp.M900081-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stadtman ER. Oxidation of free amino acids and amino acid residues in proteins by radiolysis and by metal-catalyzed reactions. Annu Rev Biochem. 1993;62:797–821. doi: 10.1146/annurev.bi.62.070193.004053. [DOI] [PubMed] [Google Scholar]
- 87.Stein G, Swallow AJ. Reduction by x- and gamma-rays of some substances of biological interest. Nature. 1954;173:937–939. doi: 10.1038/173937b0. [DOI] [PubMed] [Google Scholar]
- 88.Ebert M, Keene JP, Swallow AJ. Pulse radiolysis studies of substances of biological interest. J Physiol. 1968;197:48P. [PubMed] [Google Scholar]