

Abstract

A direct ruthenium-catalyzed radical chloroalkylation of N-acyl oxazolidinones capitalizing on valence tautomerism of titanium enolates has been developed. The chloroalkylation method served as the centerpiece in the enantioselective total synthesis of trichloroleucine-derived marine natural product neodysidenin.
Among more than four thousand halogenated natural products identified to date,1 neodysidenin and other trichloroleucine-derived marine metabolites comprise a unique group.2 For the majority of chlorinated natural products, a reasonable biosynthetic pathway involving an electrophilic chlorination can be proposed.3 On the other hand, the trichloromethyl group in neodysidenin and related compounds arises from a remarkable direct chlorination of the pro-R methyl group of l-leucine carried out by nonheme FeII halogenases requiring oxygen, chloride, and α-ketoglutarate for their activity.4 In contrast, availability of synthetic methods for stereoselective trichloromethylation is highly limited,5 whereas chlorinated natural products are attracting increasing attention as targets for chemical synthesis.6
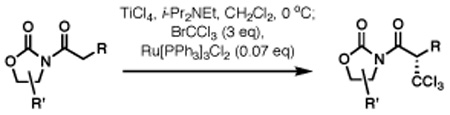
In this communication we describe a practical, efficient method for highly stereoselective direct chloroalkylation of titanium enolates and its application in the total synthesis of neodysidenin that can be readily adopted for the synthesis of other bioactive natural products in this class.7 Guided by an extension of the classic Kharasch reaction8 described by Eguchi and co-workers,9 our early efforts involved Ru(II)-catalyzed10 redox trichloromethylation of trimethylsilyl enol ethers generated from chiral N-acyl oxazolidinones such as 1 (Scheme 1).11 Although encouraging results were obtained with silyl ketene acetals (~50% yields, ds 3:1), the recent characterization of valence tautomerism in titanium enolates provided a conceptual foundation for the development of a direct radical chloroalkylation of N-acyl oxazolidinones. 12,13
Scheme 1.

Biradical character of titanium enolates in radical addition reactions catalyzed by ruthenium(II).

The unconventional biradical character of titanium enolates described by Moreira and co-workers suggests that these intermediates should be efficient radical acceptors.7 Indeed, when the Ti enolate derived from 114 was treated with BrCCl3 in the presence of [Ph3P]3RuCl2 as a readily available redox catalyst (7 mol%), product 2 was obtained in an essentially quantitative yield with exquisite stereocontrol (Scheme 1). The the mechanistic hypothesis in Scheme 1 is based on well-established redox activity of [Ph3P]3RuCl2 widely used in atom-transfer radical polymerization (ATRP)15 and is similar to that proposed by Eguchi for a related process with silyl enol ethers.9 A major advantage, however, is that the radical addition product should be stabilized by electron delocalization onto titanium, not feasible with silyl enol ethers. Thus, in the first step of the process, the CCl3 radical is formed upon an electron transfer from Ru(II) complex to BrCCl3. Addition of the electrophilic radical to biradical B should be particularly favorable because the product is a titanium (III) complex C, which is likely to be substantially more stable that a carbon-centered radical (such as D) expected from a radical addition to silyl enol ethers or other enolates. Reduction of Ru(III) species with the Ti(III) intermediate should regenerate the catalyst and provide the initial product as a Ti(IV) chelate E. In addition, the chelated nature of the titanium enolates ensures conformational rigidity required for stereocontrol.14
Next, a series of experiments were carried out to evaluate the scope of N-acyl oxazolidinone and of chloroalkylating components in this Ru-catalyzed process. Table 1 illustrates that functionalized N-acyl oxazolidinones are excellent substrates. Different Evans-type chiral auxiliaries can be used without loss of stereoselectivity, but the highest yields are achieved with the 5,5-dimethyl oxazolidinones.16 Aromatic, heterocyclic, and ethereal substituents are compatible with the reaction conditions. As evident from entry 5, a direct competition with Kharasch reaction reveals that the more sterically demanding titanium enolate is a superior trichloromethyl radical acceptor than a terminal double bond. With 3 equiv of BrCCl3, the bis-trichloromethylation product was isolated in a virtually quantitative yield. The stereoselectivity is generally very high; in no case were we able to observe or isolate the minor diastereomer.
Table 1.
Radical trichloromethylation: N-acyloxazolidinone scope a
 | |||
|---|---|---|---|
| entry | product (dr, yield)b | entry | product (dr, yield)b |
| 1 | 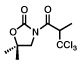 |
5 | 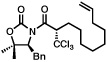 |
| 95% | 63% (91%, brsm), >98:2c | ||
| 2 | 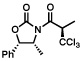 |
6 | 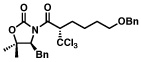 |
| 89%, >98:2 | 87%, >98:2 | ||
| 3 | 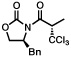 |
7 | 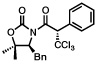 |
| 86%, >98:2 | 91%, >98:2 | ||
| 4 | 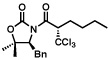 |
8 | 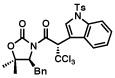 |
| 99%, >98:2 | 61%, >98:2 | ||
See Supporting Information for experimental details.
All yields are of isolated products, dr determined by 500 MHz 1H NMR of the crude mixture of products.
1.0 equiv of BrCCl3 was used, with 3 equiv the double addition product was isolated in 97% yield (dr >98:2)
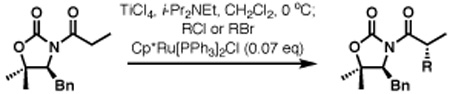
The scope of the haloalkylating reagent investigated in this study is illustrated in Table 2. For most of these examples, the more active commercially available ATRP redox catalyst Cp*Ru[PPh3]2Cl was required to attain optimal yields.15 Since a number of chlorinated natural products contain a 5,5-dichloroleucine subunit, it is notable that a direct dichloromethylation can be performed in useful yields using bromodichloromethane (Table 2, entry 2). A range of other functionalized haloalkylating reagents proved to be suitable with this catalyst, including dichloro- and trichloroacetates, 1,1,1-trichloroethane, 1-chloro-1-bromo-2,2,2-trifluoroethane, and Cl3CCH2OCO2Bn.. The remainder of the mass balance for the reactions in Table 2 is the substrate, with all yields based on recovered starting material exceeding 90%.
Table 2.
Radical haloalkylation: haloalkylating agent scope.
 | |||||
|---|---|---|---|---|---|
| entry | product | yield dra |
entry | product | yield dra |
| 1 | 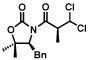 |
64% >98:2 |
4 | 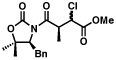 |
75% 1.3:1b |
| 2 | 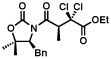 |
83% >98:2 |
5 | 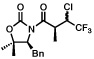 |
71% 1.6:1b |
| 3 | 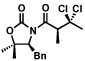 |
71% >98:2 |
6 | 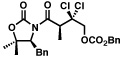 |
76% >98:2 |
All reported yields are of isolated products, dr was determined by 500 MHz 1H NMR.
at the indicated stereocenter
On the basis of this methodology, the total synthesis of neodysidenin through the intermediacy of (2S,4S)-5,5,5-trichloroleucine was accomplished (Scheme 2). Imide 2 was advanced to Ellman-type N-sufinimine 4 in 5 steps.17 Sccatalyzed Strecker synthesis with Me3SiCN followed by hydrolysis delivered (2S,4S)-5,5,5-trichloroleucine (82%, ds >98%).18 Using this modular approach, all stereoisomers of 5,5-di- or 5,5,5-trichloroleucine may be accessed in a straightforward manner. Subsequent N-acylation of the amino acid with reagent 5, also derived from nitrile 3, readily afforded dipeptide 6, which has been advanced to neodysidenin in one additional amidation step (EDC, HOAt, THF, 86% yield).19
Scheme 2.

Enantioselective total synthesis of neodysidenin.
In summary, a direct ruthenium-catalyzed radical chloroalkylation capitalizing on the valence tautomerism of titanium enolates has been developed. This method served as the centerpiece in the enantioselective total synthesis of trichloroleucine-derived marine natural product neodysidenin.
Supplementary Material
Acknowledgment
Support for this research was provided by Eli Lilly (Lilly Grantee Program) and the NIGMS (R01 GM077379). We thank Dr. Guang Wu, (UCSB), for the single-crystal X-ray analysis. We also thank Professor Molinski (UCSD) for helpful discussions.
Footnotes
Supporting Information Available:
Experimental procedures and copies of 1H and 13C NMR spectra. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Gribble GW. J. Chem. Educ. 2004;81:1441–1449. [Google Scholar]
- 2.(a) MacMillan JB, Trousdale EK, Molinski TF. Org. Lett. 2000;2:2721–2723. doi: 10.1021/ol006326u. [DOI] [PubMed] [Google Scholar]; (b) Unson MD, Rose CB, Faulkner DJ, Brinen LS, Steiner JR, Clardy J. J. Org. Chem. 1993;58:6336–6343. [Google Scholar]
- 3.(a) Butler A, Walker JV. Chem. Rev. 1993;93:1937–1944. [Google Scholar]; (b) Butler A. Curr. Opin. Chem. Biol. 1998;2:279–285. doi: 10.1016/s1367-5931(98)80070-7. [DOI] [PubMed] [Google Scholar]
- 4.(a) Galonic DP, Vaillancourt FH, Walsh CT. J. Am. Chem. Soc. 2006;128:3900–3901. doi: 10.1021/ja060151n. [DOI] [PubMed] [Google Scholar]; (b) Flatt PM, O’Connell SJ, McPhail KL, Zeller G, Willis CL, Sherman DH, Gerwick WH. J. Nat. Prod. 2006;69:938–944. doi: 10.1021/np050523q. [DOI] [PubMed] [Google Scholar]
- 5.Non-stereoselective radical trichloromethylations of silyl enol ethers and ketene acetals have been described.: Sugimoto J, Miura K, Oshima K, Utimoto K. Chem. Lett. 1991:1319–1322. Mitani M, Sakata H, Tabei H. Bull. Chem. Soc. Jpn. 2002;75:1807–1814.
- 6.(a) Shibuya GM, Kanady JS, Vanderwal CD. J. Am. Chem. Soc. 2008;130:12514–12518. doi: 10.1021/ja804167v. [DOI] [PubMed] [Google Scholar]; (b) Nilewski C, Geisser RW, Carreira EM. Nature. 2009;457:573–576. doi: 10.1038/nature07734. [DOI] [PubMed] [Google Scholar]; (c) Bedke DK, Shibuya GM, Pereira A, Gerwick WH, Haines TH, Vanderwal CD. J. Am. Chem. Soc. 2009;131:7570–7572. doi: 10.1021/ja902138w. [DOI] [PubMed] [Google Scholar]
- 7.Recent examples: Sadar MD, Williams DE, Mawji NR, Patrick BO, Wikanta T, Chasanah E, Irianto HE, Soest RV, Andersen RJ. Org. Lett. 2008;10:4947–4950. doi: 10.1021/ol802021w.
- 8.(a) Kharasch MS, Jensen EV, Urry WH. Science. 1945;102:128. doi: 10.1126/science.102.2640.128. [DOI] [PubMed] [Google Scholar]; (b) Kharasch MS, Reinmuth O, Urry UH. J. Am. Chem. Soc. 1947;69:1105–1110. [Google Scholar]
- 9.Okano T, Shimizu T, Sumida K, Eguchi S. J. Org. Chem. 1993;58:5163–5166. [Google Scholar]
- 10.First application of a Ru(II) complex for the generation of CCl3 radicals: Matsumoto H, Nakano T, Nagai Y. Tetrahedron Lett. 1973;14:5147–5150.
- 11.Besides the control of stereoselectivity, an important function of the chiral auxiliary is to suppress elimination of HCl in products in avoidance of A1,3-strain. The HCl elimination is a common problem in related methods (ref. 5, 9).
- 12.Moreira I de PR, Bofill JM, Anglada JM, Solsona JG, Nebot J, Romea P, Urpi F. J. Am. Chem. Soc. 2008;130:3242–3243. doi: 10.1021/ja076625f. [DOI] [PubMed] [Google Scholar]
- 13.A related trifluoromethylation process using photoactivated Ru(II) and Ir(I) catalysis with enamine intermediates has been recently reported: Nagib DA, Scott ME, MacMillan DWC. J. Am. Chem. Soc. 2009;131:10875–10877. doi: 10.1021/ja9053338.
- 14.(a) Evans DA, Upri F, Somers TC, Clark JS, Bilodeau MT. J. Am. Chem. Soc. 1990;112:8215–8216. [Google Scholar]; (b) Sibi MP, Sausker JB. J. Am. Chem. Soc. 2002;124:984–991. doi: 10.1021/ja016839b. [DOI] [PubMed] [Google Scholar]
- 15.Recent review: Severin K. Curr. Org. Chem. 2006;10:217–224.
- 16.Davies SG, Sanganee HJ. Tetrahedron: Asymmetry. 1999;6:671–674. [Google Scholar]
- 17.Ellman JA, Owens TD, Tang TP. Acc. Chem. Res. 2002;35:984–995. doi: 10.1021/ar020066u. [DOI] [PubMed] [Google Scholar]
- 18.Mabic S, Cordi AA. Tetrahedron. 2001;57:8861–8866. [Google Scholar]
- 19.Both diastereomers at C13 of neodysidenin have been prepared. Physical data for only the 13S isomer fully matched those reported for the natural product, resulting in the reassignment of configuration at C13 [natural: [a]D23 − 52.1° (0.165 CHCl3) ref. 2a; 13R: [a]D23 +26.1° (0.50, CHCl3); 13S: [a]D23 − 62.3° (0.20 CHCl3)]. Thus, neodysidenin and pseudodysidenin appear to be identical: Jiménez JI, Scheuer PJ. J. Nat. Prod. 2001;64:200–203. doi: 10.1021/np000462q. See Supporting Information for details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.