

Abstract
Colorectal cancer is one of the most prevalent cancers of humans. To experimentally investigate this common disease, numerous murine models have been established. These models accurately recapitulate the molecular and pathological characteristics of human colorectal cancer including activation of MYC, which has recently been suggested to be a key mediator of colorectal cancer development. This review focuses on the variety of murine models of human colorectal cancer that are available to the research community and on their use to identify common and distinct characteristics of colorectal cancer.
Keywords: murine models, colorectal cancer, review, WNT, TGFB
Introduction
Colorectal cancer (CRC) is expected to account for approximately 149,000 new cases and 50,000 deaths in the United States in 2008 (American Cancer Society statistics). Although overall five-year survival rates for CRC increased between 1975 and 2003, nearly 90% of individuals live longer than five years if diagnosis with localized CRC but only 10% if diagnosed with metastatic disease. A major clinical challenge that will improve survival from CRC is early detection. Similarly, improved treatments for metastatic CRC are needed for those cases not detected early. New approaches for CRC detection and treatment should be accelerated through insights gained from studying current and future murine models of human CRC.
Many genetic and carcinogen-based murine models have been developed that recapitulate human CRC through alteration of a variety of signaling pathways (Table 1). Most CRC models have been generated by mutating murine orthologs of human CRC genes or by discovering models that spontaneously develop cancers during unrelated studies. Two of the most highly studied signaling pathways implicated in human CRC are the WNT/CTNNB1 (wingless-related MMTV integration site/beta-catenin) and TGFB (transforming growth factor beta) pathways. The most commonly used models perturb the WNT/CTNNB1 (wingless-related MMTV integration site/beta-catenin) signaling pathway. Components of the WNT/CTNNB1 signaling pathway, originally discovered in Drosophila (Nüsslein-Volhard and Wieschaus 1980), are dysregulated in the majority of human CRCs. The predominant initiating mutation in human CRC occurs in the gene encoding the adenomatous polyposis coli (APC) tumor suppressor (Powell et al. 1992). Loss of APC function in the presence of active WNT signaling results in increased nuclear levels of the transcriptional co-activator CTNNB1. This in turn activates transcription of pro-cell cycle molecules like JUN resulting uncontrolled cell division (Mann et al. 1999).
Table 1.
Murine models of human colorectal cancer.
| Strain | Tumor Type | Location | Multiplicity | Reference | |
|---|---|---|---|---|---|
| WNT pathway | |||||
| Apc | |||||
| ApcMin | C57BL/6J | adenoma | small intestine | 3-58 | (Moser et al. 1990) |
| ApcMin | BTBR/Pas | adenoma | small intestine | 600 | (Kwong et al. 2007) |
| Apc1638 | C57BL/6J | adenoma/carcinoma | small intestine, colon | 4 | (Fodde et al. 1994) |
| ApcΔ716 | C57BL/6J | adenoma | small intestine, colon | 254 | (Oshima et al. 1995) |
| Azoxymethane | A/J | adenoma/carcinoma | distal colon | 36.4 | (Papanikolaou et al. 1998) |
| Azoxymethane | SWR/J | adenoma/carcinoma | distal colon | 16.3 | (Papanikolaou et al. 1998) |
| Azoxymethane | AKR/J | adenoma/carcinoma | distal colon | 0.12 | (Papanikolaou et al. 1998) |
| ApcMin | C57BL/6J | adenoma | small intestine | 5.7 | (Shao et al. 2007) |
| ApcMin, Pten+/- | 129/C57/BL6 | adenoma/carcinoma | small intestine | 22 | (Shao et al. 2007) |
| ApcMin | C57BL/6J | adenoma | small intestine | 18 | (Ghaleb et al. 2007) |
| Klf4-/+, ApcMin | C57BL/6J | adenoma | small intestine | 29 | (Ghaleb et al. 2007) |
| ApcMin | C57BL/6J | adenoma | small intestine | 39 | (Batlle et al. 2005) |
| Ephb2Δcy, ApcMin | C57BL/6J | carcinoma | small intestine | 13 | (Batlle et al. 2005) |
| ApcMin | C57BL/6J | adenoma | colon | 1 | (Batlle et al. 2005) |
| Ephb2Δcy, ApcMin | C57BL/6J | carcinoma | colon | 11 | (Batlle et al. 2005) |
| Pirc | F344/NTac | carcinoma | small intestine, colon | 36 | (Amos-Landgraf et al. 2007) |
| Mismatch repair | |||||
| Mlh1-/- | C57BL/6J/129/Ola (mixed) | adenoma, carcinoma | stomach, small intestine, colon | 1.1 | (Edelmann et al. 1999) |
| Mlh1-/-, Apc1638/N | Ola | adenoma, carcinoma | stomach, colon | 45.1 | (Edelmann et al. 1999) |
| Msh2-/- | C57BL/6J/129/Ola (mixed) | adenoma, carcinoma | small intestine, colon | 2.6 | (Reitmair et al. 1996) |
| Msh2-/-, ApcMin | C57BL/6J/129/Ola (mixed) | adenoma | small intestine, colon | 333 | (Reitmair et al. 1996) |
| Msh6-/- | C57BL/6J/129/Sv/SJL/J (mixed) | adenoma, carcinoma | small intestine | 0.6 | (Edelmann et al. 1997) |
| Msh3-/-, Msh6-/- | C57BL/6J/129/Sv/SJL/J (mixed) | adnoma, carcinoma | small intestine, colon | 2.75 | (Edelmann et al. 2000) |
| Non-WNT pathway | |||||
| Tgfb | |||||
| Tgfb1-/-, Rag2-/- | 129S6 × CF1 | adenoma/carcinoma | cecum, colon | NR | (Diebold et al. 1995) |
| Smad3-/- | 129/Sv | carcinoma | colon | 3.8 | (Zhu et al. 1998) |
| Inflammation | |||||
| DSS | CBA/J & BALB/C | high grade dysplasia | colon | NR | (Okayasu et al. 1990) |
| AOM/DSS | CD-1 | adenoma/carcinoma | distal colon | 5.8 | (Tanaka et al. 2003) |
| Modifiers of inflammation | |||||
| Socs1-/- | NR | adenoma/carcinoma | proximal colon | NR | (Garlanda et al. 2007; Hanada et al. 2006) |
| Socs3-/- | C57BL/6J | adenoma | colon | NR | (Rigby et al. 2007) |
| Sigirr-/- | C57BL/6J × 129/SvJ | adenoma/carcinoma | distal colon | 17 | (Xiao et al. 2007) |
NR, not reported
The second most widely studied pathway implicated in the progression of CRC is the TGFB pathway. Upon activation of TGFB receptors (TGFBR1, 2 and 3), intracellular SMAD2 and SMAD3 signal mediators become phosphorylated, bind to SMAD4 and translocate to the nucleus where the complex interacts with other transcription factors to induce down stream targets (Blobe et al. 2000). In the normal colonic epithelium and in early stages of tumorigenesis, TGFB functions as a tumor suppressor by inhibiting the cell cycle through up-regulation of CDNK1A and CDNK2B coding for cyclin-dependent protein kinase inhibitors (Derynck et al. 2001). Unlike the WNT/CTNNB1 pathway, perturbation of TGFB signaling is a later event in the process of carcinogenesis; up-regulation of this pathway is associated with increased tumor invasion and metastasis (Grady et al. 1998).
Although CRCs are heterogeneous, similarities across CRCs with different etiologies and between species are becoming apparent. Comparison of WNT and non-WNT mediated murine models implicated MYC as a key mediator of CRC, thereby linking seemingly independent pathways (Hanada et al. 2006; Kaiser et al. 2007; Rigby et al. 2007; Sansom et al. 2007). A similar role for MYC during human CRC development is also suggested from its widespread up-regulation in human CRCs (Hanada et al. 2006; Kaiser et al. 2007; Rigby et al. 2007; Sansom et al. 2007). Although murine models have been established that model the early stages of CRC, less progress has been achieved in establishing models that accurately recapitulate the later stages of invasion and metastasis.
In keeping with the goal of providing a concise narrative, we have chosen to highlight a handful of pertinent examples of murine models which have provided insight into the mechanisms of CRC development and that hold promise in informing clinical intervention of human CRC.
WNT pathway-mediated models of CRC
APC models
The ApcMin (multiple intestinal neoplasia allele of the adenomatosis polyposis coli gene) mouse model of human familial adenomatous polyposis (FAP) is the most widely used CRC model for studying tumor initiation and early progression. The ApcMin model, originally induced and fortuitously identified in a mutagenesis program (Moser et al. 1990; Su et al. 1992), bears one functional copy of the tumor suppressor Apc gene. Upon loss of the remaining wildtype copy of Apc, CTNNB1 is stabilized and transported to the nucleus where it functions as a transcriptional co-activator with the LEF/TCF family of transcription factors to stimulate cell cycle progression (Morin et al. 1997). This loss of growth control results in the development of tens-to-hundreds of polyps in the small intestine and a small number of polyps in the colon, while a similar mutation in humans results in predominantly colonic polyps (Groden et al. 1991).
Since discovery of the ApcMin allele, other mutant alleles have been described. Gene targeting was used to generate ApcΔ716 and Apc1638 alleles that display polyp distributions similar to the ApcMin mouse (Fodde et al. 1994; Oshima et al. 1995). However, the Apc1638 model develops significantly fewer polyps than ApcMin, while the ApcΔ716 model develops more. Liver metastasis has been reported using the APC1638 model, possibly due to the notably longer lifespan of these mice compared to other Apc-mediated models.
The observation that CRC predisposition in ApcMin mice is strain dependent led to the discovery of genetic loci called ‘Modifiers of Min’ (Mom) that modulate CRC susceptibility (Dietrich et al. 1993). The first Mom candidate gene cloned, secretory phospholipase A2 (Pla2g2a), was for Mom1 (MacPhee et al. 1995). Subsequently, Mom1 was found to be more complex, with at least two distinct genes contributing to the effect of Mom1 on ApcMin (Cormier et al. 2000). Since the discovery of Mom1, over a dozen additional Mom loci have been genetically mapped (McCart et al. 2008).
While the genes underlying most Mom loci have not been identified, other modifiers of ApcMin-mediated CRC have been proposed using crosses with mice carrying engineered or spontaneous mutations in specific genes. Haploinsufficiency of the Krüppel-like factor 4 (Klf4) transcription factor or reduction in EGFR activity using the Egfrwa2 hypomorphic allele combined with loss of Apc enhances and suppresses, respectively, multiplicity in the small intestine and colon while showing no role in subsequent tumor progression (Ghaleb et al. 2007; Roberts et al. 2002; Torrance et al. 2000). In contrast, ApcMin mice carrying mutations in the Pten tumor suppressor gene or the Ephb2 gene for a guidance receptor show enhanced tumor progression (Batlle et al. 2005; Shao et al. 2007). Loss of Ephb2 in mice also coincides with a shift from small intestinal adenomas to advanced colon carcinomas, potentially providing clues to the disparity in tumor distribution between humans with FAP and mice carrying ApcMin (Batlle et al. 2005).
Although Apc mutant mice have been invaluable in modeling human CRC, there remain several aspects of the human disease that are not recapitulated well with these models. Rodent colonoscopy, a technique gaining in importance since it permits longitudinal studies of colon tumors (Becker et al. 2006), is not well suited for Apc mutant mice since they develop tumors predominantly in the small intestine. Additionally, Apc mutant mice rarely develop metastases to distant organs, which is the most clinical important aspect of human CRC.
The generation of gene-specific mutations in rat by N-ethyl-N-nitrosourea (ENU) mutagenesis provides a new avenue for modeling human CRC (Zan et al. 2003). Recently, the Pirc (polyposis in rat colon) rat model of human CRC was generated that has a mutation in Apc (Amos-Landgraf et al. 2007). Unlike the ApcMin mouse model where the incidence of colon tumors is low, the Pirc model develops colon tumors with 100% incidence by four months of age. The Pirc model opens opportunities to perform experimental studies that are difficult in mice. Apc mutant mice can develop invasive cancer but typically have shortened life spans due to intestinal blockage (Boivin et al. 2003). Conversely, rats are less susceptible to intestinal blockage by tumors because of their larger intestinal diameters. In rats of at least six months of age, twenty percent of tumors in the Pirc model become invasive (Amos-Landgraf et al. 2007). Although no distant metastases were reported in the initial analysis of the Pirc model, metastasis of CRCs to the liver has been reported in a rat azoxymethane (AOM) carcinogen model (Nordlinger et al. 1991).
Mismatch repair (MMR) deficient models
Hereditary non-polyposis colon cancer (HNPCC) is an inherited condition in which inactivation of one of several DNA mismatch repair (MMR) genes, like MLH1, MSH2, MSH6, and PMS2, result in defective DNA repair (Lynch and de la Chapelle 2003). In humans this leads to the development of a variety of cancers including that of the colon, endometrium, ovary, and stomach (Lynch and de la Chapelle 2003). A number of mutant mouse lines have been generated to model the loss of function of MMR genes in humans. Mice deficient for Mhl1, Msh2 and Msh6 develop cancers of the stomach, small intestine and colon. However, due to the inherent nature of defective MMR machinery, these mice also develop cancers of the lymphatic system, skin, cervix and lung (Edelmann et al. 2000; Edelmann et al. 1999; Edelmann et al. 1997; Reitmair et al. 1996).
Analysis of MMR deficient mice carrying one functional copy of Apc showed that loss of normal MMR results in a high percentage of Apc-inactivating mutations and an elevated frequency of tumor development (Kuraguchi et al. 2001; Reitmair et al. 1996). Mice deficient for Mlh1 and heterozygous for the Apc1638N allele have a 40-fold increase in stomach and colon tumors compared to Apc1638N mice alone (Edelmann et al. 1999). Similarly, mice lacking Msh2 have accelerated development of tumors in ApcMin mice, with increased colon adenoma number and size suggesting roles for MSH2 in both tumor initiation and progression. Although loss of Msh3 does not lead to increased cancer predisposition until late in life, loss of Msh3 and Msh6 together results in an increase in gastrointestinal tumors at a much younger age, similar to mice deficient for either Mlh1 or Msh2 (Edelmann et al. 2000).
Carcinogen-induced models
While genetic models have proven useful in the investigation of cancer mechanisms, particularly for familial cancers such as FAP or HNPCC, most human CRCs are non-familial and occur sporadically. The colon-specific carcinogen dimethylhyrdrazine (DMH), as well as the down-stream metabolite AOM, has proven useful in the investigation of the molecular mechanisms underlying the development of non-familial CRCs (Druckrey et al. 1967). Mice exposed to DMH or AOM develop colorectal tumors that accurately recapitulate pathologies seen in human CRC (Kaiser et al. 2007; Uronis et al. 2007). Consistent with the ApcMin model, AOM-induced tumors result from activation of the WNT/CTNNB1 pathway (Takahashi et al. 2000). Unlike Apc-mediated models, AOM-induced tumors are primarily caused by mutations in Ctnnb1, which results in ubiquitination-resistant CTNNB1 and development of colorectal adenomas with increased expression of the key cell cycle regulators cyclin D1 (Ccnd1) and Myc (Kaiser et al. 2007; Wang et al. 1998).
Similar to the Apc-based models, modifier loci have also been identified using the DMH or AOM carcinogen models. Ptprj (a receptor-type protein tyrosine phosphatase) was shown to modify susceptibility to DMH (Ruivenkamp et al. 2002), although the significance of the orthologous gene in human CRC has not be elucidated. Although no germline PTPRJ mutations have been reported, PTPRJ frequently shows loss of heterozygosity in human colon cancers (Ruivenkamp et al. 2002). Additionally, Pref1, up-regulated in response to AOM in the distal colon of tumor susceptible A/J mice but not resistant AKR/J mice, was identified using the AOM carcinogen model (Dong et al. 2004). A direct link to WNT/CTNNB1 signaling was suggested through a putative CTNNB1/TCF response element in the promoter of Pref1 (Ruivenkamp et al. 2002).
More recently, the AOM model was used to investigate the etiology of CRCs with distinct morphologies. CRCs with flat morphologies more frequently escape detection during routine colonoscopies than their larger polypoid counterparts, and with this realization have become increasingly apparent in recent years (Owen 1996; Saitoh et al. 2001; Soetikno et al. 2006; Soetikno et al. 2008; Speake et al. 2007). The AOM model was used to show that flat and polypoid tumors arise independently, despite having a similar mutational spectrum (Uronis et al. 2007).
Non-WNT pathway-mediated models of CRC
TGFB models
The transforming growth factor beta (TGFB) signaling pathway functions in a variety of cellular processes including differentiation, growth suppression, deposition of extracellular matrix and apoptosis. CRCs often acquire resistance to TGFB signaling and at later stages of cancer progression, express increased levels of TGFB promoting invasion and metastasis (Blobe et al. 2000).
Several TGFB pathway-associated models have been used to dissect the complex role of this pathway during CRC development. Tgfb1 deficient mice die around three weeks of age due to extensive inflammation (Kulkarni et al. 1993; Shull et al. 1992). However, on a Rag2 deficient background, lacking functional B and T-cells, Tgfb1 deficient mice survive until adulthood (Diebold et al. 1995; Engle et al. 1999). Mice deficient for both Tgfb1 and Rag2 rapidly develop carcinoma of the cecum and colon suggesting that inflammation in combination with loss of TGFB1 results in predisposition to cancer (Engle et al. 1999). Interestingly, CRCs do not form with Tgfb1 deficiency unless specific bacterial pathogens are present to induce inflammation (Maggio-Price et al. 2006). Mutations in Smad2 and Smad4, but not Smad3, have been reported to occur in human CRCs (Eppert et al. 1996; Takagi et al. 1996; Thiagalingam et al. 1996). Smad2 and Smad4 deficient mice are embryonic lethal, while Smad3 deficient mice are viable and develop highly invasive CRC, which metastasizes to lymph nodes by four-to-six months of age (Zhu et al. 1998). Apc is not lost in TGFB-mediated tumors, nor do they display nuclear localization of CTNNB1 suggesting the existence of non-WNT/CTNNB1-mediated mechanism for tumor initiation (Kaiser et al. 2007). Consistent with current models of CRC and with the growth suppressive role of TGFB, mice deficient for Smad3 or heterozygous for a Smad4 mutant allele combined with mutant Apc develop an increased incidence of invasive carcinoma of the distal colon (Sodir et al. 2006; Takaku et al. 1998).
Inflammation-mediated models
The inflammatory diseases ulcerative colitis (UC) and Crohn's Disease, collectively termed inflammatory bowel disease (IBD), result in chronic inflammation of the colon and predisposition to the development of CRC (Itzkowitz and Harpaz 2004; Itzkowitz and Yio 2004). In mice the role of chronic inflammation in CRC was demonstrated by the discovery that prolonged administration of dextran sulfate sodium (DSS) results in chronic colitis and formation of high-grade dysplasia (Okayasu et al. 1990). A single dose of AOM followed by administration of DSS enhances tumor development and progression (Tanaka et al. 2003). The AOM/DSS model was used recently to show that deficiency for Sigirr (single immunoglobulin and toll-interleukin 1 receptor domain) increases susceptibility to CRC (Wald et al. 2003; Xiao et al. 2007). Similar to Tgfb1 deficient mice, bacteria-induced inflammation is probably important for SIGIRR-associated cancer.
The AOM/DSS model has also been used to demonstrated the importance of the JAK/STAT (Janus kinase/signal transducers and activators of transcription) and NFKB (nuclear factor of kappa light chain gene enhancer in B-cells) pathways for inflammation-mediated CRC (Wirtz and Neurath 2007). Consistent with a role for the JAK/STAT pathway, loss of Socs1 and Socs3 (suppressors of cytokine signaling) expression results in increased activation of STAT1, STAT3 and NFKB and development of colorectal tumors (Hanada et al. 2006; Rigby et al. 2007). A direct link between SOCS signaling and Myc exists since AOM/DSS-induced adenocarcinomas from Socs1 deficient mice have increased levels of nuclear CTNNB1 and Myc expression when compared to tumors from Socs1 wildtype mice (Hanada et al. 2006). Intestinal epithelium-specific deficient for Socs3 does not result in chronic inflammation or development tumors. However, when treated with AOM/DSS, these mice develop colon tumors preceded by inflammation suggesting that Socs3 expression in neighboring stroma may be required to suppress chronic inflammation and subsequently tumor promotion (Hanada et al. 2006; Rigby et al. 2007). While colorectal tumors from Socs3 deficient mice have not been shown to display high levels of nuclear CTNNB1 as is seen in tumors from Socs1 deficient mice, Socs3 deficiency has been shown to result in increased Myc expression in mammary tissue (Sutherland et al. 2006).
MYC as a central mediator of CRC
Recent evidence suggests that MYC functions as a global mediator of the oncogenic process, linking together a seemingly heterogeneous pool of molecular mechanisms underlying cancer development (Fig. 1) (Knoepfler 2007). The discovery that deletion of Myc rescues Apc deficiency elucidated a potential role of MYC as a key mediator of WNT/CTNNB1-initiated CRC (Sansom et al. 2007). Additionally, available evidence suggests that MYC is also involved in mediating non-WNT/CTNNB1-initiated colorectal cancers. Although WNT/CTNNB1 and non-WNT/CTNNB1-mediated CRCs can be discriminated by unique gene expression signatures, all tumors from both classes display increased Myc expression (Kaiser et al. 2007). Myc is a direct transcriptional target of the WNT/CTNNB1 pathway, while TGFB signaling is associated with Myc repression through SMAD3 binding to a repressive SMAD binding element (RSBE) within the Myc promoter (Frederick et al. 2004).
Fig. 1. Molecular pathways associated with colorectal cancer.
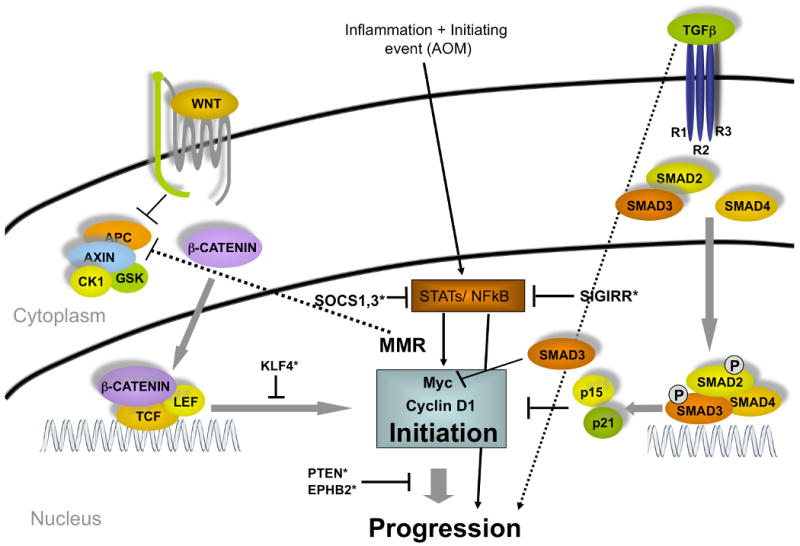
Two major pathways, WNT and TGFB, converge in the nucleus to effect Myc expression. Murine models for human CRC have been developed by genetically altering many components of these two pathways. *, Modifiers of cancer initiation and progression.
A variety of genetic and carcinogen-induced murine models have provided important reagents for investigating the complexity of human CRC. While each model has provided unique insights into human CRC, it is becoming increasingly apparent that seemingly independent pathways converge upon similar transforming genes. Numerous lines of evidence indicate Myc as a central mediator of CRC, perhaps through its role in chromatin remodeling (Knoepfler 2007; Knoepfler et al. 2006). Mouse models have been used to show that decreased Myc expression leads to reduced numbers of CRCs (Yekkala and Baudino 2007), a result confirmed by Myc inhibition in human CRCs (Hao et al. 2008; Zhang et al. 2009).
While the exact role of MYC in the development of CRC is not fully understood, it is clear that increased attention on the role of MYC during CRC development and progression warrants further study and that murine models of human CRC will be essential to fully understand MYC function. Similarly, the development of new murine models with characteristics of metastatic CRC, possibly generated using new technologies like transposon-based screens in existing models (Starr et al. 2009), should greatly accelerate the discovery of new therapies to treat advanced human CRCs.
Acknowledgments
Preparation of this review was supported in part by grants from the National Cancer Institute including the Mouse Models of Human Cancer Consortium and Specialized Program of Research Excellence in GI Cancer to DWT (U01CA105417, P50CA 106991 and R01CA092479). The intellectual environment provided by the Lineberger Cancer Center (P30CA016086) and the Center for Gastrointestinal Biology and Disease (P30DK34987) was essential.
References
- Amos-Landgraf JM, Kwong LN, Kendziorski CM, Reichelderfer M, Torrealba J, Weichert J, Haag JD, Chen KS, Waller JL, Gould MN, Dove WF. A target-selected Apc-mutant rat kindred enhances the modeling of familial human colon cancer. Proc Natl Acad Sci U S A. 2007;104:4036–4041. doi: 10.1073/pnas.0611690104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batlle E, Bacani J, Begthel H, Jonkheer S, Gregorieff A, van de Born M, Malats N, Sancho E, Boon E, Pawson T, Gallinger S, Pals S, Clevers H. EphB receptor activity suppresses colorectal cancer progression. Nature. 2005;435:1126–1130. doi: 10.1038/nature03626. [DOI] [PubMed] [Google Scholar]
- Becker C, Fantini MC, Neurath MF. High resolution colonoscopy in live mice. Nat Protoc. 2006;1:2900–2904. doi: 10.1038/nprot.2006.446. [DOI] [PubMed] [Google Scholar]
- Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med. 2000;342:1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- Boivin GP, Washington K, Yang K, Ward JM, Pretlow TP, Russell R, Besselsen DG, Godfrey VL, Doetschman T, Dove WF, Pitot HC, Halberg RB, Itzkowitz SH, Groden J, Coffey RJ. Pathology of mouse models of intestinal cancer: consensus report and recommendations. Gastroenterology. 2003;124:762–777. doi: 10.1053/gast.2003.50094. [DOI] [PubMed] [Google Scholar]
- Cormier RT, Bilger A, Lillich AJ, Halberg RB, Hong KH, Gould KA, Borenstein N, Lander ES, Dove WF. The Mom1AKR intestinal tumor resistance region consists of Pla2g2a and a locus distal to D4Mit64. Oncogene. 2000;19:3182–3192. doi: 10.1038/sj.onc.1203646. [DOI] [PubMed] [Google Scholar]
- Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nat Genet. 2001;29:117–129. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- Diebold RJ, Eis MJ, Yin M, Ormsby I, Boivin GP, Darrow BJ, Saffitz JE, Doetschman T. Early-onset multifocal inflammation in the transforming growth factor beta 1-null mouse is lymphocyte mediated. Proc Natl Acad Sci U S A. 1995;92:12215–12219. doi: 10.1073/pnas.92.26.12215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich WF, Lander ES, Smith JS, Moser AR, Gould KA, Luongo C, Borenstein N, Dove W. Genetic identification of Mom-1, a major modifier locus affecting Min-induced intestinal neoplasia in the mouse. Cell. 1993;75:631–639. doi: 10.1016/0092-8674(93)90484-8. [DOI] [PubMed] [Google Scholar]
- Dong M, Guda K, Nambiar PR, Nakanishi M, Lichtler AC, Nishikawa M, Giardina C, Rosenberg DW. Azoxymethane-induced pre-adipocyte factor 1 (Pref-1) functions as a differentiation inhibitor in colonic epithelial cells. Carcinogenesis. 2004;25:2239–2246. doi: 10.1093/carcin/bgh237. [DOI] [PubMed] [Google Scholar]
- Druckrey H, Preussmann R, Matzkies F, Ivankovic S. Selective production of intestinal cancer in rats by 1,2-dimethylhydrazine. Naturwissenschaften. 1967;54:285–286. doi: 10.1007/BF00620890. [DOI] [PubMed] [Google Scholar]
- Edelmann W, Umar A, Yang K, Heyer J, Kucherlapati M, Lia M, Kneitz B, Avdievich E, Fan K, Wong E, Crouse G, Kunkel T, Lipkin M, Kolodner RD, Kucherlapati R. The DNA mismatch repair genes Msh3 and Msh6 cooperate in intestinal tumor suppression. Cancer Res. 2000;60:803–807. [PubMed] [Google Scholar]
- Edelmann W, Yang K, Kuraguchi M, Heyer J, Lia M, Kneitz B, Fan K, Brown AM, Lipkin M, Kucherlapati R. Tumorigenesis in Mlh1 and Mlh1/Apc1638N mutant mice. Cancer Res. 1999;59:1301–1307. [PubMed] [Google Scholar]
- Edelmann W, Yang K, Umar A, Heyer J, Lau K, Fan K, Liedtke W, Cohen PE, Kane MF, Lipford JR, Yu N, Crouse GF, Pollard JW, Kunkel T, Lipkin M, Kolodner R, Kucherlapati R. Mutation in the mismatch repair gene Msh6 causes cancer susceptibility. Cell. 1997;91:467–477. doi: 10.1016/s0092-8674(00)80433-x. [DOI] [PubMed] [Google Scholar]
- Engle SJ, Hoying JB, Boivin GP, Ormsby I, Gartside PS, Doetschman T. Transforming growth factor beta1 suppresses nonmetastatic colon cancer at an early stage of tumorigenesis. Cancer Res. 1999;59:3379–3386. [PubMed] [Google Scholar]
- Eppert K, Scherer SW, Ozcelik H, Pirone R, Hoodless P, Kim H, Tsui LC, Bapat B, Gallinger S, Andrulis IL, Thomsen GH, Wrana JL, Attisano L. MADR2 maps to 18q21 and encodes a TGFbeta-regulated MAD-related protein that is functionally mutated in colorectal carcinoma. Cell. 1996;86:543–552. doi: 10.1016/s0092-8674(00)80128-2. [DOI] [PubMed] [Google Scholar]
- Fodde R, Edelmann W, Yang K, van Leeuwen C, Carlson C, Renault B, Breukel C, Alt E, Lipkin M, Khan PM, et al. A targeted chain-termination mutation in the mouse Apc gene results in multiple intestinal tumors. Proc Natl Acad Sci U S A. 1994;91:8969–8973. doi: 10.1073/pnas.91.19.8969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederick JP, Liberati NT, Waddell DS, Shi Y, Wang XF. Transforming growth factor beta-mediated transcriptional repression of c-myc is dependent on direct binding of Smad3 to a novel repressive Smad binding element. Mol Cell Biol. 2004;24:2546–2559. doi: 10.1128/MCB.24.6.2546-2559.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaleb AM, McConnell BB, Nandan MO, Katz JP, Kaestner KH, Yang VW. Haploinsufficiency of Kruppel-like factor 4 promotes adenomatous polyposis coli dependent intestinal tumorigenesis. Cancer Res. 2007;67:7147–7154. doi: 10.1158/0008-5472.CAN-07-1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady WM, Rajput A, Myeroff L, Liu DF, Kwon K, Willis J, Markowitz S. Mutation of the type II transforming growth factor-beta receptor is coincident with the transformation of human colon adenomas to malignant carcinomas. Cancer Res. 1998;58:3101–3104. [PubMed] [Google Scholar]
- Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, Joslyn G, Stevens J, Spirio L, Robertson M, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66:589–600. doi: 10.1016/0092-8674(81)90021-0. [DOI] [PubMed] [Google Scholar]
- Hanada T, Kobayashi T, Chinen T, Saeki K, Takaki H, Koga K, Minoda Y, Sanada T, Yoshioka T, Mimata H, Kato S, Yoshimura A. IFNgamma-dependent, spontaneous development of colorectal carcinomas in SOCS1-deficient mice. J Exp Med. 2006;203:1391–1397. doi: 10.1084/jem.20060436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao H, Nancai Y, Lei F, Xiong W, Wen S, Guofu H, Yanxia W, Hanju H, Qian L, Hong X. siRNA directed against c-Myc inhibits proliferation and downregulates human telomerase reverse transcriptase in human colon cancer Colo 320 cells. J Exp Clin Cancer Res. 2008;27:27. doi: 10.1186/1756-9966-27-27. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Itzkowitz SH, Harpaz N. Diagnosis and management of dysplasia in patients with inflammatory bowel diseases. Gastroenterology. 2004;126:1634–1648. doi: 10.1053/j.gastro.2004.03.025. [DOI] [PubMed] [Google Scholar]
- Itzkowitz SH, Yio X. Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: the role of inflammation. Am J Physiol Gastrointest Liver Physiol. 2004;287:G7–17. doi: 10.1152/ajpgi.00079.2004. [DOI] [PubMed] [Google Scholar]
- Kaiser S, Park YK, Franklin JL, Halberg RB, Yu M, Jessen WJ, Freudenberg J, Chen X, Haigis K, Jegga AG, Kong S, Sakthivel B, Xu H, Reichling T, Azhar M, Boivin GP, Roberts RB, Bissahoyo AC, Gonzales F, Bloom GC, Eschrich S, Carter SL, Aronow JE, Kleimeyer J, Kleimeyer M, Ramaswamy V, Settle SH, Boone B, Levy S, Graff JM, Doetschman T, Groden J, Dove WF, Threadgill DW, Yeatman TJ, Coffey RJ, Jr, Aronow BJ. Transcriptional recapitulation and subversion of embryonic colon development by mouse colon tumor models and human colon cancer. Genome Biol. 2007;8:R131. doi: 10.1186/gb-2007-8-7-r131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoepfler PS. Myc goes global: new tricks for an old oncogene. Cancer Res. 2007;67:5061–5063. doi: 10.1158/0008-5472.CAN-07-0426. [DOI] [PubMed] [Google Scholar]
- Knoepfler PS, Zhang XY, Cheng PF, Gafken PR, McMahon SB, Eisenman RN. Myc influences global chromatin structure. Embo J. 2006;25:2723–2734. doi: 10.1038/sj.emboj.7601152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, Roberts AB, Sporn MB, Ward JM, Karlsson S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci U S A. 1993;90:770–774. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuraguchi M, Yang K, Wong E, Avdievich E, Fan K, Kolodner RD, Lipkin M, Brown AM, Kucherlapati R, Edelmann W. The distinct spectra of tumor-associated Apc mutations in mismatch repair-deficient Apc1638N mice define the roles of MSH3 and MSH6 in DNA repair and intestinal tumorigenesis. Cancer Res. 2001;61:7934–7942. [PubMed] [Google Scholar]
- Kwong LN, Shedlovsky A, Biehl BS, Clipson L, Pasch CA, Dove WF. Identification of Mom7, a novel modifier of Apc(Min/+) on mouse chromosome 18. Genetics. 2007;176:1237–1244. doi: 10.1534/genetics.107.071217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348:919–932. doi: 10.1056/NEJMra012242. [DOI] [PubMed] [Google Scholar]
- Mann B, Gelos M, Siedow A, Hanski ML, Gratchev A, Ilyas M, Bodmer WF, Moyer MP, Riecken EO, Buhr HJ, Hanski C. Target genes of beta-catenin-T cell-factor/lymphoid-enhancer-factor signaling in human colorectal carcinomas. Proc Natl Acad Sci U S A. 1999;96:1603–1608. doi: 10.1073/pnas.96.4.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPhee M, Chepenik KP, Liddell RA, Nelson KK, Siracusa LD, Buchberg AM. The secretory phospholipase A2 gene is a candidate for the Mom1 locus, a major modifier of ApcMin-induced intestinal neoplasia. Cell. 1995;81:957–966. doi: 10.1016/0092-8674(95)90015-2. [DOI] [PubMed] [Google Scholar]
- Maggio-Price L, Treuting P, Zeng W, Tsang M, Bielefeldt-Ohmann H, Iritani BM. Helicobacter infection is required for inflammation and colon cancer in SMAD3-deficient mice. Cancer Res. 2006;66:828–838. doi: 10.1158/0008-5472.CAN-05-2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCart AE, Vickaryous NK, Silver A. Apc mice: models, modifiers and mutants. Pathol Res Pract. 2008;204:479–490. doi: 10.1016/j.prp.2008.03.004. [DOI] [PubMed] [Google Scholar]
- Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- Moser AR, Dove WF, Roth KA, Gordon JI. The Min (multiple intestinal neoplasia) mutation: its effect on gut epithelial cell differentiation and interaction with a modifier system. J Cell Biol. 1992;116:1517–1526. doi: 10.1083/jcb.116.6.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990;247:322–324. doi: 10.1126/science.2296722. [DOI] [PubMed] [Google Scholar]
- Nordlinger B, Panis Y, Puts JP, Herve JP, Delelo R, Ballet F. Experimental model of colon cancer: recurrences after surgery alone or associated with intraperitoneal 5-fluorouracil chemotherapy. Dis Colon Rectum. 1991;34:658–663. doi: 10.1007/BF02050346. [DOI] [PubMed] [Google Scholar]
- Nüsslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila. Nature. 1980;287:795–801. doi: 10.1038/287795a0. [DOI] [PubMed] [Google Scholar]
- Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. 1990;98:694–702. doi: 10.1016/0016-5085(90)90290-h. [DOI] [PubMed] [Google Scholar]
- Oshima M, Oshima H, Kitagawa K, Kobayashi M, Itakura C, Taketo M. Loss of Apc heterozygosity and abnormal tissue building in nascent intestinal polyps in mice carrying a truncated Apc gene. Proc Natl Acad Sci U S A. 1995;92:4482–4486. doi: 10.1073/pnas.92.10.4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen DA. Flat adenoma, flat carcinoma, and de novo carcinoma of the colon. Cancer. 1996;77:3–6. doi: 10.1002/(SICI)1097-0142(19960101)77:1<3::AID-CNCR2>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Papanikolaou A, Wang QS, Delker DA, Rosenberg DW. Azoxymethane-induced colon tumors and aberrant crypt foci in mice of different genetic susceptibility. Cancer Lett. 1998;130:29–34. doi: 10.1016/s0304-3835(98)00101-3. [DOI] [PubMed] [Google Scholar]
- Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, Vogelstein B, Kinzler KW. APC mutations occur early during colorectal tumorigenesis. Nature. 1992;359:235–237. doi: 10.1038/359235a0. [DOI] [PubMed] [Google Scholar]
- Reitmair AH, Cai JC, Bjerknes M, Redston M, Cheng H, Pind MT, Hay K, Mitri A, Bapat BV, Mak TW, Gallinger S. MSH2 deficiency contributes to accelerated APC-mediated intestinal tumorigenesis. Cancer Res. 1996;56:2922–2926. [PubMed] [Google Scholar]
- Rigby RJ, Simmons JG, Greenhalgh CJ, Alexander WS, Lund PK. Suppressor of cytokine signaling 3 (SOCS3) limits damage-induced crypt hyper-proliferation and inflammation-associated tumorigenesis in the colon. Oncogene. 2007;26:4833–4841. doi: 10.1038/sj.onc.1210286. [DOI] [PubMed] [Google Scholar]
- Roberts RB, Min L, Washington MK, Olsen SJ, Settle SH, Coffey RJ, Threadgill DW. Importance of epidermal growth factor receptor signaling in establishment of adenomas and maintenance of carcinomas during intestinal tumorigenesis. Proc Natl Acad Sci U S A. 2002;99:1521–1526. doi: 10.1073/pnas.032678499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruivenkamp CA, van Wezel T, Zanon C, Stassen AP, Vlcek C, Csikos T, Klous AM, Tripodis N, Perrakis A, Boerrigter L, Groot PC, Lindeman J, Mooi WJ, Meijjer GA, Scholten G, Dauwerse H, Paces V, van Zandwijk N, van Ommen GJ, Demant P. Ptprj is a candidate for the mouse colon-cancer susceptibility locus Scc1 and is frequently deleted in human cancers. Nat Genet. 2002;31:295–300. doi: 10.1038/ng903. [DOI] [PubMed] [Google Scholar]
- Saitoh Y, Waxman I, West AB, Popnikolov NK, Gatalica Z, Watari J, Obara T, Kohgo Y, Pasricha PJ. Prevalence and distinctive biologic features of flat colorectal adenomas in a North American population. Gastroenterology. 2001;120:1657–1665. doi: 10.1053/gast.2001.24886. [DOI] [PubMed] [Google Scholar]
- Sansom OJ, Meniel VS, Muncan V, Phesse TJ, Wilkins JA, Reed KR, Vass JK, Athineos D, Clevers H, Clarke AR. Myc deletion rescues Apc deficiency in the small intestine. Nature. 2007;446:676–679. doi: 10.1038/nature05674. [DOI] [PubMed] [Google Scholar]
- Shao J, Washington MK, Saxena R, Sheng H. Heterozygous Disruption of the PTEN Promotes Intestinal Neoplasia in APCmin/+ Mouse: Roles of Osteopontin. Carcinogenesis. 2007 doi: 10.1093/carcin/bgm186. [DOI] [PubMed] [Google Scholar]
- Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodir NM, Chen X, Park R, Nickel AE, Conti PS, Moats R, Bading JR, Shibata D, Laird PW. Smad3 deficiency promotes tumorigenesis in the distal colon of ApcMin/+ mice. Cancer Res. 2006;66:8430–8438. doi: 10.1158/0008-5472.CAN-06-1437. [DOI] [PubMed] [Google Scholar]
- Soetikno R, Friedland S, Kaltenbach T, Chayama K, Tanaka S. Nonpolypoid (flat and depressed) colorectal neoplasms. Gastroenterology. 2006;130:566–576. doi: 10.1053/j.gastro.2005.12.006. quiz 588-569. [DOI] [PubMed] [Google Scholar]
- Soetikno RM, Kaltenbach T, Rouse RV, Park W, Maheshwari A, Sato T, Matsui S, Friedland S. Prevalence of nonpolypoid (flat and depressed) colorectal neoplasms in asymptomatic and symptomatic adults. JAMA. 2008;299:1027–1035. doi: 10.1001/jama.299.9.1027. [DOI] [PubMed] [Google Scholar]
- Speake D, Biyani D, Frizelle FA, Watson AJ. Flat adenomas. ANZ J Surg. 2007;77:4–8. doi: 10.1111/j.1445-2197.2006.03847.x. [DOI] [PubMed] [Google Scholar]
- Starr TK, Allaei R, Silverstein KA, Staggs RA, Sarver AL, Bergemann TL, Gupta M, O'Sullivan MG, Matise I, Dupuy AJ, Collier LS, Powers S, Oberg AL, Asmann YW, Thibodeau SN, Tessarollo L, Copeland NG, Jenkins NA, Cormier RT, Largaespada DA. A transposon-based genetic screen in mice identifies genes altered in colorectal cancer. Science. 2009;323:1747–1750. doi: 10.1126/science.1163040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su LK, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C, Gould KA, Dove WF. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science. 1992;256:668–670. doi: 10.1126/science.1350108. [DOI] [PubMed] [Google Scholar]
- Sutherland KD, Vaillant F, Alexander WS, Wintermantel TM, Forrest NC, Holroyd SL, McManus EJ, Schutz G, Watson CJ, Chodosh LA, Lindeman GJ, Visvader JE. c-myc as a mediator of accelerated apoptosis and involution in mammary glands lacking Socs3. Embo J. 2006;25:5805–5815. doi: 10.1038/sj.emboj.7601455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi Y, Kohmura H, Futamura M, Kida H, Tanemura H, Shimokawa K, Saji S. Somatic alterations of the DPC4 gene in human colorectal cancers in vivo. Gastroenterology. 1996;111:1369–1372. doi: 10.1053/gast.1996.v111.pm8898652. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Nakatsugi S, Sugimura T, Wakabayashi K. Frequent mutations of the beta-catenin gene in mouse colon tumors induced by azoxymethane. Carcinogenesis. 2000;21:1117–1120. [PubMed] [Google Scholar]
- Takaku K, Oshima M, Miyoshi H, Matsui M, Seldin MF, Taketo MM. Intestinal tumorigenesis in compound mutant mice of both Dpc4 (Smad4) and Apc genes. Cell. 1998;92:645–656. doi: 10.1016/s0092-8674(00)81132-0. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Kohno H, Suzuki R, Yamada Y, Sugie S, Mori H. A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci. 2003;94:965–973. doi: 10.1111/j.1349-7006.2003.tb01386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiagalingam S, Lengauer C, Leach FS, Schutte M, Hahn SA, Overhauser J, Willson JK, Markowitz S, Hamilton SR, Kern SE, Kinzler KW, Vogelstein B. Evaluation of candidate tumour suppressor genes on chromosome 18 in colorectal cancers. Nat Genet. 1996;13:343–346. doi: 10.1038/ng0796-343. [DOI] [PubMed] [Google Scholar]
- Torrance CJ, Jackson PE, Montgomery E, Kinzler KW, Vogelstein B, Wissner A, Nunes M, Frost P, Discafani CM. Combinatorial chemoprevention of intestinal neoplasia. Nat Med. 2000;6:1024–1028. doi: 10.1038/79534. [DOI] [PubMed] [Google Scholar]
- Uronis JM, Herfarth HH, Rubinas TC, Bissahoyo AC, Hanlon K, Threadgill DW. Flat colorectal cancers are genetically determined and progress to invasion without going through a polypoid stage. Cancer Res. 2007;67:11594–11600. doi: 10.1158/0008-5472.CAN-07-3242. [DOI] [PubMed] [Google Scholar]
- Wald D, Qin J, Zhao Z, Qian Y, Naramura M, Tian L, Towne J, Sims JE, Stark GR, Li X. SIGIRR, a negative regulator of Toll-like receptor-interleukin 1 receptor signaling. Nat Immunol. 2003;4:920–927. doi: 10.1038/ni968. [DOI] [PubMed] [Google Scholar]
- Wang QS, Papanikolaou A, Sabourin CL, Rosenberg DW. Altered expression of cyclin D1 and cyclin-dependent kinase 4 in azoxymethane-induced mouse colon tumorigenesis. Carcinogenesis. 1998;19:2001–2006. doi: 10.1093/carcin/19.11.2001. [DOI] [PubMed] [Google Scholar]
- Wirtz S, Neurath MF. Mouse models of inflammatory bowel disease. Adv Drug Deliv Rev. 2007 doi: 10.1016/j.addr.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Xiao H, Gulen MF, Qin J, Yao J, Bulek K, Kish D, Altuntas CZ, Wald D, Ma C, Zhou H, Tuohy VK, Fairchild RL, de la Motte C, Cua D, Vallance BA, Li X. The Toll-interleukin-1 receptor member SIGIRR regulates colonic epithelial homeostasis, inflammation, and tumorigenesis. Immunity. 2007;26:461–475. doi: 10.1016/j.immuni.2007.02.012. [DOI] [PubMed] [Google Scholar]
- Yekkala K, Baudino TA. Inhibition of intestinal polyposis with reduced angiogenesis in ApcMin/+ mice due to decreases in c-Myc expression. Mol Cancer Res. 2007;5:1296–1303. doi: 10.1158/1541-7786.MCR-07-0232. [DOI] [PubMed] [Google Scholar]
- Zan Y, Haag JD, Chen KS, Shepel LA, Wigington D, Wang YR, Hu R, Lopez-Guajardo CC, Brose HL, Porter KI, Leonard RA, Hitt AA, Schommer SL, Elegbede AF, Gould MN. Production of knockout rats using ENU mutagenesis and a yeast-based screening assay. Nat Biotechnol. 2003;21:645–651. doi: 10.1038/nbt830. [DOI] [PubMed] [Google Scholar]
- Zhang X, Ge YL, Tian RH. The knockdown of c-myc expression by RNAi inhibits cell proliferation in human colon cancer HT-29 cells in vitro and in vivo. Cell Mol Biol Lett. 2009;14:305–318. doi: 10.2478/s11658-009-0001-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Richardson JA, Parada LF, Graff JM. Smad3 mutant mice develop metastatic colorectal cancer. Cell. 1998;94:703–714. doi: 10.1016/s0092-8674(00)81730-4. [DOI] [PubMed] [Google Scholar]