

Abstract
Ablation of preBötzinger complex (preBötC) neurons, critical for respiratory rhythm generation, resulted in a progressive, increasingly severe disruption of respiratory pattern, initially during sleep and then also during wakefulness in adult rats. Sleep-disordered breathing is highly prevalent in elderly humans and in some patients with neurodegenerative disease. We propose that sleep-disordered breathing results from loss of preBötC neurons and could underlie death during sleep in these populations.
Breathing is an essential regulatory behavior, continuously active throughout wakefulness and sleep from birth until death. Compared to breathing during wakefulness, however, breathing during sleep is fragile, often disordered and frequently interrupted by apneic episodes. Sleep-disordered breathing (SDB) can result in intermittent hypoxia, the cumulative effect of which includes loss of brain gray matter, impaired cognitive function and increased mortality. Most instances of SDB in children and adults are obstructive apneas, characterized by narrowing or obstruction of the airways. Less common in young adults but increasingly prevalent with age are central sleep apneas (during both rapid eye movement (REM) and non-REM (NREM) sleep)1, which are characterized by loss of respiratory rhythm.
An essential component of the respiratory circuitry in rodents is the preBötC, a small region of the ventrolateral medulla2,3. In the rat, approximately 300 preBötC neurons per side express high levels of the neurokinin 1 receptor (NK1R)4. Ablation of > 80% of preBötC NK1R neurons in adult rats leads to an ataxic breathing pattern during wakefulness5 that, in contrast to SDB, is rarely seen in humans. We hypothesized that a less extensive disruption of preBötC function could underlie SDB.
Rats were implanted with electromyographic (EMG) electrodes (diaphragmatic, abdominal and neck) and electroencephalographic (EEG) electrodes (see Supplementary Methods online). All experimental procedures were approved by the UCLA Chancellor’s Animal Research Committee. Inspiratory amplitude, respiratory frequency and disturbances on day 10 after electrode implantation surgery did not differ from control data before electrode implantation (sleep-wake cycle: ~ 3–4 min; inspiratory amplitude: 1.02 ± 0.03 versus 1.00 ± 0.01 arbitrary units (a.u.) for after implantation and before implantation, respectively; frequency: 105 ± 5.4 versus 109 ± 7.3 breaths/min; respiratory disturbances: 3.6 ± 0.9 versus 3.8 ± 1.2 episodes/h; n = 10; P > 0.05; Fig. 1a). Fourteen days after electrode implantation, either the toxin saporin conjugated to substance P (SP-SAP), which selectively ablates neurons expressing NK1R5 (n = 8), or substance P mixed with saporin (unconjugated), which does not ablate neurons (n = 4), was injected bilaterally into the preBötC. In rats injected with substance P mixed with saporin, breathing returned to pre-injection levels by day 3 post-injection (inspiratory amplitude: 1.02 ± 0.02 a.u; frequency: 110 ± 6.5 breaths/min; n = 4; P > 0.05) and respiratory disturbances at day 10 did not significantly differ from data collected pre-injection (wakefulness: 1.2 ± 0.12, NREM: 3.6 ± 0.12, REM: 2.9 ± 2.0 episodes/h; P > 0.05).
Figure 1.
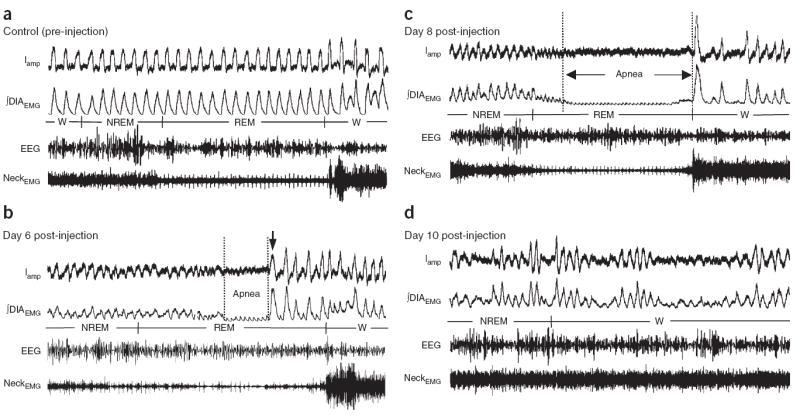
Breathing pattern is progressively disrupted, initially during sleep and then also during wakefulness. (a) Pre-injection, breathing is regular during sleep and wakefulness (W). (b) Post-injection, hypopnea and short central apneas (note the absence of ∫DIAEMG) occur initially during REM sleep. At day 6, at the end of the REM-associated apnea, a number of small breaths precede awakening (arrow). (c) Hypopnea and short central apneas subsequently increase in frequency and duration during NREM and REM sleep (day 8; Supplementary Video 1) and also during wakefulness. (d) Ultimately, an ataxic breathing pattern develops (day 10). Iamp: inspiratory amplitude. ∫DIAEMG: integrated diaphragm EMG. NeckEMG: dorsal neck EMG. EEG: electroencephalogram. Scale bar, 2 s. See Supplementary Figures 2–6 for continuous 5-min data recordings.
In contrast, in rats injected with SP-SAP at day 4 post-injection, respiratory disturbances significantly increased in number and duration only during REM sleep (66.0 ± 13.8 episodes/h, 5.9 ± 1.1 s; n = 5; P < 0.004; Fig. 2). Disturbances were characterized by markedly reduced ventilation (that is, hypopnea), occasionally leading to a short period with neither airflow nor diaphragmatic EMG inspiratory activity (that is, central apnea). However, during NREM and wakefulness, breathing was similar to that seen pre-injection (NREM 4.2 ± 0.6 episodes/h, 4.0 ± 1.4 s, wakefulness: 5.4 ± 3.6 episodes/h, 3.2 ± 0.5 s; n = 5; P > 0.05; Fig. 2). Blood gases during wakefulness did not differ from pre-injection levels (PCO2: 35.2 ± 1.5 versus 37.6 ± 2.3 mm Hg for post-injection and pre-injection, respectively; PO2: 94.4 ± 2.2 versus 96.0 ± 1.3 mm Hg; n = 3; P > 0.05).
Figure 2.
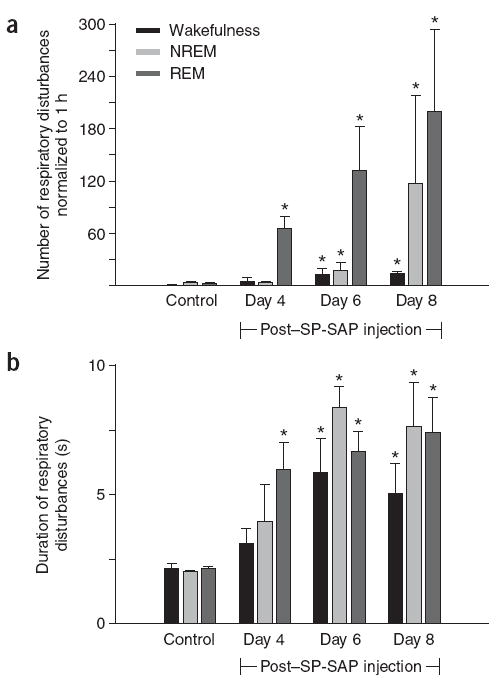
Number and duration of respiratory disturbances (apneas and hypopneas). (a) Respiratory disturbances significantly increase during REM sleep on day 4 post-injection, with no significant change during NREM and wakefulness (mean ± s.e.m.; n = 5). From day 6, respiratory disturbances increase in all three states. (b) Duration of respiratory disturbances increases in all three states up to day 6 post-injection.
* P < 0.05 compared with control.
On days 5–6 after SP-SAP injection, respiratory disturbances during REM further increased in frequency and duration (132.0 ± 50.4 episodes/h, 6.7 ± 0.8 s; n = 5; P < 0.002; Figs. 1b and 2), and during NREM there was a smaller but significant increase (19.2 ± 8.4 episodes/h, 8.3 ± 0.7 s; n = 5; P < 0.05; Figs. 1b and 2). A typical respiratory disturbance began with hypopnea during NREM that continued into REM, when apnea(s) occurred (Fig. 1b), and ended with several small breaths that preceded awakening (arrow in Fig. 1b). During wakefulness, short periods of increased frequency (194 ± 21 breaths/min; P < 0.05) and brief apneas (13.8 ± 6.0 episodes/h, 5.8 ± 1.3 s; n = 5; P < 0.05) occurred; inspiratory amplitude did not significantly change (1.00 ± 0.09 a.u; n = 5; P > 0.05; Figs. 1b and 2). Blood gases were slightly altered during wakefulness (PCO2: 38.7 ± 1.6 mm Hg; PO2: 89.7 ± 2.4 mm Hg; n = 3; P > 0.05).
From day 7 after SP-SAP injection, rats were rarely able to complete a sleep period without a marked respiratory disturbance (Figs. 1c and 2; Supplementary Video 1). During NREM, respiratory disturbances, characterized by hypopnea, noticeably increased in frequency but not duration (117.6 ± 100.8 episodes/h, 7.6 ± 1.7 s; n = 5), with apnea developing upon transition to REM (199.8 ± 94.2 episodes/h, 7.4 ± 1.4 s; n = 5) and breathing resuming only upon waking (Fig. 1c). This resulted in a highly fragmented sleep pattern and decreased total sleep time compared with that seen pre-injection (Fig. 3). Before day 7, arousals were a gentle transition to wakefulness, with slight head movement and very little body movement; post day 7, the rat abruptly awakened from each apnea and moved around the plethysmograph several times before settling down. During wakefulness, breathing became increasingly irregular, with high-frequency breathing (frequency: 176 ± 16.9 breaths/min; n = 5; P < 0.05; inspiratory amplitude: 1.05 ± 0.07 a.u; n = 5; P > 0.05) interspersed with hypopnea and short apneas (15.0 ± 2.4 episodes/h, 5.1 ± 1.1 s; n = 5; P < 0.05). Rats were hypoxic and hypercapnic during wakefulness (PCO2: 40.2 ± 1.7 mm Hg; PO2: 76.7 ± 3.2 mm Hg; n = 3; P < 0.05).
Figure 3.
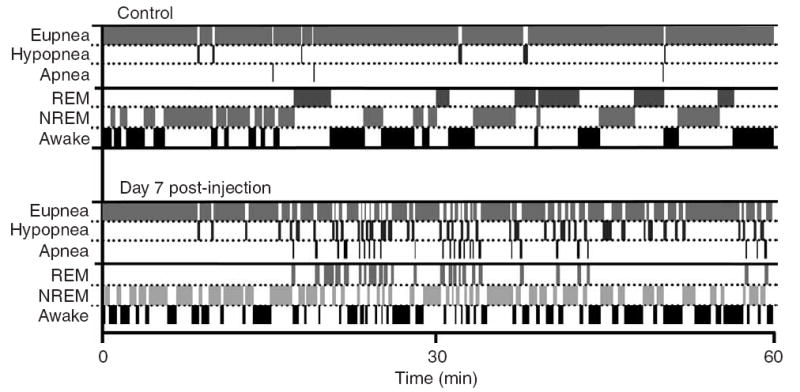
Respiration and sleep become highly fragmented. In the control (pre-injection) period (top), short respiratory disturbances occur infrequently during sleep. On day 7 post-injection, respiratory disturbances occur during REM and NREM, accounting for the highly fragmented sleep pattern and decreased total sleep time. Respiratory disturbances are also more frequent during wakefulness. Data analyzed and plotted in 5-s epochs for one continuous hour of recording.
Beginning on day 9–10 after SP-SAP injection, a severely ataxic breathing pattern developed during wakefulness5, consisting of an irregular sequence of high-frequency, large breaths (inspiratory amplitude: 1.32 ± 0.24 a.u; frequency: 206 ± 19 breaths/min; P < 0.05 compared with pre-injection) interspersed with numerous hypopneas and apneas (18.0 ± 6.0 episodes/h, 4.9 ± 0.1 s; n = 5; Fig. 1d). Breathing terminated immediately upon sleep onset and did not restart until the rat (abruptly) awakened. In two cases, a periodic Cheyne-Stokes–like pattern was intermingled with the ataxic breathing pattern.
The accumulating loss of preBötC neurons led to progressive disturbances in breathing that proceeded in clearly demarcated stages, leading to an ataxic breathing pattern only when >80% of these neurons were ultimately ablated5 (as determined by histological analysis after day 10 post-injection). Lesions were confined to the preBötC5. NK1R staining was significantly reduced or completely absent in histological sections representing ~ 15% of the preBötC (2 ± 1.3 NK1R+/side; n = 5; P < 0.05; Supplementary Fig. 1) compared with non-injected controls (39 ± 7 NK1R+/side; Supplementary Fig. 1) or controls injected with substance P mixed with saporin (32 ± 5 NK1R+/side). In the surrounding rectangular area, NK1R staining was reduced (35 ± 15 NK1R+/side) compared with non-injected controls (76 ± 11 NK1R+/side; Supplementary Fig. 1) or controls injected with substance P mixed with saporin (70 ± 6 NK1R+/ side; Supplementary Fig. 1).
In vivo, neuronal death after SAP-induced lesions occurs over a period of days; here, preBötC lesions caused repeated episodes of hypoxia that would likely have resulted in further neuronal loss. This effect was likely minor during the early stages of SDB (days 3–6) because the disturbances were limited, and blood gases taken during wakefulness were normal. Beyond day 6, however, the cumulative effect of intermittent hypoxia resulting from prolonged apneas could have induced neuronal death (Supplementary Note) not necessarily restricted to the preBötC, further disrupting respiratory pattern.
In both rats6 and humans (particularly the elderly)1, breathing is more irregular and spontaneous central apneas are most frequent during REM sleep. Notably, neurons that release serotonin or norepinephrine are relatively inactive during REM7. As serotonin and norepinephrine excite preBötC neurons, any decrease in their release would disfacilitate preBötC neurons, rendering breathing during REM vulnerable to preBötC insults (for instance, SP-SAP–induced lesions or neurodegeneration). Thus, as preBötC dysfunction progresses, disturbances in breathing would be expected initially in REM, next in NREM, and, if sufficiently severe, even during wakefulness.
SDB is common and can go unnoticed in patients in later stages of amyotrophic lateral sclerosis (ALS)8, multiple systems atrophy (MSA)9 or Parkinson disease10, whereas breathing during wakefulness is considered otherwise normal (Supplementary Note). Many of these patients die during sleep8-10. In the ventrolateral medulla (including the presumptive preBötC), NK1R neurons are depleted by ~60% in individuals with Parkinson disease and by ~89% in individuals with MSA11, suggesting substantial damage to the preBötC. In ALS, neurons with low levels of Ca2+ buffers, in particular motoneurons, are particularly vulnerable to degeneration12. In the ventrolateral respiratory column3, the only neurons lacking significant levels of Ca2+ buffers are preBötC neurons13, suggesting that preBötC neurons are also vulnerable. If preBötC neurons were to degenerate, then significant disturbances of breathing during sleep would be expected. The slow cumulative loss (compared with loss induced by SP-SAP) of preBötC neurons in these diseases could lead to extended, recurring sleep apneas with consequent intermittent hypoxias, with seemingly normal breathing during wakefulness. This SDB will likely cause an increase in the threshold for arousal from apnea14, leading to a vicious cycle in which central apneas become more frequent and longer. Additionally, SP-SAP–induced preBötC NK1R neuron ablation in rats significantly reduces the frequency of sighs (data not shown) and blunts regulatory responses to hypoxia and hypercapnea5; in humans this could further increase the arousal threshold14. Ultimately, a central sleep apnea not terminated by arousal could result in anoxia and death. We suggest that this sequence of events may also occur in the elderly, in whom the prevalence of central sleep apnea rises precipitously with age1, from the cumulative loss of the small number of preBötC neurons over a lifetime. If this occurs, death due to preBötC damage would happen before neuronal loss becomes sufficiently extensive to substantially alter breathing during wakefulness, as ataxic breathing in humans is rare. Furthermore, during sleep apneas, the induced hypoxia often results in exceptionally high systolic blood pressures that can induce fatal cardiovascular or cerebrovascular incidents15. We suggest that in individuals with preBötC neuron loss and with significantly compromised health, respiratory failure during sleep is the precipitating cause of death.
Supplementary Material
Acknowledgments
We are grateful to J.M. Siegel for his insightful advice; we thank G.S. Mitchell, C.A. Del Negro, L. Kruger, N. Dale and P.A. Gray for comments on the manuscript and G. Li for histological assistance. This work was supported by US National Institutes of Health grant HL70029.
Footnotes
COMPETING INTERESTS STATEMENT The authors declare that they have no competing financial interests.
Note: Supplementary information is available on the Nature Neuroscience website.
References
- 1.Krieger J, Turlot JC, Mangin P, Kurtz D. Sleep. 1983;6:108–120. doi: 10.1093/sleep/6.2.108. [DOI] [PubMed] [Google Scholar]
- 2.Smith JC, Ellenberger HH, Ballanyi K, Richter DW, Feldman JL. Science. 1991;254:726–729. doi: 10.1126/science.1683005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feldman JL, Mitchell GS, Nattie EE. Annu Rev Neurosci. 2003;26:239–266. doi: 10.1146/annurev.neuro.26.041002.131103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gray PA, Rekling JC, Bocchiaro CM, Feldman JL. Science. 1999;286:1566–1568. doi: 10.1126/science.286.5444.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gray PA, Janczewski WA, Mellen N, McCrimmon DR, Feldman JL. Nat Neurosci. 2001;4:927–930. doi: 10.1038/nn0901-927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mendelson WB, et al. Physiol Behav. 1988;43:229–234. doi: 10.1016/0031-9384(88)90243-0. [DOI] [PubMed] [Google Scholar]
- 7.Siegel JM. J Clin Psychiatry. 2004;65(suppl):4–7. [PMC free article] [PubMed] [Google Scholar]
- 8.Ferguson KA, Strong MJ, Ahmad D, George CF. Chest. 1996;110:664–669. doi: 10.1378/chest.110.3.664. [DOI] [PubMed] [Google Scholar]
- 9.Munschauer FE, Loh L, Bannister R, Newsom-Davis J. Neurology. 1990;40:677–679. doi: 10.1212/wnl.40.4.677. [DOI] [PubMed] [Google Scholar]
- 10.Maria B, et al. Respir Med. 2003;97:1151–1157. doi: 10.1016/s0954-6111(03)00188-4. [DOI] [PubMed] [Google Scholar]
- 11.Benarroch EE, Schmeichel AM, Low PA, Parisi JE. Brain. 2003;126:2183–2190. doi: 10.1093/brain/awg220. [DOI] [PubMed] [Google Scholar]
- 12.Alexianu ME, et al. Ann Neurol. 1994;36:846–858. doi: 10.1002/ana.410360608. [DOI] [PubMed] [Google Scholar]
- 13.Alheid GF, Gray PA, Jiang MC, Feldman JL, McCrimmon DR. J Neurocytol. 2002;31:693–717. doi: 10.1023/a:1025799830302. [DOI] [PubMed] [Google Scholar]
- 14.Berry RB, Gleeson K. Sleep. 1997;20:654–675. doi: 10.1093/sleep/20.8.654. [DOI] [PubMed] [Google Scholar]
- 15.Leung RS, Bradley TD. Am J Respir Crit Care Med. 2001;164:2147–2165. doi: 10.1164/ajrccm.164.12.2107045. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.