

Abstract
A rat model of low myocardial blood flow was established to test the hypothesis that post-translational changes to proteins of the thin and thick muscle filaments correlate with decreased cardiac contractility. Following 3 days of low blood flow by constriction of the left anterior descending artery, rat hearts demonstrated a reduction in fractional shortening at rest and a relative decline in fractional shortening when challenged with high dose versus low dose dobutamine, reflecting reduced energy reserves. Permeabilized fibers from low blood flow hearts demonstrated a decline in maximum force per cross-section and Ca2+ sensitivity as compared to their sham operated counterparts. An examination of sarcomeric proteins by twodimensional gel electrophoresis, mass spectrometry, and phospho-specific antibodies provided evidence for Ser23/24 and Ser43/45 phosphorylation of troponin I (TnI). Total TnI phosphorylation was not different between the groups, but Ser23/24 phosphorylation declined with low blood flow, implying an accompanying increase in phosphorylation at other sites of TnI. Affinity chromatography demonstrated that TnI from low blood flow myocardium had reduced relative affinity to Ca2+ bound troponin C compared to TnI from sham operated hearts, providing a mechanism for reduced Ca2+ sensitivity of force production in low blood flow fibers. These findings suggest that altered TnI function, due to changes in the distribution of phosphorylated sites, is an early contributor to reduced contractility of the heart.
Keywords: Troponin, Force, Phosphorylation, Myosin
Introduction
The contraction of striated muscles is regulated by the troponin complex, which controls the association of actin and myosin in a calcium-dependent manner. Following membrane depolarization, the rise in intracellular [Ca2+] is sensed directly by the Ca2+-binding subunit of the troponin complex, troponin C (TnC). Calcium bound TnC undergoes a conformational change increasing its apparent affinity for the inhibitory subunit of the troponin complex, troponin I (TnI). This movement of TnI, along with an accompanying shift of the tropomyosin strand, removes both a kinetic and a steric hindrance for myosin's binding to actin, facilitating crossbridge cycling and force generation (Tobacman 1996).
In various models of myocardial injury, a consistent observation has been the decrease in force production by permeabilized cardiac muscle fibers, along with a progression from Ca2+ desensitization to Ca2+ sensitization of the myocardium with advancing heart disease (Gao et al. 1995; Belin et al. 2006; Chen and Ogut 2006; Bito et al. 2007; Messer et al. 2007). Since ion channels and membrane associated signaling are largely absent from permeabilized preparations, the changes in force production and Ca2+ sensitivity under these pathophysiological conditions suggests that the function of thin and thick filament proteins may be altered through post-translational modification. Recently, TnI has garnered increased attention for its role in cardiac dysfunction since it is an in vivo target for protein kinase A (PKA) and, putatively, protein kinase C (PKC) (Kobayashi and Solaro 2005). Two adjacent serine residues, Ser23 and 24, are targets for PKA activated by β-adrenergic stimulation (Zhang et al. 1995). The extent of phosphorylation at these sites, and hence TnI function, may change when the overall activity of the β-adrenergic pathway declines (Bristow et al. 1982; Zakhary et al. 1999). The progression of cardiac dysfunction is also noted for an increase in PKC isoenzyme activity (Bowling et al. 1999), implying that increased phosphorylation of TnI at Ser43/45 by members of this protein kinase family may also participate in altered contractility (Noland et al. 1995; Kobayashi and Solaro 2005).
Although Ser23/24 phosphorylation of TnI has been verified directly (Zabrouskov et al. 2008), evidence for TnI phosphorylation at putative PKC sites has been confined to in vitro experiments (Jideama et al. 1996; Noland et al. 1996), with implicit evidence in transgenic mouse models (Pyle et al. 2002). To further explore changes in the state of contractile proteins during altered blood flow to the heart (Chen and Ogut 2006), we established a rat model of low myocardial blood flow to expand our analyses of the contractile apparatus in response to impaired myocardial perfusion, with a focus on the state of TnI phosphorylation. Permeabilized fibers from the low flow rat hearts demonstrated reduced force production and reduced Ca2+ sensitivity as compared to sham operated rats, suggesting an altered function at the level of the contractile filaments. Detailed examination of TnI phosphorylation demonstrated reduced Ser23/24 phosphorylation while providing novel evidence for Ser43/45 phosphorylation in vivo. Importantly, the data suggest that altered phosphorylation of myofilament proteins plays a role in the decline of force and loss of Ca2+ sensitivity during low blood flow to the heart.
Materials and methods
Rat model of low flow
The rat model of low blood flow was adapted from a previously published method (Chen et al. 2006). The surgical operation, pre- and post-operative palliative procedures were approved by the Institutional Animal Care and Use Committee of Case Western Reserve University, in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23). Following surgery, rats in both groups were euthanized 3 days later and the hearts were removed for study. For a detailed protocol description, please refer to the Supplemental Materials. Both sham operated and experimental animals underwent echocardiography to assess left ventricular function 3 days after surgery, as previously described (Chen et al. 2006). Measurements were taken at rest, following 1.5 μg dobutamine/g rat weight and 4.5 μg dobutamine/g rat weight. Results are presented as average ± SD and differences between groups were deemed statistically significant if P < 0.05.
Permeabilized muscle fiber contraction measurements
Once the low blood flow model was established, new cohorts of animals were prepared for fiber contraction measurements and protein analyses. Left ventricular trabeculae were dissected from the area at risk of sham-operated and low flow rat hearts, and the ends were fixed with glutaraldehyde and clamped between aluminum foil T-clips prior to permeabilization as previously described (Chen and Ogut 2006). The permeabilized trabeculae were transferred to a mechanics workstation that allowed control by either the force produced or the muscle length. Muscle length, width and thickness were measured while the fiber was in pCa9 (1 nM free Ca2+) solution. For pCa solutions, the free Ca2+ concentration was calculated by an iterative program based on published dissociation constants (Fabiato 1988). Ionic strength was kept constant at 200 mM, final pH was 7.0 and all experiments were done at 15°C. To determine the force–Ca2+ relationship, each trabeculae was cycled through the entire pCa range. The concentration of calcium required for half-maximal force production (EC50) was determined following individual Hill fits to the force versus free [Ca2+] data as previously described (Chen and Ogut 2006). Results are presented as average ± standard deviation and differences between groups were deemed statistically significant if P < 0.05.
Protein analyses
Two-dimensional SDS-PAGE was used to resolve TnT, TnI and MLC-2 isoelectric variants. Proteins were extracted from tissues obtained from the at risk area of the left ventricle by homogenization on ice in a micro tissue grinder using a buffer of 7 M urea, 2 M thiourea, 4% (w/v) 3-([3-cholamidopropyl] dimethylammonio)-2-hydroxy-1-propanesulfonate (CHAPS), 0.5% (v/v) pH 3–10 immobilized pH gradient (IPG) buffer, 1 mM EDTA, and EDTA-free Complete Protease Inhibitor (Roche, Indianapolis, IN). Following homogenization, the tissue was allowed to remain on ice for 5 min followed by centrifugation to remove insoluble debris. The homogenates were further processed with the 2D CleanUp Kit as necessary. For resolution of acidic proteins (pI < 7), homogenates were added to a rehydration solution containing 7 M urea, 2 M thiourea, 2% (w/v) CHAPS, 0.5% (v/v) 3.5–5 IPG buffer, 0.002% (w/v) bromophenol blue and protease inhibitor. Rehydration of 7 cm pH 3–5.6 NL IPG gel strips was for 7 h prior to the first dimension focusing. To resolve the basic TnI isoelectric variants, best results were obtained by cutting a 13 cm 7–11NL strip into two, and rehydrating the cathode half overnight, without left ventricular extracts, in 7 M urea, 2 M thiourea, 2% CHAPS, 0.5% 7–11 NL IPG buffer, 0.002% bromophenol blue, 12 μl/ml Destreak reagent, and protease inhibitor, as described (Rabilloud 1998). Appropriate amounts of the protein homogenate were then dissolved in the basic rehydration buffer and loaded at the anode using a sample cup. The IPG strips were focused in the “face-up” mode on an Ettan IPGphor II Isoelectric Focusing Unit. For basic strips, the filter paper placed at the cathode was pre-wetted with deionized water containing 12 μl/ml Destreak reagent. After the first-dimension, the gel strips were consecutively equilibrated for 15 min in 6 M urea, 50 mM Bis-Tris, pH 6.4, 30% glycerol, 2% SDS, and 0.002% bromophenol blue containing first 10 mM dithiothreitol and then 2.5% (w/v) iodoacetamide. Proteins on equilibrated IPG gel strips were then resolved by second dimension SDS–PAGE using the Bis-Tris buffering system (Invitrogen, LaJolla, CA). Resolved gels were stained by silver stain, Deep Purple total protein fluorescent stain, Pro-Q Diamond phosphoprotein stain (Invitrogen, Carlsbad, CA), or used for subsequent Western blotting. The positions of the MLC-2 isoelectric variants were identified by comparison to control experiments that resolved chicken smooth muscle MLC-2 protein purified from bacteria (data not shown), whereas the positions of TnT and TnI were confirmed by Western blotting. Densitometry was performed using a Personal Densitometer SI or a Typhoon 9410 and accompanying ImageQuant TL software. Results are presented as average ± standard deviation and differences between groups were deemed statistically significant if P < 0.05.
To determine the phosphorylation stoichiometry of TnT, Pro-Q Diamond phosphoprotein staining followed by Deep Purple total protein staining was used. A rat left ventricular homogenate was resolved, in triplicate, by two-dimensional SDS–PAGE using 3–5.6 NL IPG strips. The gels were stained with phosphoprotein stain to provide a ratio of the phosphate content for the two predominant TnT isoelectric variants. After destaining, gels were stained with total protein stain to provide a ratio of the total protein content of the two adjacent TnT isoelectric variants. The phosphoprotein ratio compared to the total protein ratio provides the relative stoichiometry of phosphorylation for the two phosphoforms. To determine the stoichiometry of TnI phosphorylation, five left ventricular total homogenates with known amounts of TnT phosphorylation were resolved by SDS–PAGE and stained for phosphoprotein. As TnT and TnI are equimolar in cardiac muscle, the phosphoprotein signal for TnT was used as an internal standard to determine the phosphorylation stoichiometry of TnI in the same sample. Based on the constraints imposed by the total TnI phosphorylation content, the resolved TnI isoelectric variants in two-dimensional SDS–PAGE could be reasonably assigned.
To determine the extent of total myosin binding protein C (MYBP-C) phosphorylation, homogenates were resolved by 29:1 7% SDS–PAGE and stained with either total protein stain or phosphoprotein stain. Total MYBP-C content across samples was normalized, and this normalization ratio was applied to the associated phosphoprotein signal.
Western blot analyses
To identify total TnI and Ser 23/24 phosphorylated TnI, all ten left ventricular extracts were resolved on a single gel by 29:1 (w/v) acrylamide : bisacrylamide 10% SDS–PAGE using the Bis-Tris buffering system. Following electrophoresis, the proteins were transferred to Hybond nitrocellulose membrane (GE Healthcare, Piscataway, NJ) and probed for total TnI expression using a polyclonal antibody generated against the central region of cardiac muscle TnI (amino acids 88–106, Fitzgerald Industries, Concord, MA) as well as Ser 23/24 phosphorylated TnI (Cell Signaling Technology, Danvers, MA). The blots were developed using the ECL Plus detection system (GE Healthcare, Piscataway, NJ) and two different exposure time points were collected for each blot. Signals were quantified by densitometry on a Personal Densitometer SI and were well within the linear range of the instrument. To provide a reliable comparison of relative Ser23/24 phosphorylated TnI content, all ten samples were analyzed on the same nitrocellulose membrane. In addition, two normalization steps were undertaken: (1) the total TnI signal was normalized across the ten samples, providing relative adjustment factors for each sample, (2) these adjustment factors were then applied to the Ser 23/24 phosphorylated TnI densitometry signals to adjust for total TnI content in each sample. Results are presented as average ± standard deviation and the difference between groups was deemed statistically significant if P < 0.05.
Troponin C affinity chromatography of Troponin I
A cDNA encoding rat cardiac troponin C, based on Genbank accession DQ062205, was cloned by reverse-transcription—PCR using standard molecular biology techniques. The TnC cDNA was cloned into the pAED4 expression vector and sequenced to ensure accurate cloning. E. coli BL21 Star (Invitrogen, LaJolla, CA) were transformed with the TnC expression construct, and used to inoculate 4 L of Terrific Broth culture media containing 100 μg/ml ampicillin and 25 μg/mL chloramphenicol. Bacteria were shaken at 37°C until O.D.600 nm = 0.4, and TnC protein expression was induced for 3 hours by the addition of isopropyl β-D-1-thiogalactopyranoside to 0.2 mM. Following induction, cells were harvested by centrifugation, resuspended in 50 ml of 20 mM potassium phosphate buffer, pH 7.0, 1 mM EDTA, 10 mM β-mercaptoethanol, protease inhibitor, and stored at −80°C overnight. Following sonication of the freeze-thawed bacterial suspension, the majority of contaminant proteins were cleared from the bacterial lysate by ammonium sulfate fractionation to 60% saturation, or ∼ 3 M (NH4)2SO4, which left the TnC in the supernatant. This supernatant was exchanged into 20 mM Tris-HCl, pH 7.8, 1 mM EDTA, 10 mM β-mercaptoethanol buffer by Sephadex G25 gel filtration using a HiPrep 26/10 desalting column and loaded on a similarly equilibrated 10 ml Source 15Q column. The anion exchange column was developed by a linear NaCl gradient up to 1 M in the equilibration buffer. Pure TnC eluting from the column was thoroughly dialyzed against 0.5% (w/v) ammonium bicarbonate and lyophilized.
Purified TnC was covalently coupled to 0.5 ml of CNBr-activated Sepharose 4B Fast Flow (GE Healthcare, Piscataway, NJ) according to the manufacturer's instructions, and the coupled beads were packed into a Tricorn 5/20 chromatography column (GE Healthcare, Piscataway, NJ). Left ventricular lysates were individually prepared from the five sham operated and five low flow rat hearts by homogenizing 35–40 mg of tissue in 1 ml of 1 M KCl, 20 mM HEPES, pH 7, 0.1 mM CaCl2, Roche EDTA-free Complete Protease Inhibitor and PhosStop phosphatase inhibitor. After centrifugation to remove insoluble materials, an aliquot of the lysate containing ∼ 5 mg total protein was diluted into a final solution of 300 mM KCl, 20 mM HEPES, pH 7, 0.1 mM CaCl2, containing Roche Protease and Phosphatase inhibitors. This mixture was incubated on ice for 30 min, followed by centrifugation to remove insoluble materials. The soluble fraction was loaded on the TnC affinity column and allowed to mix, end over end, for 3 h at room temperature. The column was connected to an AktaFPLC system running Unicorn software, and was washed with 300 mM KCl, 20 mM HEPES, pH 7, 0.1 mM CaCl2 until the absorbance of the eluate reached baseline. The relative TnI binding affinity was determined by a linear gradient that eventually substituted 1 M KCl for the 300 mM KCl in the washing buffer. To determine the relative affinity of the TnI to the TnC affinity column, the TnI elution profile was resolved by 12% SDS–PAGE and the fraction containing the peak of TnI elution was identified by densitometry. The midpoint of the conductivity range of the peak fraction was accepted as the value at which peak TnI elution occurred. All ten samples were run in random order and all experiments were done with the same batch of column buffers to ensure reproducible binding conditions and a reproducible linear KCl gradient. For the five sham operated versus five low flow rat samples, the conductivity at the start of the linear gradient was 35.8 ± 0.3 mS/cm (average ± S.D.) versus 35.7 ± 0.4 mS/cm, respectively, whereas the final conductivity at the end of the gradient was 92.1 ± 1.1 mS/cm versus 90.9 ± 3.0 mS/cm, respectively. The differences in conductivity at the start and end of the gradients were not statistically significant (P > 0.3).
Enrichment of troponin I from normal and low flow hearts
Fractions of enriched TnI were prepared from the low blood flow rat hearts for mass spectrometry. Briefly, 200 mg of ventricular tissue from each of five low flow rat hearts were pooled and extracted with homogenization solution (0.2 M KCl, 50 mM HEPES, pH 7.6, 1 mM EDTA, 1 mM EGTA, 10 mM DTT including protease and phosphatase inhibitors). The homogenate was centrifuged for 10 min at 50,000g and the precipitate was resuspended in a washing solution that substituted 50 mM KCl for 200 mM KCl. After centrifugation as before, the pellet was extracted with 1 M KCl in the above homogenization solution, followed by centrifugation that yielded TnI in the supernatant. This supernatant was buffer exchanged into 6 M urea, 50 mM HEPES, pH 7.6, 0.1 mM EDTA, 0.1 mM EGTA using a HiPrep 26/10 desalting column, and the resultant protein mixture was fractionated on a Mono S column with a gradient from 0 to 750 mM NaCl. Fractions containing TnI were identified by SDS–PAGE. Densitometry of pooled fractions demonstrated that TnI was 22% of the total protein in the pooled fractions (data not shown). Troponin I samples were analyzed by mass spectrometry at two independent facilities (Mayo Medical School Mass Spectrometry Facility and 21st Century Biochemicals). For protocol details, please refer to the Supplemental Materials.
Results
Rat model of low flow
A rat model of reduced blood flow was established through partial occlusion of the left anterior descending artery for 3 days. As shown in Fig. 1, the fractional shortening of hearts from the model was significantly reduced as compared to the sham operated rats (33.5 ± 1.1% vs. 38.6 ± 4.8%, respectively; P < 0.05). The change in fractional shortening was further characterized by dobutamine echocardiography to determine the response to inotropic stimulation. In this low blood flow model, low dose dobutamine produced an initial increase in fractional shortening over baseline (62.6 ± 4.0% vs. 33.5 ± 1.1%; P < 0.05), but a higher dobutamine dose resulted in a relative decline in fractional shortening (53.7 ± 2.7% vs. 62.6 ± 4.0%; P < 0.05). By contrast, sham operated rat hearts demonstrated a stepwise increase in fractional shortening to 56.1 ± 8.2% with low dose and a further increase to 71.6 ± 7.6% with high dose dobutamine.
Fig. 1.
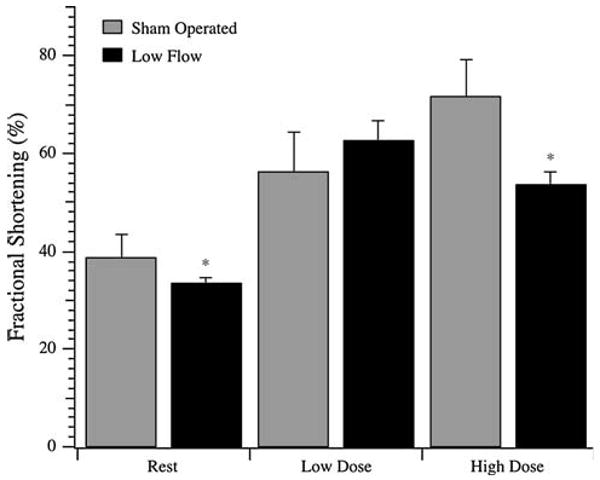
Dobutamine challenge of sham operated and low flow rat hearts. Fractional shortening was measured by echocardiography at rest and following dosing to 1.5 μg/g rat weight (Low Dose) and 4.5 μg/g rat weight (High Dose) dobutamine. Data are average ± SD. N = 5 rats for all groups. * denotes P < 0.05 compared to the corresponding sham operated counterpart
Contractile parameters of permeabilized muscle fibers
Left ventricular trabeculae were recovered from an independent cohort of sham operated and low flow rat hearts and permeabilized by Triton X-100 to measure the maximum force per cross-sectional area and the Ca2+-concentration required to generate half-maximal force (Fig. 2). The Fmax of fibers from the low flow rat hearts was significantly depressed in comparison to fibers from sham operated hearts (28.67 ± 4.25 vs. 20.23 ± 1.72 mN/mm2; P < 0.05). This was coupled with an increase in the EC50 for fibers from the low flow rat hearts compared to sham operated rat hearts (2.15 ± 0.67 × 10−6 M vs. 1.59 ± 0.13 × 10−6 M; P < 0.05).
Fig. 2.
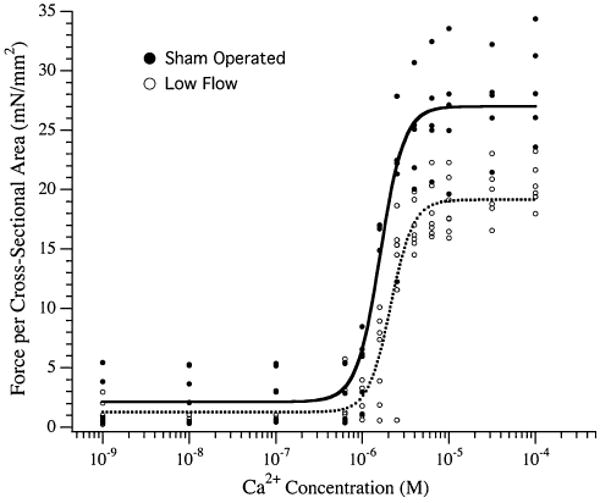
Force–calcium relationship of permeabilized trabeculae from sham operated and low flow rat hearts. Left ventricular trabeculae from the hearts of sham operated (●) and low flow (○) rats were permeabilized and the force per cross-sectional area as a function of free Ca2+ was measured. The lines through the points represent Hill fits to data accumulated from the sham operated (solid line) or low flow (dotted line) rat hearts. N = 5 and 7 rats for sham operated and low blood flow groups
Phosphorylation state of contractile proteins
Adult rat cardiac muscles demonstrate expression of a lower abundance high Mr TnT isoform in addition to the highly abundant lower Mr isoform (Biesiadecki et al. 2002). When resolved by two-dimensional SDS–PAGE, these TnT isoforms additionally resolve into multiple isoelectric variants. Despite this complexity, there are two abundant isoelectric variants of the lower Mr isoform that predominate (Fig. 3a). To determine the relative phosphorylation stoichiometry of these two isoelectric variants, a single rat left ventricular homogenate was resolved in triplicate and stained with phosphoprotein and total protein stain. Densitometry of the phosphoprotein versus the total protein signal demonstrated that the rightmost, basic isoelectric variant (T1) was phosphorylated at half the amount of the relatively acidic isoelectric variant (T2), suggesting these are mono- and diphosphorylated forms of TnT (Fig. 3b). Subsequently, the sham operated and low flow rat heart samples were resolved by two-dimensional SDS-PAGE to quantify the isoelectric variants of TnT and MLC-2 (Fig. 3c). Comparing the abundant lower Mr phosphorylated variants, there was no statistically significant difference in TnT phosphorylation between groups, but a trend towards dephosphorylation was observed with low blood flow (P = 0.21). Myosin light chain-2 in the rat heart is predominantly of a single isoform and was resolved into three isoelectric variants corresponding to a non-phosphorylated form and two phosphorylated forms (Hidalgo et al. 2006). There was no difference in the phosphorylation states of MLC-2 between groups, although there was a modest trend towards dephosphorylation during low flow (P = 0.19; Table 1). Total phosphorylation of MYBP-C was examined by phosphoprotein staining of sham operated and low blood flow myocardium homogenates, but did not reveal a difference between the two groups (Fig. 4).
Fig. 3.
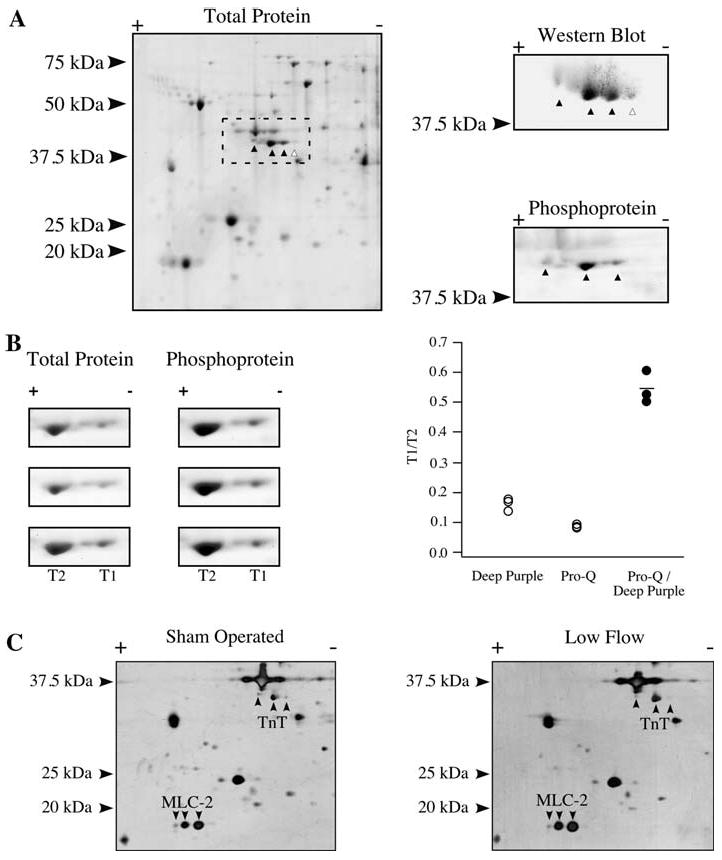
Phosphorylation of Troponin T and MLC-2. a Total protein stain of a homogenate resolved by 2D SDS-PAGE using a 3-5.6NL strip. The putative TnT isoelectric variants are marked (left). Western blotting using a TnT antibody (right, top) or phosphoprotein staining (right, bottom) were carried out to identify phosphorylated (▲) or nonphosphorylated (△) forms of TnT. b Panels focusing on the two predominant TnT isoelectric variants of the more abundant lower Mr isoform, marked T1 and T2, stained in triplicate by total protein stain and phosphoprotein stain (left). T1/T2 ratio for the total protein and phosphoprotein stain, as well as the quotient of the phosphoprotein T1/T2 value over the total protein T1/T2 value (right). c Two-dimensional SDS–PAGE of representative total homogenates from a sham operated and low flow heart, identifying TnT and MLC-2 isoelectric variants
Table 1.
Phosphorylation state of select sarcomeric proteins from sham operated and low flow rat hearts
| Sham operated (%) | Low flow (%) | |
|---|---|---|
| Troponin I | ||
| 1-P | 7.1 ± 3.2 | 16.1 ± 11.0 |
| 2-P | 2.9 ± 3.4 | 1.4 ± 2.0 |
| 3-P | 83.2 ± 7.5 | 69.0 ± 10.9* |
| 4-P | 4.9 ± 3.8 | 8.0 ± 2.7 |
| 5-P | 1.9 ± 1.7 | 5.5 ± 2.0* |
| Troponin T | ||
| 1-P | 8.9 ± 6.0 | 13.3 ± 3.4 |
| 2-P | 91.1 ± 6.0 | 86.7 ± 3.4 |
| MLC-2 | ||
| 0-P | 59.4 ± 4.0 | 63.8 ± 9.5 |
| 1-P | 36.7 ± 2.9 | 32.5 ± 7.8 |
| 2-P | 3.9 ± 1.8 | 3.8 ± 2.5 |
Values are average ± SD from 5 sham operated and 5 low blood flow rat hearts; 0-P through 5-P denotes the corresponding number of phosphorylation sites
P < 0.05 versus sham operated
Fig. 4.
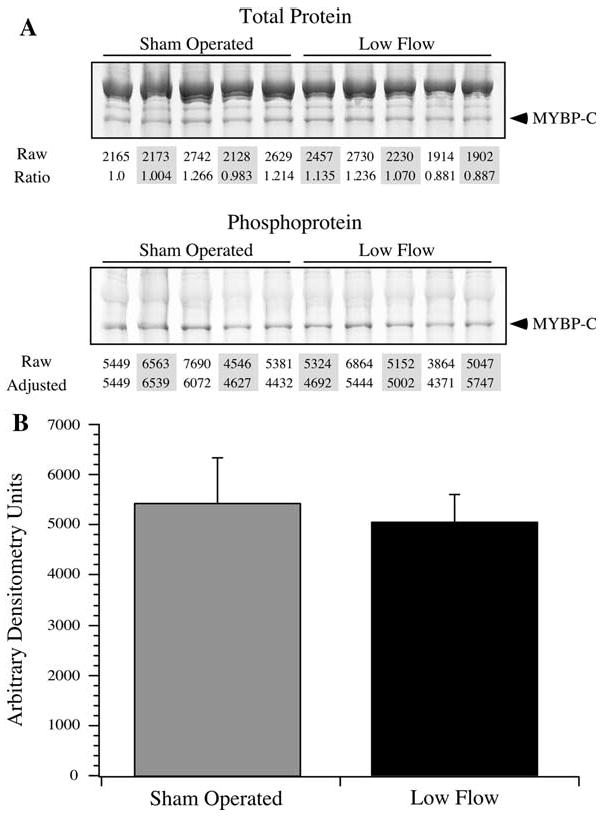
Phosphorylation of MYBP-C. The extent of MYBP-C phosphorylation was determined by phosphoprotein staining. a Sham operated and low flow samples were resolved by SDS-PAGE and stained for total protein and phosphoprotein. The MYBP-C signals were quantified by densitometry (marked “Raw”), and were normalized to the first sham operated sample, providing a normalization factor across samples (marked “Ratio”). This ratio was applied to the unadjusted MYBP-C phosphoprotein signals (marked “Raw”) to provide a normalized value of the extent of MYBP-C phosphorylation. b The average ± SD of the normalized phosphorylated MYBP-C densitometry values are presented. There was no statistically significant difference between sham operated and low flow samples
For TnI, the position and phosphorylation status of the resolved isoelectric variants were initially identified by Western blotting and phosphoprotein staining (Fig. 5a). The overall phosphorylation stoichiometry of TnI was initially determined using phosphoprotein staining of five homogenates with known TnT phosphorylation levels serving as internal controls (Fig. 5b). As TnT and TnI are present in equimolar amounts in cardiac muscle, the TnT phosphoprotein signal in each homogenate was compared to the TnI phosphoprotein signal in the same sample to deduce the TnI phosphorylation stoichiometry. Generally, the phosphorylation level of TnI is higher than for TnT, in agreement with published data (Jacques et al. 2008). This analysis yielded an average of 2.72 mol phosphate per mol TnI for the five samples examined, and this value was used as a guide to identify the individual TnI isoelectric variants. Two-dimensional SDS–PAGE resolved up to five isoelectric variants of TnI, all of which were phosphoproteins (Figs. 5a, c). Using an average stoichiometry of phosphorylation of 2.72 mol phosphate per mol TnI as a guide, the relatively high abundance isoelectric variant would therefore have three phosphorylated sites. Under these circumstances, adjacent TnI isoelectric variants were assumed to differ by one phosphorylation, with the most basic variant having being 1-P and the most acidic being 5-P. Detailed assessment of TnI phosphorylation demonstrated that the 3-P and 5-P isoelectric variants showed significant differences between groups (Table 1). Total TnI phosphorylation was determined from these data by multiplying the fractional content of each isoelectric variant by its number of phosphorylated sites. In sham operated hearts, there was an average of 2.92 mol phosphate per mol TnI, which was not different compared to the low flow samples that averaged 2.86 mol phosphate per mol protein. These phosphorylation stoichiometries were in good agreement with the range determined by phosphoprotein staining (Fig. 5b), suggesting that the isoelectric variants were appropriately identified.
Fig. 5.
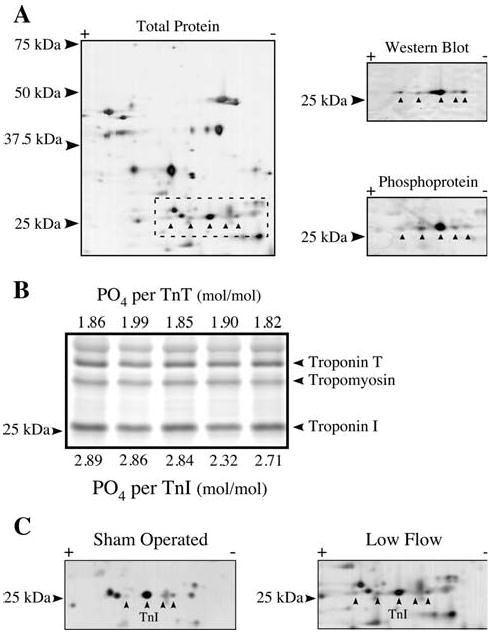
Phosphorylation of Troponin I. a Total protein stain of a homogenate resolved by 2D SDS–PAGE using the cathodic half of a 7–11NL strip. The putative TnI isoelectric variants are marked (left). Western blotting using a TnI antibody (right, top) or phosphoprotein staining (right, bottom) were carried out to identify the TnI variants, all of which were phosphorylated. b Five rat heart homogenates with known amounts of TnT phosphorylation were resolved by SDS–PAGE and stained with Pro-Q Diamond phosphoprotein stain. The TnI phosphoprotein signal from each sample was measured and the stoichiometry determined based on a comparison to the TnT phosphoprotein signal. c Two-dimensional SDS–PAGE of representative sham operated and low flow rat heart homogenates stained for total protein, identifying the isoelectric variants of TnI
Troponin I phosphorylation was further examined by the use of antibodies that identified total TnI or TnI phosphorylated at Ser 23/24, two prominent residues for PKA phosphorylation. The relative amount of Ser23/24 phosphorylated TnI in low flow hearts was significantly decreased (Fig. 6). To identify the additional sites of phosphorylation, TnI enriched from the low flow rat hearts was analyzed by mass spectrometry using LC-MS and tandem MS at two facilities. Coverage of TnI was 75 and 72% in these analyses. Tandem mass spectrometry confirmed phosphorylation at Ser23 and Ser24, identifying two of the phosphorylation sites (Fig. 7a). LC-MS identified peptides of masses consistent with none, single or double Ser43/45 phosphorylation, but tandem data were not captured (Fig. 7b). Peptides containing the putative phosphorylation sites at Thr144 and Ser150 were recovered but did not provide evidence of phosphorylation (Buscemi et al. 2002; Vahebi et al. 2005). At this time, peptides containing the confirmed phosphorylation site at Ser77/Thr78 were not captured (Zabrouskov et al. 2008).
Fig. 6.
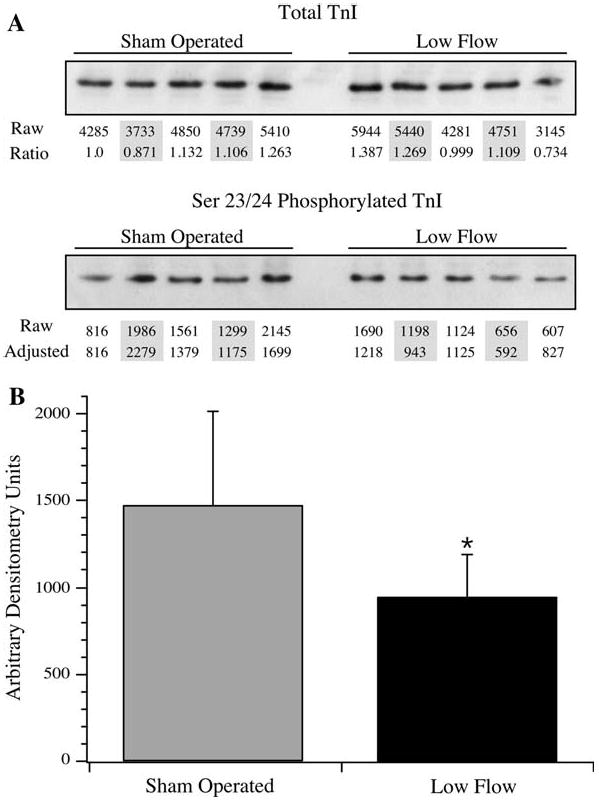
Phosphorylation at Ser23/24 of TnI. To determine the extent of phosphorylation at Ser 23/24 of TnI, sham operated and low flow samples were resolved by SDS–PAGE and Western blotted using antibodies that identified total TnI or Ser23/24 phosphorylated TnI. a For total TnI, samples were quantified by densitometry (marked “Raw”), and were normalized to the first sham operated sample, providing a normalization factor in between samples (marked “Ratio”). This ratio was applied to the unadjusted Ser23/24 phosphorylated TnI signals (marked “Raw”) to provide a normalized value of the Ser23/24 signal (marked “Adjusted”). b The average ± SD of the adjusted Ser23/24 densitometry values are presented. * The differences in Ser23/24 phosphorylation were statistically significant (P < 0.05)
Fig. 7.
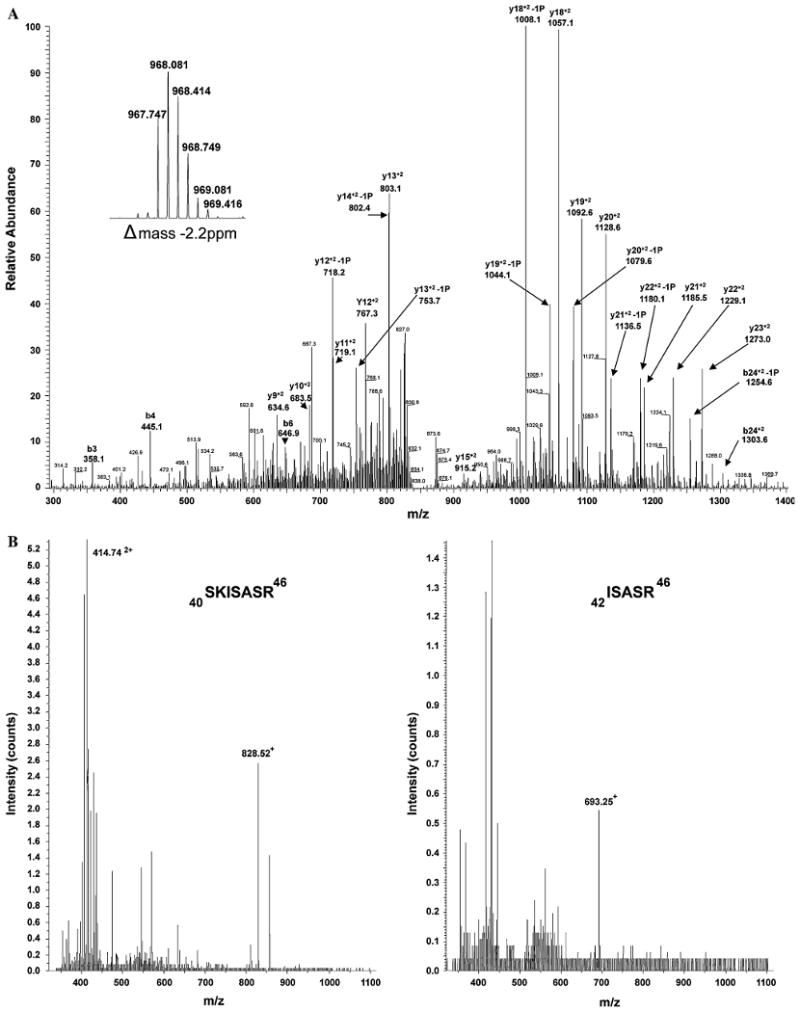
Mass Spectrometry analyses of TnI. To determine the sites of phosphorylation of TnI, mass spectrometry was used to analyze an enriched low blood flow TnI preparation. a Tandem MS spectra of the chymotrypsin generated parent ion 967.7 [M + 3H] 3+ highlighting b and y ions corresponding to phosphorylated Ser23 and 24. The inset scan is part of the Orbitrap FT full scan at 60,000 resolution, showing the parent ion. The delta mass of the parent ion is −2.2 ppm. b LC-MS spectra of trypsin digested TnI representing m/z consistent with the monophosphorylated peptide 40SKISASR46 with net +1 and +2 charge (Left) and the diphosphorylated peptide 42ISASR46 with net +1 charge (Right)
Troponin C affinity chromatography
The relative affinities of TnI populations from sham operated and low blood flow myocardium for Ca2+-bound TnC were tested by affinity chromatography. Troponin I in left ventricular homogenates was allowed to interact with Ca2+-bound TnC, assuring that TnC was in a conformational state that increases the protein's apparent affinity for the inhibitory and COOH-terminal regions of TnI (Farah et al. 1994). Under these conditions, it was observed that TnI from low blood flow myocardium homogenates demonstrated peak elution earlier in the column profile (41.3 ± 4.3 mS/cm) as compared to TnI from sham operated rat hearts (50.3 ± 6.1 mS/cm), suggesting a higher relative affinity of the sham operated TnI population for Ca2+-bound TnC (Fig. 8). This difference in the conductivity at peak elution was statistically significant (P < 0.05).
Fig. 8.
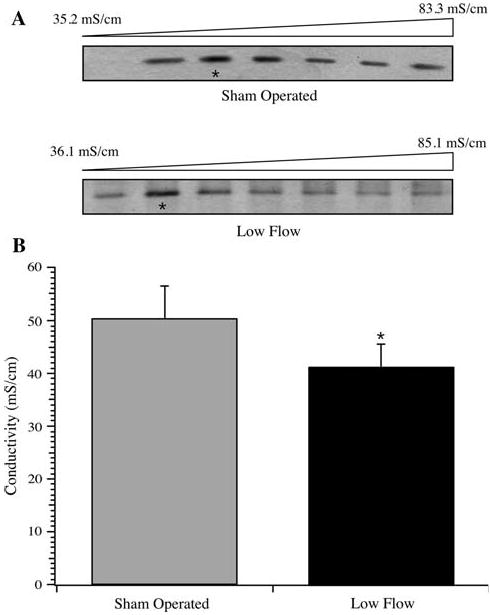
TnC affinity chromatography of TnI. Left ventricular homogenates from sham operated (n = 5) and low flow rat hearts (n = 5) were loaded on a TnC affinity column in the presence of 0.1 mM Ca2+, and the bound TnI was eluted by a linear gradient of increasing KCl. a Troponin I eluting in fractions from the gradients of two runs are shown, with the peak fractions denoted by *. b Average ± SD of the conductivity of the peak fraction of TnI eluting from the TnC affinity column. The difference in peak elution conductivity was statistically significant (* P < 0.05)
Discussion
Altered contractile function in a rat model of low blood flow
Reduced blood flow to the heart, either sudden and transient or gradual and chronic, leads to a reduction in contractility and contributes to the progression of cardiac dysfunction (Bolli and Marban 1999). We have hypothesized that post-translational modifications to proteins of the contractile filaments contribute to the observed decline. To complement a transient ischemia-reperfusion model (Chen and Ogut 2006), we established a rat model whereby low blood flow was induced through constriction of the left anterior descending artery for 3 days. Compared to sham operated counterparts, low myocardial blood flow altered contractility as manifest by a reduction in fractional shortening at rest (Fig. 1), and a biphasic response to dobutamine challenge, indicative of an impaired energy reserve (Afridi et al. 1995). In permeabilized fibers, we observed a decrease in the Fmax and an increase in EC50 with low flow (Fig. 2). Similar reductions in Fmax have been reported from multiple models of altered blood flow to the heart (Hofmann et al. 1993; Gao et al. 1995; Belin et al. 2006; Chen and Ogut 2006; Bito et al. 2007), which poses the question of whether the declines in Fmax have similar root causes. Ultimately, a decline in force production may arise from a reduction in the number of active crossbridges (Chen and Ogut 2006), a change in the distribution of attached crossbridges to low or non-force producing states, or both (Gordon et al. 2000). These can be caused by direct changes in myosin function (Sweeney and Stull 1990) or a change in thin filament function that in turn impacts force production (Montgomery et al. 2002; Pyle et al. 2002; Scruggs et al. 2006). Therefore, to explore the underlying molecular basis for reduced contractility, we compared the phosphorylation states of MLC-2, MYBP-C, TnT and TnI in the two groups.
Myosin light chain-2 phosphorylation in striated muscle may be regulated in a spatially dependent manner, allowing MLC-2 to contribute to stretch activation (Davis et al. 2001; Hidalgo et al. 2006). Our current study did present a trend towards MLC-2 dephosphorylation, but it remains possible that MLC-2 dephosphorylation may be limited in this short duration low blood flow model. Similar to MLC-2, MYBP-C phosphorylation is proposed to change during the progression of cardiac dysfunction. Transgenic models have demonstrated that MYBP-C may be involved in regulating left ventricular function (Brickson et al. 2007) in a manner responsive to PKC activity (Xiao et al. 2007). A canine model of myocardial stunning showed a shift in overall MYBP-C phosphorylation and a follow-up rat model demonstrated a site-specific increase in phosphorylation at Ser288 (Yuan et al. 2006). In our model of low blood flow, there was no statistically significant change in overall MYBP-C phosphorylation as measured by phosphoprotein staining (Fig. 4). However, MYBP-C phosphorylation is complex with up to nine unique isoelectric variants (Yuan et al. 2006). Therefore, interpretations of altered MYBP-C phosphorylation under physiological conditions remain challenging.
Phosphorylation of troponin T
The phosphorylation state of TnT was determined in an effort to define its contribution to reduced contractility during low blood flow. We determined that the predominant TnT isoelectric variants in the rat heart were single and double phosphorylated forms of the lower Mr isoform (Fig. 3). This results in a total phosphorylation <2 mol PO4/mol TnT, significantly less than the 3.05 mol PO4/mol previously described (Messer et al. 2007), and deviates from reports that nonphosphorylated and single phosphorylated TnT predominate in cardiac muscle (Bito et al. 2007). Nonetheless, we did not observe a statistically significant change in the relative content of single or double phosphorylated TnT between groups but did note a trend towards dephosphorylation through an increase in the single phosphorylated form. Considering that TnT may have up to four phosphorylation sites (Sumandea et al. 2003), a change in the distribution of phosphorylation among different sites may be occurring while remaining undetected.
Phosphorylation of troponin I
Subsequently, the phosphorylation state of TnI was examined, and suggests a complex cellular interplay in defining the state of thin filament phosphorylation. We determined that total TnI phosphorylation was ∼2.8 mol PO4/mol TnI in this model, which is not greatly discordant with the ∼2.3 mol PO4/mol reported by others (Messer et al. 2007). Based on the resolved phosphoforms, total TnI phosphorylation was not different between groups. When individual isoelectric variants were examined, a significant reduction in the relative percentage of 3-P TnI, and an increase in 5-P TnI were observed in low flow animals as compared to sham operated animals. This was accompanied by a relative decline in Ser23/24 phosphorylation of TnI (Fig. 6), which is consistent with documented reductions in the content of PKA regulatory subunits (Zakhary et al. 1999), reduction in β-adrenergic receptor density (Brickson et al. 2007), and desensitization of β-adrenergic receptors (Lymperopoulos et al. 2007) during cardiac dysfunction. As two-dimensional SDS-PAGE demonstrated up to five isoelectric variants, we attempted to identify these sites by mass spectrometry. Phosphorylation at Ser23 and 24 was observed by tandem MS, confirming the phosphospecific antibody results. LC-MS provided evidence for Ser43 and 45 being third and fourth sites (Fig. 7b), but the data are qualitative and not quantitative. Evidence for a fifth site of phosphorylation was insufficient, although multiple candidates remain. These include Thr144, through a Rho-kinase (Vahebi et al. 2005) or PKCβII dependent mechanism (Wang et al. 2006), Ser150 through p21-activated kinase (Buscemi et al. 2002), or Ser77/Thr78 phosphorylation by an unidentified kinase (Zabrouskov et al. 2008). Absent quantitative data, we cannot determine how phosphorylation at these additional sites may have contributed, if at all, to our observations. Further, our data do not account for novel post-translational modifications, such as o-linked modification by monosaccharides (Ramirez-Correa et al. 2008). Detailed analyses will require quantitative models to assess, in parallel, the various types of o-linked modifications.
As total TnI phosphorylation was constant between groups, the data suggest a relative increase of phosphorylation at other sites to offset the decline in Ser23/24 phosphorylation with low flow. This change in phosphorylation coincided with the decreased relative affinity of TnI for Ca2+ bound TnC (Fig. 8). Mechanistically, a conformational change in TnC following Ca2+ binding increases its apparent affinity for TnI, allowing relief of TnI-based inhibition of the thin filament (Farah et al. 1994). For low flow TnI, reduced relative affinity measured by chromatography is therefore consistent with increased EC50 (Fig. 2). It suggests that this population of TnI is less apt to associate with Ca2+ bound TnC and would thereby persist longer in its inhibitory position in the thin filament. Previous in vitro reports have demonstrated that Ser43/45 phosphorylation of TnI increased EC50 of contraction and decreased maximum actin-activated myosin ATPase activity (Noland and Kuo 1991; Pyle et al. 2002). Affinity chromatography results suggest that increased Ser43/45 phosphorylation of TnI in the low flow hearts may have contributed to the increased EC50, which opens the possibility that this phosphorylation may be involved in commensurately decreasing the measured Fmax. Given that TnI phosphorylation is complex, and absent quantitative data for Ser43/45 phosphorylation, it remains unclear how phosphorylation at sites other than Ser23/24 contribute to function under physiological or pathophysiological conditions. Importantly, the impact of individual phosphorylation events may be very sensitive to cellular context. For example, increased phosphorylation of Ser23/24 in response to β-adrenergic activation predicts reduced Ca2+ sensitivity, yet our model demonstrated reduced Ca2+ sensitivity with reduced Ser23/24 phosphorylation. However, it remains unclear if a given change in the magnitude of Ser23/24 phosphorylation elicits the same functional response regardless of the starting level of phosphorylation. In other words, reducing Ser23/24 phosphorylation from resting, physiological levels may not have a direct functional impact, but rather render the thin filament more sensitive to changes in phosphorylation at other residues of TnI, TnT or Tm. These mechanisms can only be deciphered by determining the absolute levels of phosphorylation at multiple sites under varying physiological and pathophysiological conditions.
In conclusion, we present evidence that the decline in contractility during low blood flow may be explained, in part, by changes to the post-translational state of contractile proteins. Identification of these novel post-translational changes in vivo, combined with their physiological ranges of variation, will provide an important framework to rationalize consequent changes in contractility. Importantly, limitations on contractility imposed by post-translational changes may have broad clinical corollaries. For example, although thrombolytic therapy improves survival by restoring blood flow through an infarct related artery, this eventual benefit occurs with little or no change in left ventricular ejection fraction [reviewed in (Solomon and Gersh 1998)]. Our findings generally suggest that subsequent to the restoration of blood flow, the limited change in ejection fraction may be related to intracellular signaling that persists to limit fiber contractility through post-translational modification of thin filament proteins such as TnI. Additional studies are currently underway to identify additional molecular events that underlie the progression to cardiac dysfunction and to determine their functional consequences.
Supplementary Material
Acknowledgments
We thank Dr. Frank Brozovich for support and for critical reading of the manuscript.
Funding This work was supported by grants from the NHLBI (R01 HL078845 to O·O). B·C. was supported by NIH grant T32 GM07250.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s10974-009-9180-2) contains supplementary material, which is available to authorized users.
Conflict of interest None.
Contributor Information
Bridgette Christopher, Department of Physiology and Biophysics, Case Western Reserve University, Cleveland, OH 44106, USA.
Gresin O. Pizarro, Division of Cardiovascular Diseases, Mayo Clinic, 200 First Street S.W., Rochester, MN 55902, USA
Bryson Nicholson, Department of Physiology and Biophysics, Case Western Reserve University, Cleveland, OH 44106, USA.
Samantha Yuen, Division of Cardiovascular Diseases, Mayo Clinic, 200 First Street S.W., Rochester, MN 55902, USA.
Brian D. Hoit, Department of Medicine, University Hospitals of Cleveland, Cleveland, OH 44106, USA
Ozgur Ogut, Email: ogut.ozgur@mayo.edu, Division of Cardiovascular Diseases, Mayo Clinic, 200 First Street S.W., Rochester, MN 55902, USA.
References
- Afridi I, Kleiman NS, Raizner AE, et al. Dobutamine echocardiography in myocardial hibernation. Optimal dose and accuracy in predicting recovery of ventricular function after coronary angioplasty. Circulation. 1995;91:663–670. doi: 10.1161/01.cir.91.3.663. [DOI] [PubMed] [Google Scholar]
- Belin RJ, Sumandea MP, Kobayashi T, et al. Left ventricular myofilament dysfunction in rat experimental hypertrophy and congestive heart failure. Am J Physiol Heart Circ Physiol. 2006;291:H2344–H2353. doi: 10.1152/ajpheart.00541.2006. [DOI] [PubMed] [Google Scholar]
- Biesiadecki BJ, Elder BD, Yu ZB, et al. Cardiac troponin T variants produced by aberrant splicing of multiple exons in animals with high instances of dilated cardiomyopathy. J Biol Chem. 2002;277:50275–50285. doi: 10.1074/jbc.M206369200. [DOI] [PubMed] [Google Scholar]
- Bito V, van der Velden J, Claus P, et al. Reduced force generating capacity in myocytes from chronically ischemic, hibernating myocardium. Circ Res. 2007;100:229–237. doi: 10.1161/01.RES.0000257829.07721.57. [DOI] [PubMed] [Google Scholar]
- Bolli R, Marban E. Molecular and cellular mechanisms of myocardial stunning. Physiol Rev. 1999;79:609–634. doi: 10.1152/physrev.1999.79.2.609. [DOI] [PubMed] [Google Scholar]
- Bowling N, Walsh RA, Song G, et al. Increased protein kinase C activity and expression of Ca(2+)-sensitive isoforms in the failing human heart. Circulation. 1999;99:384–391. doi: 10.1161/01.cir.99.3.384. [DOI] [PubMed] [Google Scholar]
- Brickson S, Fitzsimons DP, Pereira L, et al. In vivo left ventricular functional capacity is compromised in cMyBP-C null mice. Am J Physiol Heart Circ Physiol. 2007;292:H1747–H1754. doi: 10.1152/ajpheart.01037.2006. [DOI] [PubMed] [Google Scholar]
- Bristow MR, Ginsburg R, Minobe W, et al. Decreased catecholamine sensitivity and beta-adrenergic-receptor density in failing human hearts. N Engl J Med. 1982;307:205–211. doi: 10.1056/NEJM198207223070401. [DOI] [PubMed] [Google Scholar]
- Buscemi N, Foster DB, Neverova I, et al. p21-activated kinase increases the calcium sensitivity of rat triton-skinned cardiac muscle fiber bundles via a mechanism potentially involving novel phosphorylation of troponin I. Circ Res. 2002;91:509–516. doi: 10.1161/01.res.0000035246.27856.53. [DOI] [PubMed] [Google Scholar]
- Chen FC, Ogut O. Decline of contractility during ischemiareperfusion injury: actin glutathionylation and its effect on allosteric interaction with tropomyosin. Am J Physiol Cell Physiol. 2006;290:C719–C727. doi: 10.1152/ajpcell.00419.2005. [DOI] [PubMed] [Google Scholar]
- Chen FC, Ogut O, Rhee AY, et al. Captopril prevents myosin light chain phosphatase isoform switching to preserve normal cGMP-mediated vasodilatation. J Mol Cell Cardiol. 2006;41:488–495. doi: 10.1016/j.yjmcc.2006.05.018. [DOI] [PubMed] [Google Scholar]
- Davis JS, Hassanzadeh S, Winitsky S, et al. The overall pattern of cardiac contraction depends on a spatial gradient of myosin regulatory light chain phosphorylation. Cell. 2001;107:631–641. doi: 10.1016/s0092-8674(01)00586-4. [DOI] [PubMed] [Google Scholar]
- Fabiato A. Computer programs for calculating total from specified free or free from specified total ionic concentrations in aqueous solutions containing multiple metals and ligands. Methods Enzymol. 1988;157:378–417. doi: 10.1016/0076-6879(88)57093-3. [DOI] [PubMed] [Google Scholar]
- Farah CS, Miyamoto CA, Ramos CH, et al. Structural and regulatory functions of the NH2- and COOH-terminal regions of skeletal muscle troponin I. J Biol Chem. 1994;269:5230–5240. [PubMed] [Google Scholar]
- Gao WD, Atar D, Backx PH, et al. Relationship between intracellular calcium and contractile force in stunned myocardium. Direct evidence for decreased myofilament Ca(2+) responsiveness and altered diastolic function in intact ventricular muscle. Circ Res. 1995;76:1036–1048. doi: 10.1161/01.res.76.6.1036. [DOI] [PubMed] [Google Scholar]
- Gordon AM, Homsher E, Regnier M. Regulation of contraction in striated muscle. Physiol Rev. 2000;80:853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- Hidalgo C, Wu Y, Peng J, et al. Effect of diastolic pressure on MLC2v phosphorylation in the rat left ventricle. Arch Biochem Biophys. 2006;456:216–223. doi: 10.1016/j.abb.2006.06.026. [DOI] [PubMed] [Google Scholar]
- Hofmann PA, Miller WP, Moss RL. Altered calcium sensitivity of isometric tension in myocyte-sized preparations of porcine postischemic stunned myocardium. Circ Res. 1993;72:50–56. doi: 10.1161/01.res.72.1.50. [DOI] [PubMed] [Google Scholar]
- Jacques AM, Briceno N, Messer AE, et al. The molecular phenotype of human cardiac myosin associated with hypertrophic obstructive cardiomyopathy. Cardiovasc Res. 2008;79:481–491. doi: 10.1093/cvr/cvn094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jideama NM, Noland TAJ, Raynor RL, et al. Phosphorylation specificities of protein kinase C isozymes for bovine cardiac troponin I and troponin T and sites within these proteins and regulation of myofilament properties. J Biol Chem. 1996;271:23277–23283. doi: 10.1074/jbc.271.38.23277. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Solaro RJ. Calcium, thin filaments, and the integrative biology of cardiac contractility. Annu Rev Physiol. 2005;67:39–67. doi: 10.1146/annurev.physiol.67.040403.114025. [DOI] [PubMed] [Google Scholar]
- Lymperopoulos A, Rengo G, Funakoshi H, et al. Adrenal GRK2 upregulation mediates sympathetic overdrive in heart failure. Nat Med. 2007;13:315–323. doi: 10.1038/nm1553. [DOI] [PubMed] [Google Scholar]
- Messer AE, Jacques AM, Marston SB. Troponin phosphorylation and regulatory function in human heart muscle: dephosphorylation of Ser23/24 on troponin I could account for the contractile defect in end-stage heart failure. J Mol Cell Cardiol. 2007;42:247–259. doi: 10.1016/j.yjmcc.2006.08.017. [DOI] [PubMed] [Google Scholar]
- Montgomery DE, Wolska BM, Pyle WG, et al. alpha-Adrenergic response and myofilament activity in mouse hearts lacking PKC phosphorylation sites on cardiac TnI. Am J Physiol Heart Circ Physiol. 2002;282:H2397–H2405. doi: 10.1152/ajpheart.00714.2001. [DOI] [PubMed] [Google Scholar]
- Noland TAJ, Kuo JF. Protein kinase C phosphorylation of cardiac troponin I or troponin T inhibits Ca(2+)-stimulated actomyosin MgATPase activity. J Biol Chem. 1991;266:4974–4978. [PubMed] [Google Scholar]
- Noland TAJ, Guo X, Raynor RL, et al. Cardiac troponin I mutants. Phosphorylation by protein kinases C and A and regulation of Ca(2+)-stimulated MgATPase of reconstituted actomyosin S-1. J Biol Chem. 1995;270:25445–25454. doi: 10.1074/jbc.270.43.25445. [DOI] [PubMed] [Google Scholar]
- Noland TAJ, Raynor RL, Jideama NM, et al. Differential regulation of cardiac actomyosin S-1 MgATPase by protein kinase C isozyme-specific phosphorylation of specific sites in cardiac troponin I and its phosphorylation site mutants. Biochemistry. 1996;35:14923–14931. doi: 10.1021/bi9616357. [DOI] [PubMed] [Google Scholar]
- Pyle WG, Sumandea MP, Solaro RJ, et al. Troponin I serines 43/45 and regulation of cardiac myofilament function. Am J Physiol Heart Circ Physiol. 2002;283:H1215–H1224. doi: 10.1152/ajpheart.00128.2002. [DOI] [PubMed] [Google Scholar]
- Rabilloud T. Use of thiourea to increase the solubility of membrane proteins in two-dimensional electrophoresis. Electrophoresis. 1998;19:758–760. doi: 10.1002/elps.1150190526. [DOI] [PubMed] [Google Scholar]
- Ramirez-Correa GA, Jin W, Wang Z, et al. O-linked GlcNAc modification of cardiac myofilament proteins: a novel regulator of myocardial contractile function. Circ Res. 2008;103:1354–1358. doi: 10.1161/CIRCRESAHA.108.184978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scruggs SB, Walker LA, Lyu T, et al. Partial replacement of cardiac troponin I with a non-phosphorylatable mutant at serines 43/45 attenuates the contractile dysfunction associated with PKCepsilon phosphorylation. J Mol Cell Cardiol. 2006;40:465–473. doi: 10.1016/j.yjmcc.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Solomon A, Gersh B. The open-artery hypothesis. Annu Rev Med. 1998;49:63–76. doi: 10.1146/annurev.med.49.1.63. [DOI] [PubMed] [Google Scholar]
- Sumandea MP, Pyle WG, Kobayashi T, et al. Identification of a functionally critical protein kinase C phosphorylation residue of cardiac troponin T. J Biol Chem. 2003;278:35135–35144. doi: 10.1074/jbc.M306325200. [DOI] [PubMed] [Google Scholar]
- Sweeney HL, Stull JT. Alteration of cross-bridge kinetics by myosin light chain phosphorylation in rabbit skeletal muscle: implications for regulation of actin-myosin interaction. Proc Natl Acad Sci U S A. 1990;87:414–418. doi: 10.1073/pnas.87.1.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobacman LS. Thin filament-mediated regulation of cardiac contraction. Annu Rev Physiol. 1996;58:447–481. doi: 10.1146/annurev.ph.58.030196.002311. [DOI] [PubMed] [Google Scholar]
- Vahebi S, Kobayashi T, Warren CM, et al. Functional effects of rho-kinase-dependent phosphorylation of specific sites on cardiac troponin. Circ Res. 2005;96:740–747. doi: 10.1161/01.RES.0000162457.56568.7d. [DOI] [PubMed] [Google Scholar]
- Wang H, Grant JE, Doede CM, et al. PKC-betaII sensitizes cardiac myofilaments to Ca(2+) by phosphorylating troponin I on threonine-144. J Mol Cell Cardiol. 2006;41:823–833. doi: 10.1016/j.yjmcc.2006.08.016. [DOI] [PubMed] [Google Scholar]
- Xiao L, Zhao Q, Du Y, et al. PKCepsilon increases phosphorylation of the cardiac myosin binding protein C at serine 302 both in vitro and in vivo. Biochemistry. 2007;46:7054–7061. doi: 10.1021/bi700467k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan C, Guo Y, Ravi R, et al. Myosin binding protein C is differentially phosphorylated upon myocardial stunning in canine and rat hearts—evidence for novel phosphorylation sites. Proteomics. 2006;6:4176–4186. doi: 10.1002/pmic.200500894. [DOI] [PubMed] [Google Scholar]
- Zabrouskov V, Ge Y, Schwartz J, et al. Unraveling molecular complexity of phosphorylated human cardiac troponin I by top down electron capture dissociation/electron transfer dissociation mass spectrometry. Mol Cell Proteomics. 2008;7:1838–1849. doi: 10.1074/mcp.M700524-MCP200. [DOI] [PubMed] [Google Scholar]
- Zakhary DR, Moravec CS, Stewart RW, et al. Protein kinase A (PKA)-dependent troponin-I phosphorylation and PKA regulatory subunits are decreased in human dilated cardiomyopathy. Circulation. 1999;99:505–510. doi: 10.1161/01.cir.99.4.505. [DOI] [PubMed] [Google Scholar]
- Zhang R, Zhao J, Potter JD. Phosphorylation of both serine residues in cardiac troponin I is required to decrease the Ca(2+) affinity of cardiac troponin C. J Biol Chem. 1995;270:30773–30780. doi: 10.1074/jbc.270.51.30773. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.