

Abstract
Chronic allograft rejection (CR) is the main barrier to long-term transplant survival. CR is a progressive disease defined by interstitial fibrosis, vascular neointimal development, and graft dysfunction. The underlying mechanisms responsible for CR remain poorly defined. Transforming growth factor β (TGFβ) has been implicated in promoting fibrotic diseases including CR, but is beneficial in the transplant setting due to its immunosuppressive activities. To assess the requirement for T cell TGFβ signaling in allograft acceptance and the progression of CR, we used mice with abrogated T cell TGFβ signaling as allograft recipients. We compared responses from recipients that were transiently depleted of CD4+ cells (that develop CR and express intragraft TGFβ) to responses from mice that received anti-CD40L mAb therapy (that do not develop CR and do not express intragraft TGFβ). Allograft acceptance and suppression of graft-reactive T and B cells were independent of T cell TGFβ signaling in mice treated with anti-CD40L mAb. In recipients transiently depleted of CD4+ T cells, T cell TGFβ signaling was required for the development of fibrosis associated with CR, long-term graft acceptance, and suppression of graft-reactive T and B cell responses. Further, IL-17 was identified as a critical element in TGFβ driven allograft fibrosis. Thus, IL-17 may provide a therapeutic target for preventing graft fibrosis, a measure of CR, while sparing the immunosuppressive activities of TGFβ.
Keywords: T cells, Th1/Th2, transplantation, suppression
Introduction
With the advent of immunosuppressive therapies, a decline in graft loss due to acute rejection has established chronic allograft rejection (CR)3 as the leading cause of late graft failure (1). CR is an irreversible disease characterized by deteriorating graft function, interstitial fibrosis, and occlusive neointima (2-5). Despite continued investigation, the underlying mechanisms responsible for these disease manifestations remain poorly defined and no therapies exist to prevent or treat CR except re-transplantation (6).
The immune system evolved to combat pathogens while maintaining tolerance to self, and TGFβ plays a pivotal role in regulating immune responses (7). The importance of TGFβ in immune regulation is underscored by the severe autoimmunity observed in knockout mice that lack TGFβ or are unable to signal through the TGFβ receptor (8-11). TGFβ controls T cell mediated self-reactivity by regulating lymphocyte proliferation and survival, inhibiting Th1/Th2-cell differentiation, and dampening effector function (reviewed in (12). TGFβ signaling in B cells inhibits proliferation and survival, prevents activation, and inhibits IgG class switching (12). TGFβ is also critical in both the development and function of T regulatory cells (Treg) (12-17). TGFβ signaling in Treg is essential for peripheral maintenance of this cell subset (7, 18) and for the induction of FoxP3 expression and Treg function in CD4+CD25- T cells (19).
In addition to TGFβ's anti-inflammatory activities, TGFβ also mediates pro-inflammatory as well as pro-fibrotic activities. A reciprocal developmental pathway exists for the generation of pathogenic effector Th17 and Treg (20-22). TGFβ in association with IL-6 or IL-21 favors the commitment of CD4+ T cells to the Th17 lineage (20, 21, 23-26). IL-17 stimulates stromal cells, such as fibroblasts, endothelial cells, and epithelial cells to produce IL-6, IL-8, granulocyte CSF (G-CSF), and PGE2 and up-regulates critical chemoattractants, such as CXCL1 and CXCL2 (27-29). IL-17 serves to amplify the inflammatory responses and has also recently been implicated as a pro-fibrotic cytokine (30-37).
A critical role for TGFβ in transplant acceptance has been described (38, 39), and early studies revealed the importance of TGFβ in donor-specific transfusions and allograft acceptance (40). Subsequent studies investigating skin allograft acceptance have also identified TGFβ as a protective factor against allograft rejection (41, 42). Further, transduction of cardiac allografts with active TGFβ prolongs graft survival (43) and is associated with the induction of graft-reactive Treg (44).
While TGFβ mediates many beneficial anti-inflammatory activities in the immune system (7), we have previously reported an association between TGFβ and fibrosis associated with CR using the mouse vascularized cardiac allograft model (45). Intragraft transcript levels of TGFβ were readily detectable in the CR grafts from recipients transiently depleted of CD4+ T cells, but not in the grafts of anti-CD40L treated recipients, which remain free of CR (45). Allograft transduction with active TGFβ resulted in CR in anti-CD40L treated recipients that do not normally exhibit CR, but was not observed in TGFβ transduced syngeneic grafts. This supported a critical role for TGFβ and alloantigen in the progression of CR. In this study, we sought to determine the role of TGFβ signaling on alloreactive effector cells and IL-17 induction by using T cell-specific dominant negative TGFβ receptor type II (CD4-DNR) (10) and IL-17 deficient (IL-17-/-) mice (46) as transplant recipients. Further, we identify both TGFβ dependent and independent pathways to allograft acceptance.
Materials and Methods
Mice
C57BL/6 CD4-DNR (10), C57BL/6 wild type (WT) and BALB/c mice were purchased from the Jackson Laboratory. CD4-DNR mice express a dominant-negative form of the human TGFβ receptor II (TGFβRII) under the direction of the mouse CD4 promoter, which lacks a CD8 silencer. TGFβ signaling is abrogated in both CD4+ and CD8+ T cells in these transgenic mice. CD4-DNR were propagated by breeding C57BL/6 WT females with CD4-DNR males. The genotyping of transgene expressing CD4-DNR mice was carried out using the following PCR primers: primer WT forward 5′CTAGGCCACAGAATTGAAAGATCT-3′; primer WT reverse 5′-TAGGTGGAAATTCTAGCATCATCC-3′; primer CD4-DNR transgene (TG) forward 5′-GCTGCACAT CGTCCTGTG-3′; primer TG reverse 5′-ACT TGACTGCACCGT TGTTG-3′. Primers WT forward and WT reverse were used to detect the internal control, IL-2 (324 bp). Primers TG forward and TG reverse were used to detect the transgenic allele (100 bp). CD4-DNR mice exhibit an autoimmune phenotype and immunopathology with age, resulting in the development of multi-focal inflammation best characterized by inflammatory bowel disease between 3-5 months of age (10). To avoid potential complications of age-related autoimmunity, mice were transplanted at 6 weeks of age and at the termination of each experiment the colons were examined macroscopically and histologically for autoimmune manifestations. None of the mice used in this study exhibited an autoimmune phenotype. IL-17-/- mice (46) were generated by Dr. Yoichiro Iwakura and provided by Dr. Weiping Zou in collaboration. All mice were housed under specific pathogen-free conditions in the Unit for Laboratory Animal Medicine at the University of Michigan. These experiments were approved by the University Committee on Use and Care of Animals at the University of Michigan.
Culture medium
Culture medium consisted of the following: RPMI 1640 supplemented with 2% FCS, 1 mM sodium pyruvate, 100 U/mL penicillin, 100 ug/mL streptomyin, 1.6 mM L-glutamine, 10 mM HEPES buffer (all from Invitrogen), 0.27 mM L-asparagine, 1.4 mM L-arginine HCl, 14 uM folic acid, and 50 uM 2-ME (all from Sigma-Aldrich).
Vascularized cardiac transplantation
CD4-DNR, IL-17-/-, and WT mice were transplanted with intact BALB/c cardiac allografts, as described (47). Briefly, the aorta and pulmonary artery of the donor heart were anastomosed end-to-side to the recipient's abdominal aorta and inferior vena cava, respectively. Upon perfusion with the recipient's blood, the transplanted heart resumes contraction. Graft function is monitored by abdominal palpation.
In vivo mAb treatment
The hybridoma secreting anti-CD4 (clone GK1.5) was obtained from American Type Culture Collection. The hybridoma secreting anti-CD40L (clone MR1) was provided by Dr. Randy Noelle (Dartmouth, Lebanon, NH). Anti-CD4 and anti-CD40L mAb were purified and resuspended in PBS by Bio X Cell (West Lebanon, NH). Mice received 1 mg i.p. of anti-CD4 mAb on days -1, 0, 7 (45, 48, 49). In recipients transiently depleted of CD4+ T cells, CD4+ T cells begin to repopulate the periphery 3-4 weeks post-transplantation (48-50). Mice received 1 mg of anti-CD40L i.p. days 0, 1, and 2 (45). All doses are relative to day of transplant.
Histology
Allografts were recovered at the times indicated post-transplantation, fixed in formalin, and embedded in paraffin. Sections were stained with hematoxylin-eosin (H & E) to assess myocyte viability (presence of cross striation and myocyte nuclei), and the nature and intensity of graft infiltrating cells.
Recovery of graft infiltrating cells (GIC)
Groups of three transplanted hearts were removed, pooled, minced, and digested with 1 mg/mL collagenase A (Roche) for 30 min at 37 °C. Tissue debris were allowed to settle at 1 × g and the suspension containing GIC was harvested by pipette. RBC were lysed by hypotonic shock, GIC were passed though a 30-μm pore size nylon mesh, and viable leukocytes were enumerated by trypan blue exclusion.
ELISPOT assays for cytokine-producing cells
ELISPOT assays were performed as previously described (51). Capture and detection antibodies specific for IFNγ (R4-6A2, XMG1.2), IL-4 (11B11, BVD6-24G2) and IL-17 (TC11-18H10.1, TC11-8H4.1) were purchased from Pharmingen (San Diego, CA). PVDF-backed microtiter plates (Millipore, Bedford, MA) were coated with unlabeled mAb and blocked with 1% BSA in PBS. Irradiated (1000 rad) donor splenocytes (4×105) and 1×106 recipient splenocytes were added to the plates. After washing, a 1:1000 dilution of anti-biotin alkaline phosphatase (AP) conjugate (Vector Laboratories, Burlingame, CA) was added to IFNγ and IL-17 plates, and a 1:2000 dilution of horseradish peroxidase-conjugated streptavidin (SA-HRP; Dako, Carpinteria, CA) was added to IL-4 plates. Plates were washed and spots visualized by addition of nitroblue tetrazolium (NBT; Biorad, Hercules, CA) / 3 bromo-4-chloro-inolyl-phosphate (BCIP; Sigma) to IFNγ and IL-17 plates, or 3-amino-9-ethylcarbazole (AEC; Pierce, Rockford, IL) to IL-4 plates. Color development continued until spots were visible and was stopped by adding H2O. Plates were dried and spots quantified with an Immunospot Series 1 ELISPOT analyzer (Cellular Technology Ltd., Cleveland, OH).
Flow cytometry
Splenocytes were isolated by mechanical dissociation followed by lysis via hypotonic shock and blocked in PBS containing 0.1% BSA, 0.025% NaN3 and 10% FBS. After washing, 1 × 106 cells were stained with fluorochrome-conjugated anti-mouse CD4 (clone GK1.5), CD3 (clone 145-2C11) and CD8 (clone 53-6.7) (all from BD Biosciences, San Jose, CA). Three-color flow cytometry was performed with a FACS Calibur (BD Biosciences) equipped with Cell Quest software.
RNA isolation and RT-PCR
Cardiac allografts were homogenized in 1 mL TRIzol® (Invitrogen Life Technologies, Carlsbad, CA) and RNA was isolated as per manufacturers protocol. 5 μg of total RNA were reverse transcribed using 10× PCR buffer (Roche), 10 mM dNTPs, Oligo (dt), M-MLV-RT (all from Invitrogen), and RNAsin (Promega). Products were then cleaned with 1:1 phenol/chloroform/isoamyl (25:24:1) and re-precipitated with 7.5 M NH4OAC in pure EtOH overnight at -80 °C.
Real-time PCR was performed on cDNA using a Rotor-Gene 3000 TM (Corbett Life Science, CA). Primer binding to DNA was detected by SYBR Green ITM dye (Roche, Indianapolis, IN). Relative expression of the gene of interest was expressed as the comparative concentration of the gene product to the GAPDH product as calculated by accompanying Rotor-Gene software. Significance was determined with an unpaired Student t-test.
Primer sequences:
IL-17 sense: 5′ GGACTCTCCACCGCAATGA
IL-17 anti-sense: 5′GACCAGGATCTCTTGCTGGA
FoxP3 sense: 5′CCAAGGTGAGCGAGTGTC
FoxP3 anti-sense: 5′AAGGCAGAGTCAGGAGAAGT
GAPDH sense: 5′CTGGTGCTGAGTATGTCGTG
GAPDH anti-sense: 5′CAGTCTTCTGAGTGGCAGTG
Donor-reactive Ab determination
As described (48, 50, 52) P815 cells (H-2d) were stained for flow cytometric analysis using diluted (1:50) sera obtained from mice as the primary antibody, followed by FITC-conjugated isotype specific anti-mouse IgG, IgG1, or IgG2a secondary antibodies (The Binding Site, San Diego, CA, USA) at a 1:50 dilution. Data are reported as the mean channel fluorescence determined on a Becton Dickinson FACSCaliber (San Jose, CA, USA).
Immunohistochemistry
To detect IgG deposition within the graft, frozen sections of grafts were fixed in cold acetone and incubated with 1:150 dilution of goat anti-mouse IgG-HRP (Southern Biotech, Birmingham, AL) followed by AEC staining (50). To detect C3d and C4d deposition (50), sections of paraffin embedded tissue were fixed in methanol. A 1:20 dilution of goat anti-mouse C3d (R&D Systems, Minneapolis, MN) was added followed by secondary detection antibodies added as per R&D System's anti-goat cell and tissue staining kit. Slides were stained with rabbit anti-mouse C4d (kindly provided by Dr. William Baldwin, Cleveland Clinic) at a 1:500 dilution, followed by DAB development using the SuperPicTure™ Polymer Detection Kit (Zymed). Specificity of staining was ensured by staining of native hearts.
Morphometric analysis of cardiac allograft fibrosis
Graft fibrosis was quantified by morphometric analysis of Masson's trichrome stained tissues using iPLab software (Scanalytics Inc., Fairfax, VA) (53). Mean fibrotic areas were calculated from 10 to 12 areas per heart section analyzed at 200× magnification. A minimum of 5 individual hearts were analyzed per group.
Statistical analysis
Data were analyzed with GraphPad Prism 4.0c software using unpaired Student t-tests. p values of ≤ 0.05 were considered statistically significant.
Results
Experimental System
In the mouse cardiac allograft model, prolonged allograft survival can be achieved by transiently depleting recipients of CD4+ cells or by disrupting CD40-CD40L interactions. However, these two inductive therapies differ with respect to the development of CR. Allografts in mice treated with anti-CD40L mAb remain free of CR, while grafts in mice transiently depleted of CD4+ cells develop CR, which is associated with intragraft expression of TGFβ (45). While graft-reactive T cells remain in a hyporesponsive state in both settings, recall responses of cells from these groups differ in that dominant Th2 responses are mounted by mice that are depleted of CD4+ cells, while Th1 responses are mounted by recipients given anti-CD40L mAb therapy (54). The current study assessed the requirement for TGFβ signaling in T cells for allograft acceptance, graft-reactive T and B cell hyporesponsiveness, and graft fibrosis as a parameter of CR.
Effect of T cell TGFβ signaling on transplant acceptance
TGFβ mediates beneficial immunosuppressive activities in the transplant setting (38-40). To assess the requirement for T cell TGFβ signaling in allograft acceptance and the progression of graft fibrosis associated with CR, we used CD4-DNR mice with abrogated T cell TGFβ signaling (10) as allograft recipients. CD4-DNR mice express a dominant-negative form of the human TGFβRII under the direction of the mouse CD4 promoter, which lacks a CD8 silencer. Hence, TGFβ signaling is abrogated in both CD4+ and CD8+ T cells in these transgenic mice. To rule out alloantigen independent cellular infiltration and tissue damage in CD4-DNR recipients, both WT and CD4-DNR mice were transplanted with syngeneic grafts. Grafts from both groups functioned until the experiment was terminated on day 50 post-transplant (Figure 1A). Histologically, syngeneic grafts from both groups were free of infiltrate, exhibited minimal fibrosis, normal arteries and viable myocytes (data not shown). Similar observations were made in CD4-DNR recipients of syngeneic grafts treated with inductive CD4+ T cell depletion (data not shown). These results indicate that there were no differences in graft survival or histology between the WT and the CD4-DNR recipients of syngeneic grafts ruling out non-antigen specific inflammatory responses in graft loss in CD4-DNR mice.
Figure 1. TGFβ dependent and independent mechanisms of allograft acceptance.
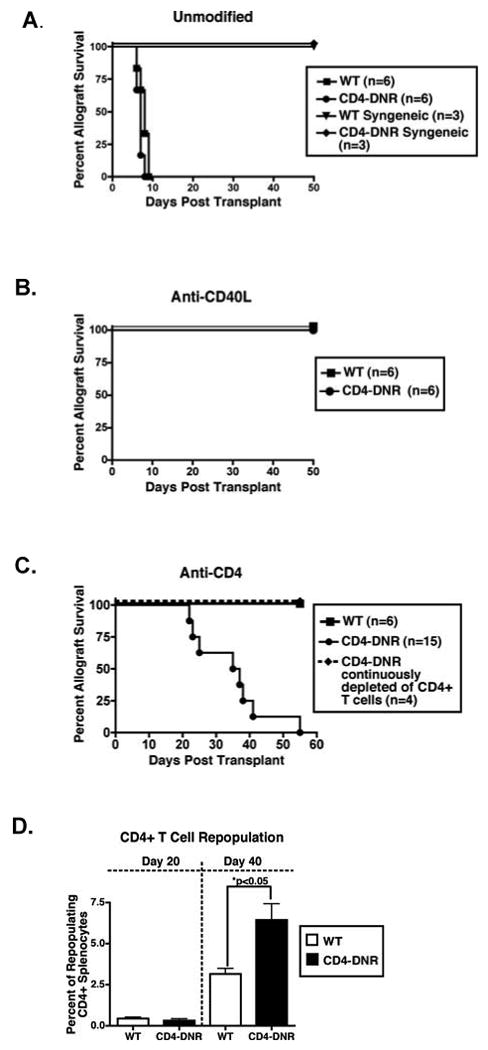
WT (squares and triangles) and CD4-DNR (circles and diamonds) mice were transplanted with allogeneic or syngeneic cardiac grafts and were either left untreated (A), given inductive anti-CD40L mAb (B), or transiently depleted of CD4+ cells (C). Graft function was monitored by palpation and recipients were recovered either at the time of rejection or 50 days post-transplant. (D) Flow cytometric analysis of CD4+ splenocytes that were harvested from WT or CD4-DNR allograft recipients on day 20 and day 40 post-transplant. Allograft recipients were treated inductively with anti-CD4 mAb.
Due to the immunosuppressive activities of TGFβ, we predicted that CD4-DNR recipients would mount exacerbated rejection responses when compared to WT mice. Allografts in both WT and CD4-DNR recipients were acutely rejected by day 9 post-transplant (Figure 1A) and histological examination revealed evidence of rejection (data not shown). While the tempo of rejection was not different between these two groups, a more intense infiltration of the grafts was observed in CD4-DNR recipients (total number of GICs per graft: WT = 0.33 × 106 +/- 0.01; CD4-DNR = 1.4 × 106 +/- 0.23 (p<0.01)) indicating that T cell TGFβ signaling dampens cell proliferation and/or cellular infiltration in unmodified recipients.
Treatment of both WT and CD4-DNR recipients with inductive anti-CD40L mAb resulted in long-term graft survival (Figure 1B), demonstrating that allograft acceptance following anti-CD40L therapy is independent of TGFβ signaling in T cells. WT allograft recipients treated with inductive CD4+ T cell depletion also exhibited long-term graft survival (Figure 1C). In contrast, 90% of the allografts from CD4-DNR mice transiently depleted of CD4+ T cells were rejected by day 40 post-transplantation (Figure 1C). The majority of the allografts were rejected between days 35-40, correlating with CD4+ T cell repopulation of the periphery (48, 50). A greater percentage of CD4+ T cells were present in the spleens of CD4-DNR recipients compared to WT suggesting that TGFβ signaling controls CD4+ T cell proliferation in recipients inductively depleted of CD4+ T cells (Figure 1D). Rejection could be attenuated in CD4-DNR recipients when CD4+ T cells were continuously depleted by weekly injections of anti-CD4 mAb (Figure 1C). These results indicate that T cell TGFβ signaling is essential for allograft acceptance in recipients transiently depleted of CD4+ T cells and that repopulation of CD4+ cells is required for rejection in CD4-DNR mice.
TGFβ regulation of donor-reactive Th1, Th2 and Th17 responses
T cell TGFβ signaling inhibits differentiation of Th1 and Th2 cells through the suppression of T-bet/STAT4 and Gata-3/NFAT (7, 12). To determine if T cell TGFβ signaling regulates donor-reactive cellular immune responses, we employed ELISPOT to quantify the number of in vivo primed donor-reactive Th1 (IFNγ), Th2 (IL-4) and Th17 (IL-17) responses (Figure 2). Elevated donor-reactive Th1 responses were observed in unmodified WT and CD4-DNR recipients indicating acute rejection is associated with a dominant Th1 response (Figure 2A). While abrogation of TGFβ signaling in recipients did not result in elevated Th1 responses compared to WT recipients, CD4-DNR recipients did exhibit enhanced IL-4 priming in response to donor antigen. However, this was not statistically significant. Donor-reactive Th17 responses were negligible in both WT and CD4-DNR recipients.
Figure 2. Donor-reactive Th1, Th2 and Th17 responses in WT and CD4-DNR recipients.
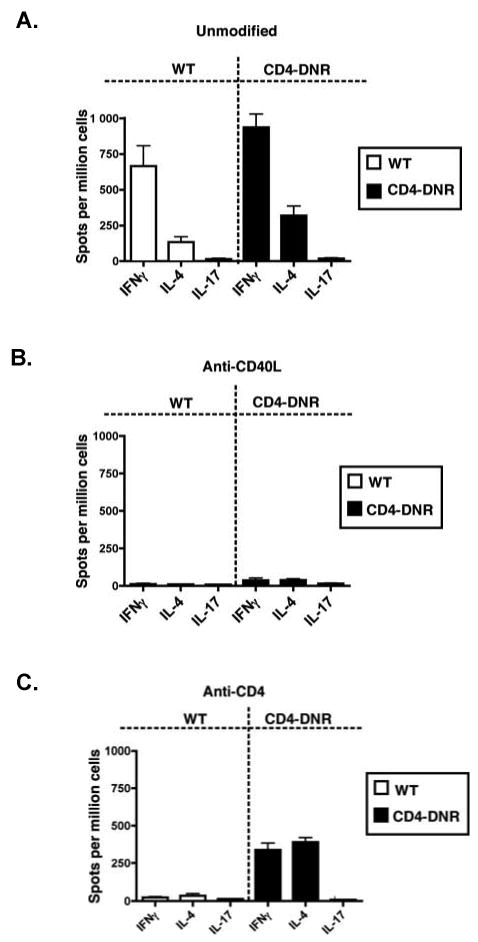
WT (open bars) or CD4-DNR (shaded bars) allograft recipients were left untreated (A), treated with inductive anti-CD40L mAb (B), or treated with inductive anti-CD4 mAb (C). Graft function was monitored by palpation and recipients were recovered either at the time of rejection, 50 days post-transplant (for anti-CD40L mAb treated WT and CD4-DNR recipients), or 40 days post-transplant (for anti-CD4 mAb treated WT recipients in order to directly compare to CD4-DNR recipients, which reject between days 35-40). At the termination of the experiment, splenocytes were harvested and processed for ELISPOT assays to quantify primed, donor-reactive IFN-γ, IL-4, or IL-17-producing cells. Bars represent the mean number of cytokine producing cells (+/- S.E.M.) from at least six recipients per group.
Donor-reactive Th responses were not observed in either WT or CD4-DNR recipients treated with anti-CD40L (Figure 2B). These findings indicate that T cell TGFβ signaling is not required for the suppression of donor-reactive responses (55) and is dispensable for allograft acceptance following anti-CD40L mAb treatment.
As previously reported (48, 49, 54), donor-reactive Th1 and Th2 responses are suppressed in recipients transiently depleted of CD4+ T cells. Consistent with these findings, inductive anti-CD4 mAb therapy resulted in hyporesponsiveness in WT recipients compared to unmodified recipients (Figure 2A and 2C). In contrast, elevated donor-reactive Th1 and Th2 responses were detected in the CD4-DNR recipients transiently depleted of CD4+ T cells (Figure 2C). These data indicate that TGFβ plays a pivotal role in suppressing immune responses and in maintaining allograft acceptance in recipients initially depleted of CD4+ T cells.
Cardiac fibrosis is reduced in mice with abrogated T cell TGFβ signaling
TGFβ has been strongly implicated in many fibrotic diseases (30, 56), including CR (3, 4, 57, 58). To investigate the relationship between T cell TGFβ signaling and the development of graft fibrosis as a measure of CR, quantitative morphometric trichrome analysis was employed to evaluate allograft fibrosis in WT and CD4-DNR recipients treated with inductive anti-CD40L or anti-CD4 mAb. Allografts from WT and CD4-DNR recipients treated with inductive anti-CD40L mAb revealed minimal fibrosis (Figure 3B). Allografts from WT recipients transiently depleted of CD4+ T cells revealed significant fibrosis compared to allografts from CD4-DNR recipients (Figure 3C). These findings indicate that in recipients transiently depleted of CD4+ cells, allograft fibrosis is dependent on T cell TGFβ signaling.
Figure 3. Allograft fibrosis in WT versus CD4-DNR recipients treated with anti-CD4 mAb.
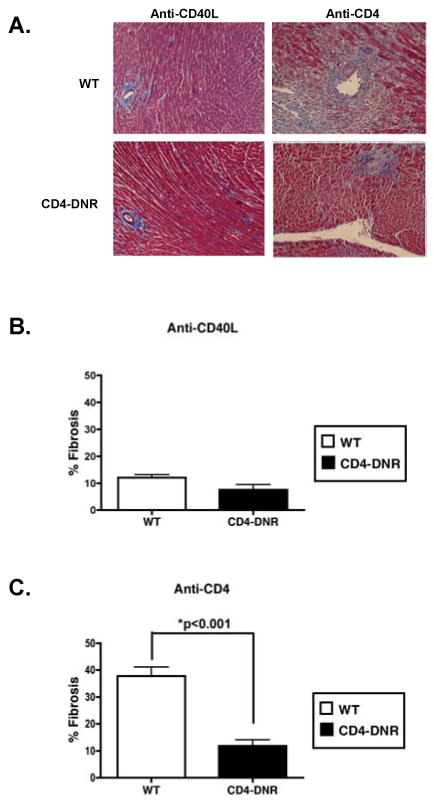
(A) Sections of grafts from recipients treated with inductive anti-CD40L (day 50 post-transplant) or anti-CD4 mAb (between days 35-40 post-transplant due to rejection in CD4-DNR recipients) were stained with Masson's trichrome, which stains fibrotic tissue blue. Frames are of grafts from WT and CD4-DNR recipients and are representative of at least 6 anti-CD40L mAb treated mice and 6-8 mice transiently depleted of CD4+ T cells. 200× magnification. (B) Quantification of mean fibrotic area by morphometric analysis in inductive anti-CD40L and (C) anti-CD4 mAb treated WT and CD4-DNR recipients. Bars represent the average percentage (+/- S.E.M.) of graft area positive for fibrosis in 5 anti-CD40L treated recipients and 6 anti-CD4 treated recipients. WT (open bars) and CD4-DNR (shaded bars).
Donor-reactive IgG production in WT and CD4-DNR recipients
Since donor-reactive antibodies have been implicated in both acute and chronic rejection (59, 60), we analyzed WT and CD4-DNR recipients for donor-reactive IgG antibodies (Figure 4). At the time of rejection, both WT and CD4-DNR unmodified recipients exhibited comparable donor-reactive IgG production (Figure 4A). These results indicate that TGFβ signaling does not overtly influence IgG alloantibody production in unmodified recipients.
Figure 4. Effect of TGFβ unresponsiveness on donor-reactive alloantibody levels.
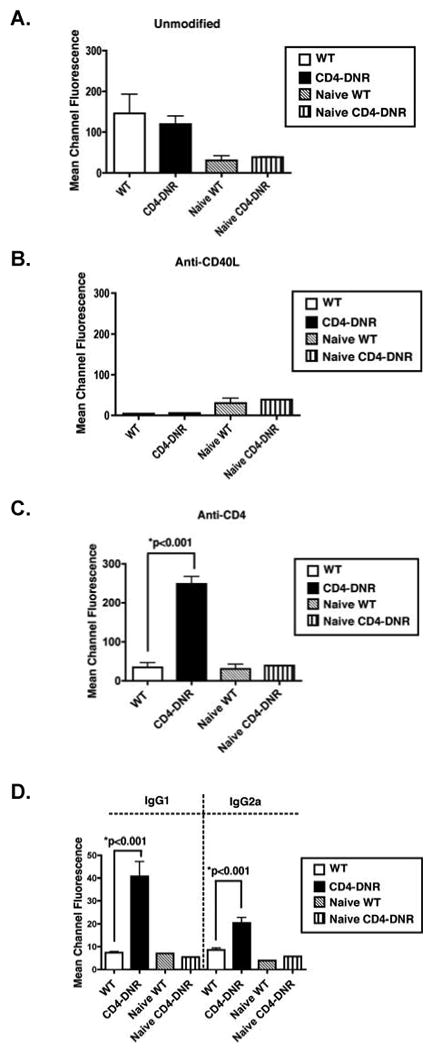
Sera were obtained from WT (open bars) or CD4-DNR (shaded bars) allograft recipients that were left untreated (A), treated with inductive anti-CD40L mAb (B), or treated with inductive anti-CD4 mAb (C). Sera were obtained at the time of rejection for unmodified recipients), 50 days post-transplant (for anti-CD40L mAb treated WT and CD4-DNR recipients), or 40 days post-transplant (for anti-CD4 mAb treated WT recipients). P815 (H-2d) cells were incubated with 1:50 dilution of sera and bound donor-reactive Ab were detected by incubation with FITC-tagged anti-IgG, anti-IgG1 or anti-IgG2a Abs. The mean channel fluorescence is indicative of the relative amount of donor-reactive antibodies. Bars represent the average mean channel fluorescence of at least 6 WT and 6 CD4-DNR recipient samples (+/- S.E.M.). IgG1 and IgG2a donor-reactive antibody levels were analyzed in anti-CD4 mAb treated WT and CD4-DNR recipients (D).
It is well established that CD40-CD40L interactions are critical for antibody isotype switch (61). Consistent with these findings, both WT and CD4-DNR recipients treated with anti-CD40L mAb did not generate donor-reactive IgG production compared to unmodified recipients (Figure 4B). Hence, CD40-CD40L interactions are required for IgG isotype switching in the presence or absence of TGFβ signaling in T cells.
CD4-DNR recipients inductively depleted of CD4+ T cells produced significantly elevated levels of donor-reactive IgG compared to WT controls (Figure 4C). To further characterize the donor-reactive antibody production in these recipients, both Th1- and Th2- dependent IgG2a and IgG1 isotypes were measured (62). Consistent with an increase in total IgG level, the CD4-DNR recipients exhibited significantly elevated levels of both IgG1 and IgG2a alloantibody production compared to WT recipients (Figure 4D). Collectively, these findings demonstrate that in the absence of TGFβ signaling, T cells acquire effector functions, secrete both Th1 and Th2 cytokines, and provide help for alloreactive B cells in recipients transiently depleted of CD4+ T cells.
Evidence of antibody-mediated rejection in CD4-DNR recipients treated with anti-CD4 mAb
The diagnostic criteria for antibody-mediated rejection include histologic evidence of acute tissue injury, serologic evidence of circulating donor-reactive antibodies, and the deposition of complement C3d and C4d within allografts (reviewed in (59, 63, 64)). Hence, we investigated the deposition of IgG and C3d and C4d within the grafts from WT and CD4-DNR recipients transiently depleted of CD4+ T cells (Figure 5). Intense IgG staining was localized mainly to the capillaries and arteries of allografts from CD4-DNR recipients, while allografts from WT recipients were free of IgG deposition (Figure 5A). Further, both C3d and C4d were deposited within the vasculature structures of allografts from CD4-DNR recipients, but were not present in control allografts (Figure 5B). These data indicate that in recipients transiently depleted of CD4+ T cells, antibody mediated rejection of allografts occurs when T cells are unresponsive to TGFβ.
Figure 5. IgG, C3d and C4d deposition in allografts of WT and CD4-DNR recipients.
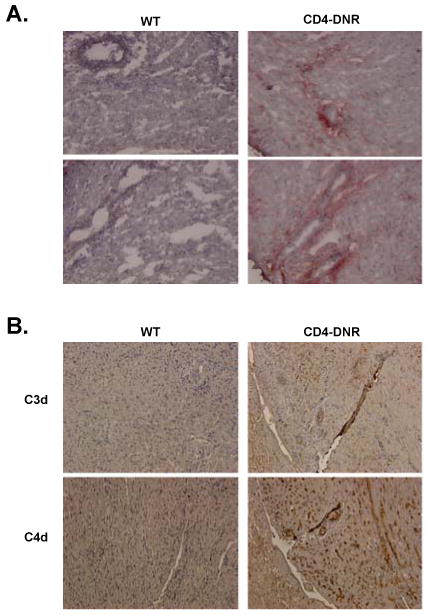
WT (left column) or CD4-DNR (right column) allograft recipients were treated with inductive anti-CD4 mAb therapy. Grafts were recovered at either the time of rejection or 40 days after transplantation. Graft sections were fixed and incubated with goat anti-mouse IgG (A) or the goat anti-mouse C3d or C4d (B) followed by development with 3-amino-9-ethylcarbazole or DAB to visualize mouse Ab and complement deposition. Results are representative of grafts from 6-10 recipients. Magnification, 400× (A) and 200× (B).
Anti-CD4 treated CD4-DNR recipients exhibit reduced intragraft FoxP3 and IL-17 levels compared to WT controls
TGFβ induces expression of FoxP3 (65), leading to the differentiation of CD4+CD25+ Treg cells from CD4+CD25- T cells (19, 66, 67). However, a dichotomy exists in which TGFβ in the context of IL-6 or IL-21 promotes pathogenic Th17 cell development (20, 21, 23, 24, 26, 68). To investigate the role of TGFβ signaling in Treg and Th17 induction and the contribution of these subsets in CR, we assessed intragraft FoxP3 and IL-17 transcript levels from WT and CD4-DNR recipients inductively depleted of CD4+ T cells (Figure 6). Allografts from the CD4-DNR recipients exhibited significantly reduced intragraft FoxP3 gene expression compared to WT (Figure 6). This may indicate a failure to induce Treg from naïve CD4+ T cells in the absence of T cell TGFβ signaling, poor maintenance of peripheral Treg, and/or reduced Treg trafficking into the graft (reviewed in (69)). In addition, intragraft IL-17 was detected in allografts from WT but not CD4-DNR recipients (Figure 6). IL-17 was not detected by ELISPOT in the spleens of the WT mice (Figure 2), which may suggest a compartmentalized immune response against donor antigen. Indeed, similar polarization of Th17 responses solely within target tissue has been observed in murine models of allergic lung disease and hypersentitivity pneumonitis and lung fibrosis (37, 70). IL-17 is best recognized as a cytokine that mobilizes neutrophils and coordinates local tissue inflammation through the induction of various pro-inflammatory cytokines (27, 28). Numerous recent reports implicate IL-17 as a pro-fibrotic cytokine in the context of T cell dependent fibrosis (31-37, 71), capable of inducing fibroblast proliferation and collagen deposition in cardiac tissue (31, 32). Our observation that allografts from CD4-DNR recipients treated with anti-CD4 exhibited minimal fibrosis (Figure 3) led us to hypothesize that IL-17 plays a pro-fibrotic role in CR.
Figure 6. Reduction of TGFβ-dependent intragraft gene expression from CD4-DNR recipients transiently depleted of CD4+ T cells.
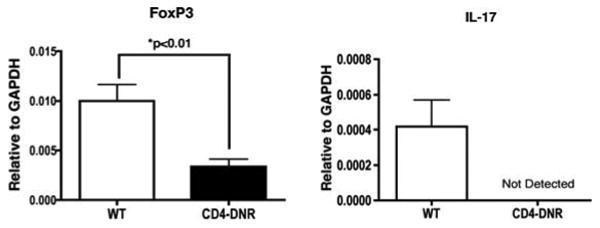
RNA was harvested from allografts of WT and CD4-DNR recipients transiently depleted of CD4+ T cells. Intragraft transcript levels of FoxP3 and IL-17 were assessed by real-time RT-PCR. Allografts were recovered between days 35-40 post-transplant. Bars depict the means of RNA expression from 6 WT and 9 CD4-DNR grafts.
Allograft fibrosis is reduced in anti-CD4 mAb treated IL-17-/- recipients
To investigate the role of IL-17 production in fibrosis associated with CR, IL-17-/- mice (46) were used as transplant recipients. Both WT and IL-17-/- allograft recipients treated with anti-CD4 exhibited long-term graft survival, while unmodified recipients rejected their grafts by day 9 post-transplant (Figure 7A). In IL-17-/- recipients transiently depleted of CD4+ T cells, fibrosis was markedly reduced relative to WT counterparts (Figure 7B and 7C). This reduction of allograft fibrosis in IL-17-/- recipients implicates IL-17 as a cytokine responsible for TGFβ-mediated fibrosis and CR.
Figure 7. In recipients that fail to produce IL-17, allografts are protected from fibrosis.
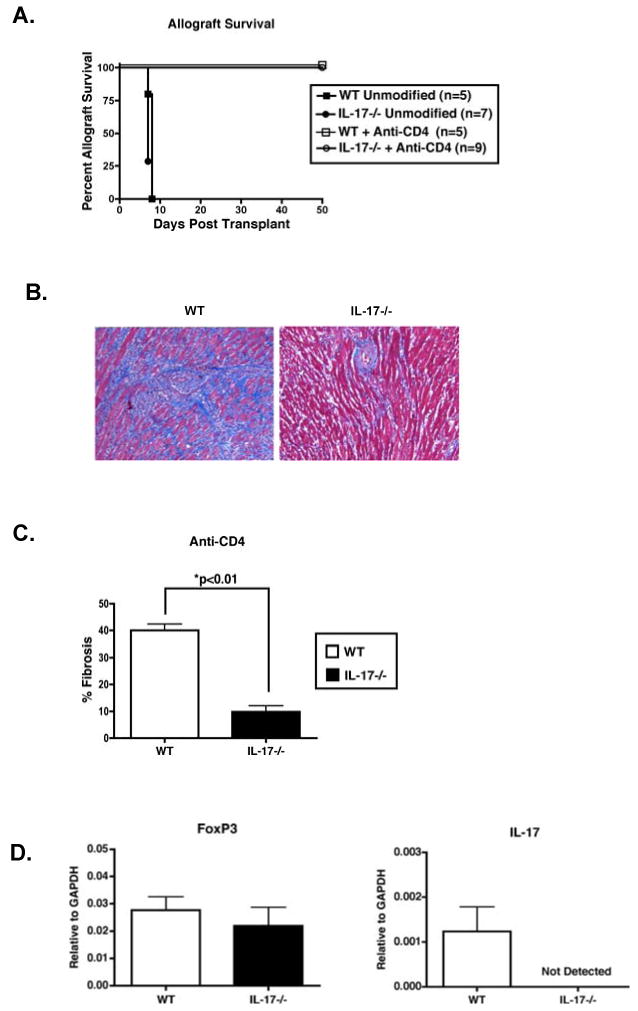
(A) WT (squares) and IL-17-/- (circles) mice were transplanted with BALB/c cardiac allografts and were either left untreated (closed symbols) or transiently depleted of CD4+ cells (open symbols). Graft function was monitored by palpation. Unmodified recipients were harvested at time of rejection, while inductive anti-CD4 mAb treated recipients were harvested at day 50 post-transplantation. Numbers in parentheses represent the number of recipients in each group. (B) Sections of grafts from recipients transiently depleted of CD4+ T cells (day 50 post-transplant) were stained with Masson's trichrome stain. Frames are of grafts from WT and IL-17-/- recipients and are representative of at least 6 WT and 9 IL-17-/- recipient allografts. 200× magnification. (C) Quantification of fibrosis by morphometric analysis. Bars represent the average percentage (+/- S.E.M.) of graft area positive for fibrosis in at least 5 WT and IL-17-/- recipients treated with anti-CD4 mAb. WT (open bars) and IL-17-/- (closed bars). (D) On day 50 post-transplant, RNA was harvested from allografts of WT and IL-17-/- recipients transiently depleted of CD4+ T cells. Intragraft transcript levels of FoxP3 and IL-17 were assessed by real-time RT-PCR. Bars depict the means of RNA from 5 WT and 9 IL-17-/- grafts.
To investigate the effect of IL-17 deficiency on intragraft gene expression, we assessed both IL-17 and FoxP3 transcript levels from WT and IL-17-/- recipients that were transiently depleted of CD4+ T cells. Allografts from IL-17-/- recipients exhibited comparable FoxP3 gene expression to WT counterparts, while IL-17 transcripts were not detected in the IL-17-/- recipients (Figure 7D). These results suggest that reduced fibrosis in IL-17-/- recipients transiently depleted of CD4+ T cells is not a result of enhanced Treg number as assessed by intragraft FoxP3 transcript levels.
Discussion
We have previously reported an association between TGFβ and graft fibrosis associated with CR using the mouse vascularized cardiac model (45). Intragraft transcript levels of TGFβ are readily detectable in the CR grafts from recipients transiently depleted of CD4+ T cells, but not in the grafts of anti-CD40L treated recipients, which remain free of CR. In the current study, we used inductive anti-CD4 or anti-CD40L mAb therapy and CD4-DNR recipients to evaluate the role of T cell TGFβ signaling in graft acceptance and the progression of fibrosis associated with CR. Collectively, our data suggest that in WT allograft recipients transiently depleted of CD4+ cells, allograft acceptance, T and B cell hyporesponsiveness, and fibrosis of the graft are dependent on TGFβ signaling in T cells. We further demonstrate that IL-17 is involved in the development of graft fibrosis in recipients transiently depleted of CD4+ T cells.
T cell TGFβ signaling is not required for long term allograft acceptance following anti-CD40L therapy (Figure 1). It is not fully understood how blockade of CD40L results in allograft acceptance, but a number of mechanisms, including donor-reactive T cell anergy and/or deletion, and the induction of Treg have been proposed (69, 72). We have reported that anti-CD40L mAb therapy allows for a transient appearance of primed donor-reactive cells (54). Hence, it is possible that these primed T cells express CD40L, which targets these cells for deletion and/or silencing by anti-CD40L mAb therapy. Our study demonstrates that these processes do not require T cell TGFβ signaling.
In recipients transiently depleted of CD4+ T cells, our findings support a TGFβ-dependent mechanism of graft acceptance. Inductive anti-CD4 mAb treatment of recipients results in transient depletion of CD4+ T cells at the time of transplant (48-50). CD4+ T cells begin to repopulate the periphery 3-4 weeks post-transplantation (48-50). As CD4+ T cells return, donor-reactive T cells are functionally distinct from naïve cells in that these repopulating CD4+ are hyporesponsive toward the graft but mount Th2 recall responses (54). This altered functional T cell capacity is associated with intragraft expression of TGFβ. Inductive anti-CD4 mAb treatment of CD4-DNR recipients results in allograft rejection between day 35-40 post-transplantation (Figure 1). Th1 and Th2 were maintained in a quiescent state in WT recipients transiently depleted of CD4+ cells (Figure 2). In contrast, CD4-DNR recipients treated with inductive anti-CD4 mAb mount donor-reactive Th1 and Th2 responses, revealing that the induction of hyporesponsiveness requires that T cells be responsive to TGFβ (Figure 2). Repopulation of CD4+ T cells in the periphery is required for rejection in that CD4-DNR transplant recipients that are continuously depleted of CD4+ T cells did not reject their grafts (Figure 1) and did not mount Th1 and Th2 responses (data not shown). These observations support an essential role for TGFβ in cellular hyporesponsiveness and allograft acceptance in recipients inductively depleted of CD4+ T cells.
In the absence of TGFβ signaling, T cells differentiate into effector cells, secrete cytokines and provide help to B cells in CD4-DNR recipients transiently depleted of CD4+ cells. CD4-DNR recipients transiently depleted of CD4+ cells mounted significantly elevated donor-reactive alloantibody levels of both Th2 induced non-complement fixing, IgG1, and Th1 induced complement fixing, IgG2a, compared to wild type controls (Figure 4) (73). Both subclassses have been documented to synergize to cause rejection of cardiac allografts (73). IgG and complement split product C3d and C4d capillary deposition in both human and mouse myocardium is significantly associated with graft loss (74) and these products were detected in the vessels and surrounding the cardiac myocytes in the CD4-DNR recipient allografts that were rejected (Figure 5). Hence, T cells that are unable to respond to TGFβ mount cellular responses and provide help to activate alloreactive B cells in recipients inductively depleted of CD4+ T cells.
Donor-specific hyporesponsiveness is observed in both human and mouse transplant recipients that exhibit prolonged allograft acceptance (15, 66, 75). In many of these studies, allograft acceptance is strongly associated with Treg infiltration into the graft as detected by high FoxP3 transcript levels (15, 66, 75, 76). Treg are believed to play a critical role within allografts by inhibiting alloreactive T cells responses (15). Studies in skin allograft models reveal Treg enrichment in accepted grafts (77) is dependent on TGFβ and that this cytokine is important for long-term acceptance (42, 66). Consistent with these observations, inductive anti-CD4 mAb treatment of WT recipients exhibit enhanced intragraft FoxP3 transcript levels compared to CD4-DNR (Figure 6). In CD4-DNR recipients transiently depleted of CD4+ T cells, reduced FoxP3 expression may indicate impaired maintenance of peripheral Treg, reduced Treg localization within the graft, or a failure in Treg induction (reviewed in (69). Donor-reactive Th responses observed in CD4-DNR recipients may result from decreased induction of Treg and/or the failure of Treg to control effector cells in the absence of TGFβ signaling (78). Our data suggest an active regulatory mechanism in which Treg migration into the grafts (Figure 6) and inhibition of alloreactive responses (Figure 2) require T cell TGFβ signaling (79, 80).
While functional T cell TGFβ signaling prevented allograft rejection in recipients inductively depleted of CD4+ T cells, it promoted fibrosis associated with CR (Figure 3). In contrast, T cell unresponsiveness to TGFβ resulted in minimal fibrosis of grafts (Figure 3). It has been reported that sustained production of TGFβ in tissues is a main contributor to the development of fibrosis (57, 81), but we have observed that gene transfer of TGFβ in syngeneic grafts fail to develop CR (45). Fibrosis was observed only in allografts that adenovirally expressed TGFβ and were transplanted into anti-CD40L mAb treated recipients (45). This indicates that TGFβ alone is insufficient to induce fibrosis of the graft, and that alloantigen and elements of the immune system are required for fibrosis induction.
As T cells infiltrate the allograft and respond to TGFβ, they secrete multiple cytokines, which may influence the local environment to become pro-fibrotic. One TGFβ-induced cytokine that could potentially mediate fibrosis in this setting is IL-17 (31, 32). IL-17 is important in coordinating local tissue inflammation through the induction of various pro-inflammatory cytokines (27, 28). IL-17 has also been implicated as a contributor to fibrosis in a number of diseases (30, 33-35, 37). Allografts from IL-17-/-recipients transiently depleted of CD4+ T cells showed a significant reduction in fibrosis relative to their WT counterparts (Figure 7). These findings are consistent with TGFβ induced IL-17 promoting interstitial fibrosis in CR allografts.
IL-17 may induce collagen deposition within CR allografts through multiple mechanisms. IL-17 upregulates collagen gene expression in primary mouse cardiac fibroblasts (32). Indirectly, IL-17 induces the production of IL-6 (27, 28), which enhances the accumulation of collagen (53, 82-84). IL-17 may also play a role in fibrosis by acting as a potent pro-inflammatory cytokine that induces endothelial cells and fibroblasts to secrete additional pro-inflammatory cytokines and chemokines (27, 28). These factors may enhance the recruitment of APC and alloreactive T cells into the graft resulting in myocardial damage and extracellular matrix remodeling that favors fibrosis (4).
In summary, TGFβ is critical for the induction of fibrosis in this model of CR and in a number of fibrotic diseases, including diabetic nephropathy, rheumatoid arthritis, myocarditis, Crohn's disease and radiation-induced fibrosis (reviewed in (30)). As in most immune-mediated diseases, TGFβ can have both exacerbating and ameliorating actions making global inhibition of TGFβ unacceptable and local targeting of TGFβ or its downstream mediators an attractive therapy. This is evident in recipients transiently depleted of CD4+ cells in which T cell responsiveness to TGFβ is critical in maintaining alloreactive T cells in a hyporesponsive state. Our findings that TGFβ in CR grafts correlates with localized Th17 induction supports findings in other chronic inflammatory diseases (30) and provides a therapeutic target for preventing CR, while sparing the immunosuppressive activities of TGFβ.
Acknowledgments
The authors would like to thank Dr. Bryna E. Burrell, Dr. Jose Diaz and Sherri C. Wood for their assistance with protocols and comments throughout the course of these investigations. The authors would also like to thank Kimberly C. Gates for outstanding colony maintenance.
Footnotes
Disclosures: The authors declare no conflict of interest.
This work was supported by R01 HL070613 (DKB), R01 AI061469 (DKB) and by T90 DK070071 (SMF).
Abbreviations used in this paper: anti-CD40L, anti-CD40 ligand; CD4-DNR, transgenic mice with a T cell specific dominant negative TGFβ receptor; CR, chronic allograft rejection; GIC, graft infiltrating cells; IL-17-/-, IL-17 deficient; TGFβ, transforming growth factor-beta; TGFβRII, transforming growth factor-beta receptor II; Treg, T regulatory cell; WT, wild type.
References
- 1.Weiss MJ, Madsen JC, Rosengard BR, Allan JS. Mechanisms of chronic rejection in cardiothoracic transplantation. Front Biosci. 2008;13:2980–2988. doi: 10.2741/2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Waaga AM, Gasser M, Laskowski I, Tilney NL. Mechanisms of chronic rejection. Curr Opin Immunol. 2000;12:517–521. doi: 10.1016/s0952-7915(00)00132-1. [DOI] [PubMed] [Google Scholar]
- 3.Mannon RB. Therapeutic targets in the treatment of allograft fibrosis. Am J Transplant. 2006;6:867–875. doi: 10.1111/j.1600-6143.2006.01261.x. [DOI] [PubMed] [Google Scholar]
- 4.Orosz CG, Pelletier RP. Chronic remodeling pathology in grafts. Curr Opin Immunol. 1997;9:676–680. doi: 10.1016/s0952-7915(97)80048-9. [DOI] [PubMed] [Google Scholar]
- 5.Womer KL, Vella JP, Sayegh MH. Chronic allograft dysfunction: mechanisms and new approaches to therapy. Semin Nephrol. 2000;20:126–147. [PubMed] [Google Scholar]
- 6.Garrity ER, Jr, Mehra MR. An update on clinical outcomes in heart and lung transplantation. Transplantation. 2004;77:S68–74. doi: 10.1097/01.tp.0000126930.57516.28. [DOI] [PubMed] [Google Scholar]
- 7.Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2006;24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- 8.Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kulkarni AB, Karlsson S. Transforming growth factor-beta 1 knockout mice. A mutation in one cytokine gene causes a dramatic inflammatory disease. Am J Pathol. 1993;143:3–9. [PMC free article] [PubMed] [Google Scholar]
- 10.Gorelik L, Flavell RA. Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity. 2000;12:171–181. doi: 10.1016/s1074-7613(00)80170-3. [DOI] [PubMed] [Google Scholar]
- 11.Lucas PJ, Kim SJ, Melby SJ, Gress RE. Disruption of T cell homeostasis in mice expressing a T cell-specific dominant negative transforming growth factor beta II receptor. J Exp Med. 2000;191:1187–1196. doi: 10.1084/jem.191.7.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rubtsov YP, Rudensky AY. TGFbeta signalling in control of T-cell-mediated self-reactivity. Nat Rev Immunol. 2007;7:443–453. doi: 10.1038/nri2095. [DOI] [PubMed] [Google Scholar]
- 13.Nakamura K, Kitani A, Strober W. Cell contact-dependent immunosuppression by CD4(+)CD25(+) regulatory T cells is mediated by cell surface-bound transforming growth factor beta. J Exp Med. 2001;194:629–644. doi: 10.1084/jem.194.5.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levings MK, Bacchetta R, Schulz U, Roncarolo MG. The role of IL-10 and TGF-beta in the differentiation and effector function of T regulatory cells. Int Arch Allergy Immunol. 2002;129:263–276. doi: 10.1159/000067596. [DOI] [PubMed] [Google Scholar]
- 15.Wood KJ, Sakaguchi S. Regulatory T cells in transplantation tolerance. Nat Rev Immunol. 2003;3:199–210. doi: 10.1038/nri1027. [DOI] [PubMed] [Google Scholar]
- 16.Ming JE, Russell KL, McDonald-McGinn DM, Zackai EH. Autoimmune disorders in Kabuki syndrome. Am J Med Genet A. 2005;132A:260–262. doi: 10.1002/ajmg.a.30332. [DOI] [PubMed] [Google Scholar]
- 17.Andersson J, Tran DQ, Pesu M, Davidson TS, Ramsey H, O'Shea JJ, Shevach EM. CD4+ FoxP3+ regulatory T cells confer infectious tolerance in a TGF-beta-dependent manner. J Exp Med. 2008;205:1975–1981. doi: 10.1084/jem.20080308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marie JC, Liggitt D, Rudensky AY. Cellular mechanisms of fatal early-onset autoimmunity in mice with the T cell-specific targeting of transforming growth factor-beta receptor. Immunity. 2006;25:441–454. doi: 10.1016/j.immuni.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 19.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 21.Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 22.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 23.Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, Shen Y, Du J, Rubtsov YP, Rudensky AY, Ziegler SF, Littman DR. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. 2008;453:236–240. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, Basham B, Smith K, Chen T, Morel F, Lecron JC, Kastelein RA, Cua DJ, McClanahan TK, Bowman EP, de Waal Malefyt R. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. 2007;8:950–957. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- 25.Korn T, Oukka M, Kuchroo V, Bettelli E. Th17 cells: effector T cells with inflammatory properties. Semin Immunol. 2007;19:362–371. doi: 10.1016/j.smim.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, Oukka M, Kuchroo VK. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bettelli E, Korn T, Oukka M, Kuchroo VK. Induction and effector functions of T(H)17 cells. Nature. 2008;453:1051–1057. doi: 10.1038/nature07036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. 2008;28:454–467. doi: 10.1016/j.immuni.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25:821–852. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- 30.Pohlers D, Brenmoehl J, Loffler I, Muller CK, Leipner C, Schultze-Mosgau S, Stallmach A, Kinne RW, Wolf G. TGF-beta and fibrosis in different organs - molecular pathway imprints. Biochim Biophys Acta. 2009;1792:746–756. doi: 10.1016/j.bbadis.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 31.Cortez DM, Feldman MD, Mummidi S, Valente AJ, Steffensen B, Vincenti M, Barnes JL, Chandrasekar B. IL-17 stimulates MMP-1 expression in primary human cardiac fibroblasts via p38 MAPK- and ERK1/2-dependent C/EBP-beta, NF-kappaB, and AP-1 activation. Am J Physiol Heart Circ Physiol. 2007;293:H3356–3365. doi: 10.1152/ajpheart.00928.2007. [DOI] [PubMed] [Google Scholar]
- 32.Venkatachalam K, Mummidi S, Cortez DM, Prabhu SD, Valente AJ, Chandrasekar B. Resveratrol inhibits high glucose-induced PI3K/Akt/ERK-dependent interleukin-17 expression in primary mouse cardiac fibroblasts. Am J Physiol Heart Circ Physiol. 2008;294:H2078–2087. doi: 10.1152/ajpheart.01363.2007. [DOI] [PubMed] [Google Scholar]
- 33.Burlingham WJ, Love RB, Jankowska-Gan E, Haynes LD, Xu Q, Bobadilla JL, Meyer KC, Hayney MS, Braun RK, Greenspan DS, Gopalakrishnan B, Cai J, Brand DD, Yoshida S, Cummings OW, Wilkes DS. IL-17-dependent cellular immunity to collagen type V predisposes to obliterative bronchiolitis in human lung transplants. J Clin Invest. 2007;117:3498–3506. doi: 10.1172/JCI28031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Molet S, Hamid Q, Davoine F, Nutku E, Taha R, Page N, Olivenstein R, Elias J, Chakir J. IL-17 is increased in asthmatic airways and induces human bronchial fibroblasts to produce cytokines. J Allergy Clin Immunol. 2001;108:430–438. doi: 10.1067/mai.2001.117929. [DOI] [PubMed] [Google Scholar]
- 35.Dubin PJ, McAllister F, Kolls JK. Is cystic fibrosis a TH17 disease? Inflamm Res. 2007;56:221–227. doi: 10.1007/s00011-007-6187-2. [DOI] [PubMed] [Google Scholar]
- 36.Fukami N, Ramachandran S, Saini D, Walter M, Chapman W, Patterson GA, Mohanakumar T. Antibodies to MHC class I induce autoimmunity: role in the pathogenesis of chronic rejection. J Immunol. 2009;182:309–318. doi: 10.4049/jimmunol.182.1.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simonian PL, Roark CL, Wehrmann F, Lanham AK, Diaz del Valle F, Born WK, O'Brien RL, Fontenot AP. Th17-polarized immune response in a murine model of hypersensitivity pneumonitis and lung fibrosis. J Immunol. 2009;182:657–665. [PMC free article] [PubMed] [Google Scholar]
- 38.Walsh PT, Taylor DK, Turka LA. Tregs and transplantation tolerance. J Clin Invest. 2004;114:1398–1403. doi: 10.1172/JCI23238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yong Z, Chang L, Mei YX, Yi L. Role and mechanisms of CD4+CD25+ regulatory T cells in the induction and maintenance of transplantation tolerance. Transpl Immunol. 2007;17:120–129. doi: 10.1016/j.trim.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 40.Josien R, Douillard P, Guillot C, Muschen M, Anegon I, Chetritt J, Menoret S, Vignes C, Soulillou JP, Cuturi MC. A critical role for transforming growth factor-beta in donor transfusion-induced allograft tolerance. J Clin Invest. 1998;102:1920–1926. doi: 10.1172/JCI4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zelenika D, Adams E, Mellor A, Simpson E, Chandler P, Stockinger B, Waldmann H, Cobbold SP. Rejection of H-Y disparate skin grafts by monospecific CD4+ Th1 and Th2 cells: no requirement for CD8+ T cells or B cells. J Immunol. 1998;161:1868–1874. [PubMed] [Google Scholar]
- 42.Daley SR, Ma J, Adams E, Cobbold SP, Waldmann H. A key role for TGF-beta signaling to T cells in the long-term acceptance of allografts. J Immunol. 2007;179:3648–3654. doi: 10.4049/jimmunol.179.6.3648. [DOI] [PubMed] [Google Scholar]
- 43.Chan SY, Goodman RE, Szmuszkovicz JR, Roessler B, Eichwald EJ, Bishop DK. DNA-liposome versus adenoviral mediated gene transfer of transforming growth factor beta1 in vascularized cardiac allografts: differential sensitivity of CD4+ and CD8+ T cells to transforming growth factor beta1. Transplantation. 2000;70:1292–1301. doi: 10.1097/00007890-200011150-00006. [DOI] [PubMed] [Google Scholar]
- 44.Csencsits K, Wood SC, Lu G, Bishop DK. Transforming growth factor-beta1 gene transfer is associated with the development of regulatory cells. Am J Transplant. 2005;5:2378–2384. doi: 10.1111/j.1600-6143.2005.01042.x. [DOI] [PubMed] [Google Scholar]
- 45.Csencsits K, Wood SC, Lu G, Faust SM, Brigstock D, Eichwald EJ, Orosz CG, Bishop DK. Transforming growth factor beta-induced connective tissue growth factor and chronic allograft rejection. Am J Transplant. 2006;6:959–966. doi: 10.1111/j.1600-6143.2006.01292.x. [DOI] [PubMed] [Google Scholar]
- 46.Nakae S, Komiyama Y, Nambu A, Sudo K, Iwase M, Homma I, Sekikawa K, Asano M, Iwakura Y. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity. 2002;17:375–387. doi: 10.1016/s1074-7613(02)00391-6. [DOI] [PubMed] [Google Scholar]
- 47.Corry RJ, Winn HJ, Russell PS. Primarily vascularized allografts of hearts in mice. The role of H-2D, H-2K, and non-H-2 antigens in rejection. Transplantation. 1973;16:343–350. doi: 10.1097/00007890-197310000-00010. [DOI] [PubMed] [Google Scholar]
- 48.Bishop DK, Li W, Chan SY, Ensley RD, Shelby J, Eichwald EJ. Helper T lymphocyte unresponsiveness to cardiac allografts following transient depletion of CD4-positive cells. Implications for cellular and humoral responses. Transplantation. 1994;58:576–584. doi: 10.1097/00007890-199409150-00009. [DOI] [PubMed] [Google Scholar]
- 49.Piccotti JR, Li K, Chan SY, Eichwald EJ, Bishop DK. Cytokine regulation of chronic cardiac allograft rejection: evidence against a role for Th1 in the disease process. Transplantation. 1999;67:1548–1555. doi: 10.1097/00007890-199906270-00008. [DOI] [PubMed] [Google Scholar]
- 50.Csencsits K, Burrell BE, Lu G, Eichwald EJ, Stahl GL, Bishop DK. The classical complement pathway in transplantation: unanticipated protective effects of C1q and role in inductive antibody therapy. Am J Transplant. 2008;8:1622–1630. doi: 10.1111/j.1600-6143.2008.02295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matesic D, Lehmann PV, Heeger PS. High-resolution characterization of cytokine-producing alloreactivity in naive and allograft-primed mice. Transplantation. 1998;65:906–914. doi: 10.1097/00007890-199804150-00008. [DOI] [PubMed] [Google Scholar]
- 52.Burrell BE, Lu G, Li XC, Bishop DK. OX40 costimulation prevents allograft acceptance induced by CD40-CD40L blockade. J Immunol. 2009;182:379–390. doi: 10.4049/jimmunol.182.1.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Diaz JA, Booth AJ, Lu G, Wood SC, Pinsky DJ, Bishop DK. Critical role for IL-6 in hypertrophy and fibrosis in chronic cardiac allograft rejection. Am J Transplant. 2009;9:1773–1783. doi: 10.1111/j.1600-6143.2009.02706.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wood SC, Lu G, Burrell BE, Bishop DK. Transplant acceptance following anti-CD4 versus anti-CD40L therapy: evidence for differential maintenance of graft-reactive T cells. Am J Transplant. 2008;8:2037–2048. doi: 10.1111/j.1600-6143.2008.02372.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nathan MJ, Yin D, Eichwald EJ, Bishop DK. The immunobiology of inductive anti-CD40L therapy in transplantation: allograft acceptance is not dependent upon the deletion of graft-reactive T cells. Am J Transplant. 2002;2:323–332. doi: 10.1034/j.1600-6143.2002.20406.x. [DOI] [PubMed] [Google Scholar]
- 56.Branton MH, Kopp JB. TGF-beta and fibrosis. Microbes Infect. 1999;1:1349–1365. doi: 10.1016/s1286-4579(99)00250-6. [DOI] [PubMed] [Google Scholar]
- 57.Leask A. TGFbeta, cardiac fibroblasts, and the fibrotic response. Cardiovasc Res. 2007;74:207–212. doi: 10.1016/j.cardiores.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 58.Jain S, Furness PN, Nicholson ML. The role of transforming growth factor beta in chronic renal allograft nephropathy. Transplantation. 2000;69:1759–1766. doi: 10.1097/00007890-200005150-00001. [DOI] [PubMed] [Google Scholar]
- 59.Colvin RB. Antibody-mediated renal allograft rejection: diagnosis and pathogenesis. J Am Soc Nephrol. 2007;18:1046–1056. doi: 10.1681/ASN.2007010073. [DOI] [PubMed] [Google Scholar]
- 60.Singh N, Pirsch J, Samaniego M. Antibody-mediated rejection: treatment alternatives and outcomes. Transplant Rev (Orlando) 2009;23:34–46. doi: 10.1016/j.trre.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 61.Calderhead DM, Kosaka Y, Manning EM, Noelle RJ. CD40-CD154 interactions in B-cell signaling. Curr Top Microbiol Immunol. 2000;245:73–99. doi: 10.1007/978-3-642-59641-4_4. [DOI] [PubMed] [Google Scholar]
- 62.Wasowska BA, Qian Z, Cangello DL, Behrens E, Van Tran K, Layton J, Sanfilippo F, Baldwin WM., 3rd Passive transfer of alloantibodies restores acute cardiac rejection in IgKO mice. Transplantation. 2001;71:727–736. doi: 10.1097/00007890-200103270-00007. [DOI] [PubMed] [Google Scholar]
- 63.Baldwin WM, 3rd, Kasper EK, Zachary AA, Wasowska BA, Rodriguez ER. Beyond C4d: other complement-related diagnostic approaches to antibody-mediated rejection. Am J Transplant. 2004;4:311–318. doi: 10.1111/j.1600-6143.2004.00348.x. [DOI] [PubMed] [Google Scholar]
- 64.Rodriguez ER, Skojec DV, Tan CD, Zachary AA, Kasper EK, Conte JV, Baldwin WM., 3rd Antibody-mediated rejection in human cardiac allografts: evaluation of immunoglobulins and complement activation products C4d and C3d as markers. Am J Transplant. 2005;5:2778–2785. doi: 10.1111/j.1600-6143.2005.01074.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schubert LA, Jeffery E, Zhang Y, Ramsdell F, Ziegler SF. Scurfin (FOXP3) acts as a repressor of transcription and regulates T cell activation. J Biol Chem. 2001;276:37672–37679. doi: 10.1074/jbc.M104521200. [DOI] [PubMed] [Google Scholar]
- 66.Cobbold SP, Castejon R, Adams E, Zelenika D, Graca L, Humm S, Waldmann H. Induction of foxP3+ regulatory T cells in the periphery of T cell receptor transgenic mice tolerized to transplants. J Immunol. 2004;172:6003–6010. doi: 10.4049/jimmunol.172.10.6003. [DOI] [PubMed] [Google Scholar]
- 67.Fantini MC, Becker C, Monteleone G, Pallone F, Galle PR, Neurath MF. Cutting edge: TGF-beta induces a regulatory phenotype in CD4+CD25- T cells through Foxp3 induction and down-regulation of Smad7. J Immunol. 2004;172:5149–5153. doi: 10.4049/jimmunol.172.9.5149. [DOI] [PubMed] [Google Scholar]
- 68.Veldhoen M, Stockinger B. TGFbeta1, a “Jack of all trades”: the link with pro-inflammatory IL-17-producing T cells. Trends Immunol. 2006;27:358–361. doi: 10.1016/j.it.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 69.Waldmann H, Chen TC, Graca L, Adams E, Daley S, Cobbold S, Fairchild PJ. Regulatory T cells in transplantation. Semin Immunol. 2006;18:111–119. doi: 10.1016/j.smim.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 70.Wakashin H, Hirose K, Maezawa Y, Kagami S, Suto A, Watanabe N, Saito Y, Hatano M, Tokuhisa T, Iwakura Y, Puccetti P, Iwamoto I, Nakajima H. IL-23 and Th17 cells enhance Th2-cell-mediated eosinophilic airway inflammation in mice. Am J Respir Crit Care Med. 2008;178:1023–1032. doi: 10.1164/rccm.200801-086OC. [DOI] [PubMed] [Google Scholar]
- 71.Deleuran B, Abraham DJ. Possible implication of the effector CD4+ T-cell subpopulation TH17 in the pathogenesis of systemic scleroderma. Nat Clin Pract Rheumatol. 2007;3:682–683. doi: 10.1038/ncprheum0618. [DOI] [PubMed] [Google Scholar]
- 72.Waldmann H, Graca L, Cobbold S, Adams E, Tone M, Tone Y. Regulatory T cells and organ transplantation. Semin Immunol. 2004;16:119–126. doi: 10.1016/j.smim.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 73.Rahimi S, Qian Z, Layton J, Fox-Talbot K, Baldwin WM, 3rd, Wasowska BA. Non-complement- and complement-activating antibodies synergize to cause rejection of cardiac allografts. Am J Transplant. 2004;4:326–334. doi: 10.1111/j.1600-6143.2004.00334.x. [DOI] [PubMed] [Google Scholar]
- 74.Behr TM, Feucht HE, Richter K, Reiter C, Spes CH, Pongratz D, Uberfuhr P, Meiser B, Theisen K, Angermann CE. Detection of humoral rejection in human cardiac allografts by assessing the capillary deposition of complement fragment C4d in endomyocardial biopsies. J Heart Lung Transplant. 1999;18:904–912. doi: 10.1016/s1053-2498(99)00043-1. [DOI] [PubMed] [Google Scholar]
- 75.Bestard O, Cruzado JM, Mestre M, Caldes A, Bas J, Carrera M, Torras J, Rama I, Moreso F, Seron D, Grinyo JM. Achieving donor-specific hyporesponsiveness is associated with FOXP3+ regulatory T cell recruitment in human renal allograft infiltrates. J Immunol. 2007;179:4901–4909. doi: 10.4049/jimmunol.179.7.4901. [DOI] [PubMed] [Google Scholar]
- 76.Lee I, Wang L, Wells AD, Dorf ME, Ozkaynak E, Hancock WW. Recruitment of Foxp3+ T regulatory cells mediating allograft tolerance depends on the CCR4 chemokine receptor. J Exp Med. 2005;201:1037–1044. doi: 10.1084/jem.20041709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Graca L, Cobbold SP, Waldmann H. Identification of regulatory T cells in tolerated allografts. J Exp Med. 2002;195:1641–1646. doi: 10.1084/jem.20012097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fahlen L, Read S, Gorelik L, Hurst SD, Coffman RL, Flavell RA, Powrie F. T cells that cannot respond to TGF-beta escape control by CD4(+)CD25(+) regulatory T cells. J Exp Med. 2005;201:737–746. doi: 10.1084/jem.20040685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen D, Bromberg JS. T regulatory cells and migration. Am J Transplant. 2006;6:1518–1523. doi: 10.1111/j.1600-6143.2006.01372.x. [DOI] [PubMed] [Google Scholar]
- 80.Zhang N, Schroppel B, Lal G, Jakubzick C, Mao X, Chen D, Yin N, Jessberger R, Ochando JC, Ding Y, Bromberg JS. Regulatory T cells sequentially migrate from inflamed tissues to draining lymph nodes to suppress the alloimmune response. Immunity. 2009;30:458–469. doi: 10.1016/j.immuni.2008.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J. 2004;18:816–827. doi: 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- 82.Hirano T, Akira S, Taga T, Kishimoto T. Biological and clinical aspects of interleukin 6. Immunol Today. 1990;11:443–449. doi: 10.1016/0167-5699(90)90173-7. [DOI] [PubMed] [Google Scholar]
- 83.Uitto J. IL-6 signaling pathway in keloids: a target for pharmacologic intervention? J Invest Dermatol. 2007;127:6–8. doi: 10.1038/sj.jid.5700604. [DOI] [PubMed] [Google Scholar]
- 84.Duncan MR, Berman B. Stimulation of collagen and glycosaminoglycan production in cultured human adult dermal fibroblasts by recombinant human interleukin 6. J Invest Dermatol. 1991;97:686–692. doi: 10.1111/1523-1747.ep12483971. [DOI] [PubMed] [Google Scholar]