

Abstract
Local glucocorticoid (GC) action depends on intracellular GC metabolism by 11β-hydroxysteroid dehydrogenases (11βHSDs). 11βHSD1 activates GCs, while 11βHSD2 inactivates GCs. Adipocyte-specific amplification of GCs through transgenic overexpression of 11βHSD1 produces visceral obesity and the metabolic syndrome in mice. To determine whether adipocyte-specific inactivation of GCs protects against this phenotype, we created a transgenic model in which human 11βHSD2 is expressed under the control of the murine adipocyte fatty acid binding protein (aP2) promoter (aP2-h11βHSD2). Transgenic mice have increased 11βHSD2 expression and activity exclusively in adipose tissue, with the highest levels in subcutaneous adipose tissue, while systemic indexes of GC exposure are unchanged. Transgenic mice resist weight gain on high-fat diet due to reduced fat mass accumulation. This improved energy balance is associated with decreased food intake, increased energy expenditure, and improved glucose tolerance and insulin sensitivity. Adipose tissue gene expression in transgenic mice is characterized by decreased expression of leptin and resistin and increased expression of adiponectin, peroxisome proliferator–activated receptor γ, and uncoupling protein 2. These data suggest that reduction of active GCs exclusively in adipose tissue is an important determinant of a favorable metabolic phenotype with respect to energy homeostasis and the metabolic syndrome.
Glucocorticoids (GCs) play a critical role in multiple metabolic processes, including glucose homeostasis, insulin sensitivity, lipid metabolism, and adipogenesis. GC excess, whether endogenous (Cushing's syndrome) or exogenous, promotes visceral obesity, insulin resistance, dyslipidemia, and hypertension (1). A similar constellation of metabolic abnormalities is associated with idiopathic obesity and defines the much more prevalent metabolic syndrome (1, 2). While subtle alterations in the hypothalamic-pituitary-adrenal axis have been reported in idiopathic obesity and the metabolic syndrome, no clear role for increased circulating GCs has been established (3).
GC action in target tissues, however, not only depends on circulating GC concentrations and cellular GC receptor (GR) expression but also on tissue-specific intracellular GC metabolism by 11β-hydroxysteroid dehydrogenases (11βHSDs) (4). 11βHSDs act at the prereceptor level to catalyze the interconversion of hormonally active 11β-hydroxylated GCs (cortisol in human and corticosterone in mice) and their hormonally inactive 11β-keto metabolites (cortisone in humans and 11-dehydrocorticosterone [11DHC] in mice) (5). Two isoforms of 11βHSD have been identified. 11βHSD1 is a low-affinity NADPH-dependent reductase expressed primarily in GC target tissues, such as liver, adipose tissue, and the central nervous system, where it amplifies local GC action. 11βHSD2 is a high-affinity NAD-dependent dehydrogenase expressed primarily in mineralocorticoid target tissues, such as kidney, where it potently decreases local GC action, thus ensuring that only the nonsubstrate aldosterone can access intrinsically nonselective mineralocorticoid receptors (6,7).
Evidence is rapidly accumulating to support a role for 11βHSD1 in the pathogenesis of visceral obesity and the metabolic syndrome. 11βHSD1 is decreased in liver and enhanced in mesenteric adipose tissue (MAT) in several models of rodent obesity, including leptin-resistant Leprfa/Leprfa rats (8,9) and leptin-deficient Lepob/Lepob mice (10). Several studies have shown that 11βHSD1 activity and expression in subcutaneous adipose tissue (SCAT) are positively correlated with obesity and insulin resistance in both men and women (11–17). In addition, polymorphic variability at the 11βHSD1 locus is associated with increased waist-to-hip ratio in adults (18) and with body composition and insulin resistance in children (19).
As such, 11βHSD1 has been proposed as a novel therapeutic target for the treatment of obesity and the metabolic syndrome. Indeed, pharmacologic inhibition of 11βHSD1 in obese rodents improves glucose tolerance, insulin sensitivity, and lipid profiles (20–22). While such treatment is associated with alterations in hepatic gene expression, adipose tissue has not been evaluated (20,21). Similarly, mice with targeted disruption of 11βHSD1 in all tissues have reduced weight gain on high-fat diet (HFD), attenuated gluconeogenic response to fasting, improved glucose tolerance and insulin sensitivity, and atheroprotective lipid profiles, despite having modestly elevated serum corticosterone levels (23–25). This favorable metabolic phenotype is associated with improved GC-related metabolic functions in both liver (24) and adipose tissue (25). Nevertheless, both pharmacologic inhibition and targeted gene deletion of 11βHSD1 affect all tissues, and, hence, the relative contribution of any individual tissue to overall metabolic phenotype is unclear.
Transgenic mouse models have provided insight into the role of 11βHSD1 in individual tissues. Mice with adipocyte-specific overexpression of 11βHSD1 driven by the murine adipocyte fatty acid binding protein (aP2) promoter develop visceral obesity and the metabolic syndrome (10,26). In contrast, mice with liver-specific overexpression of 11βHSD1 driven by the human apolipoprotein E promoter develop insulin resistance, dyslipidemia, and hypertension without obesity (27). These data suggest that GC amplification in adipose tissue, more than liver, contributes to overall energy balance. Whether 11βHSD1 in adipose tissue contributes to the other features of the metabolic syndrome, independent of liver, is unknown. Furthermore, the phenotypic consequences of inhibition or absence of GC regeneration by 11βHSD1 exclusively in adipose tissue has not been explored.
We hypothesized that a reduction of active GCs exclusively in adipose tissue is an important determinant of a favorable metabolic phenotype with respect to energy homeostasis and features of the metabolic syndrome. To test this hypothesis, we generated an animal model of adipocyte-specific GC inactivation through ectopic overexpression of h11βHSD2 under the control of the murine aP2 promoter.
Research Design and Methods
Generation of the aP2-h11βHSD2 construct and transgenic mice
A 5.4-kb fragment corresponding to position −5.4 to +21 of the murine aP2 promoter (provided by B.M. Spiegelman) was ligated to a 1.9-kb fragment corresponding to position −133 to +1,764 of human placental 11βHSD2 cDNA, including its native polyadenylation signal (28,29). The linearized 7.3-kb NotI-SalI aP2-h11βHSD2 construct was injected into pronuclei of fertilized zygotes from FVB mice and transferred to pseudopregnant females. Offspring were screened for genomic integration by PCR of tail DNA using the following h11βHSD2-specific primers: forward 5′-CGGCCGTGGCGCTACT-CAT, reverse 5′-GGGAAGGGCCGGGGCTCAGGTTT.
Mice were generated by breeding F1 heterozygous transgenic males to wild-type (WT) females. Experiments were performed in F2 males heterozygous for the transgene. Mice were housed individually under standard conditions at 25°C with a 14/10-h light-dark cycle and free access to food and water. Animals were handled in accordance with the guidelines established by the National Institutes of Health. Procedures were approved by the Animal Care and Use Committee at the Beth Israel Deaconess Medical Center. For serum corticosterone and thymic weight, mice were fed chow diet (6% fat wt/wt; Harlan Teklad; RD8664) from 3 to 10 weeks (n = 8 per group). Ad libitum-fed mice were killed between 9:00 A.M. and 10:00 A.M. under non-stressed conditions. For other experiments, mice were fed chow diet or HFD (42% fat wt/wt; Harlan Teklad; TD88137) from 3 to 24 weeks (n = 18–22 per group). Body weight and food intake were determined weekly. Tail bleeds were performed in fasted mice at 20 weeks. Intraperitoneal glucose (1 g/kg) and insulin (1.125 units/kg) tolerance tests were performed at 16 and 21 weeks, respectively. At 24 weeks, ad libitum-fed mice were subjected to dual-energy X-ray absorptiometry (DEXA; Lunar PIXImus mouse densitometer). Mice of each genotype were subsequently divided into two groups with equal means ± SEs for body weight. One group (n = 10 per group) was immediately killed by CO2 inhalation. The other group (n = 8 per group) was placed in a comprehensive laboratory animal monitoring system for measurement of daily food intake and continuous oxygen consumption (Vo2) for 12 days following a 48-h acclimation period. At the time the mice were killed, trunk blood was collected by cardiac puncture, centrifuged, and stored at −20°C. Tissues were weighed, frozen in liquid nitrogen, and stored at −80°C.
RNA extraction and gene expression analysis
Total RNA was extracted from homogenized tissues using RNeasy Lipid Tissue Mini Kit with on-column DNAse treatment (Qiagen) followed by generation of cDNA using RETRO-script (Ambion). Quantitative real-time PCR was performed in triplicate with 1:100 dilution of cDNA, 1 × Taqman Universal Master Mix (Applied Biosystems), and gene-specific primer-probe sets using a Stratagene MX4000 Mulitiplex Quantitative PCR System. Reactions were run at 95°C × 10 min, followed by 40 cycles of 95°C × 15 s and 60°C × 1 min. Sequences for 11βHSD2 and 11βHSD1 primer-probe sets (Biosearch Technologies) were as follows: h11βHSD2: forward 5′-GGGCCTATGGAACCTCCAA, reverse 5′-GACCCA CGTTTCTCACTGACTCT, probe 5′-FAM-CCGTGGCGCTACTCATGGACACA-BHQ1; m11βHSD2: forward 5′-CAGCCTACGGCACCTCCA, reverse 5′-GCGTTTCTCCCAGAGGTTCA, probe 5′-FAM-TCGGCTGTGAGCTGCTTCCC TGG-BHQ1; and m11βHSD1: forward 5′-AAGCAGAGCAATGGCAGCAT, reverse 5′-GAGCAATCATAGGCTGGGTCAT, probe 5′-FAM-CGTCATCTC CTCCTTGGCTGGGAA-BHQ1. Human- and mouse-specific primers for 11βHSD2 were tested to confirm that the h11βHSD2 primer-probe set did not amplify m11βHSD2 or vice versa. Primer-probe sets for other genes were purchased as predesigned Taqman gene expression assays and run as per the manufacturer's instructions (Applied Biosystems). Gene expression was determined by the standard curve method and normalized to expression of 18S RNA (Applied Biosystems). Accuracy of RNA quantification was optimized by DNAse treatment, gene-specific primer-probe sets that span intron-exon boundaries, and lack of amplification in no-RT and no-template controls.
11βHSD1 and 11βHSD2 activity
Activities were assayed as previously described (28,30). 11βHSD2 dehydrogenase activity was determined by measuring percent conversion of tritiated corticosterone (10 nmol/l) to 11DHC in the presence of excess (400 μmol/l) enzyme-specific cofactor (NAD+ for 11βHSD2 and NADPH for 11βHSD1). Reactions contained 0.1 mg/ml protein with an incubation time of 10 min. 11βHSD1 reductase activity was determined in the same way using tritiated 11DHC as substrate but with 0.2 mg/ml protein and 30-min incubation. Protein concentration and incubation times were optimized such that activity was in the linear range for product formation. Reactions were run in duplicate with blank assays run in parallel. Steroids were extracted with ethyl acetate, separated by thin-layer chromatography, identified by migration in comparison to known standards, and quantified with a phosphorimager tritium screen (Fuji).
Blood analysis
Plasma glucose was determined using a One Touch FastTake glucometer (LifeScan). Serum insulin was determined by an Ultra Sensitive Mouse Insulin ELISA Kit (CrystalChem). Serum corticosterone was determined by a Rat Corticosterone Radioimmunoassay Kit (ICN Pharmaceuticals).
Statistical analysis
Data are expressed as means ± SE. Comparisons between groups were made by unpaired two-tailed Student's t tests or by one- or two-way ANOVA, as appropriate with genotype and/or diet as variables. Where differences were found by ANOVA, post hoc Tukey multiple comparison tests were performed. For longitudinal body weight and glucose and insulin tolerance tests, comparisons were made by two-way ANOVA with repeated measures. For energy expenditure data, comparisons were made using mixed model analysis.
Results
11βHSD2 expression and activity is exclusively increased in adipose tissue of transgenic mice
Transgenic mice were viable and fertile with progeny in the expected Mendelian ratios. Since endogenous 11βHSD2 in kidney is sufficient to fully inactivate corticosterone in that tissue, the goal was to create transgenic mice with murine “kidney” levels of h11βHSD2 in adipose tissue. Data are shown from a representative line (Fig. 1). The h11βHSD2 transgene was expressed in all adipose tissue depots of transgenic mice including SCAT, MAT, perigonadal adipose tissue (PGAT), perirenal plus retroperitoneal adipose tissue (PRRP), intrascapular white adipose tissue (ISWAT), and brown adipose tissue (BAT) (Fig. 1A). Relative expression of h11βHSD2 was significantly greater than endogenous m11βHSD1 expression with the ratio of h11βHSD2 to m11βHSD1 expression in SCAT being 2.55 ± 0.65 and 12.74 ± 4.84 in chow- and HFD-fed mice, respectively. Expression of h11βHSD2 was absent from nonadipose tissues of transgenic mice including whole brain, skeletal muscle, liver, and kidney and was also absent in all of the above tissues of WT mice (data not shown).
FIG. 1.
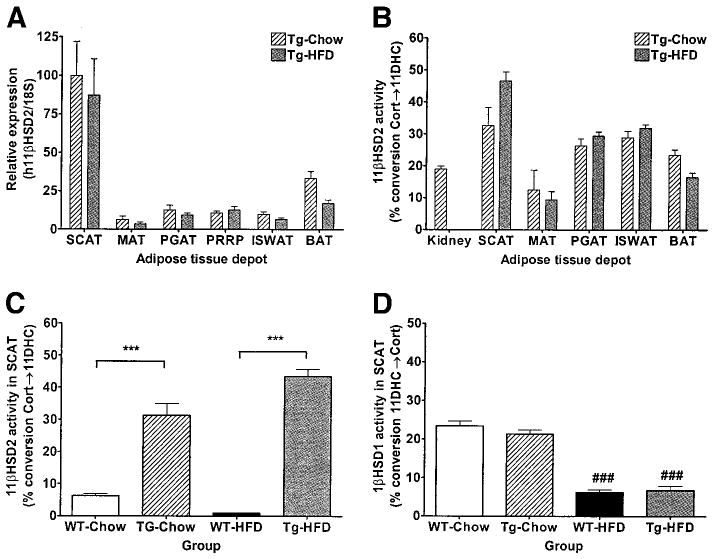
11βHSD2 and 11βHSD1 expression and activity. A: h11βHSD2 expression relative to 18S RNA in various adipose tissue depots of 24-week-old transgenic and WT mice (n = 10 per group) using quantitative real-time PCR with human kidney standard curve and a primer-probe set that detects only human and not murine 11βHSD2. B: 11βHSD2 activity (% conversion Cort→11DHC) in the above adipose tissue depots and kidney of 24-week-old transgenic mice (n = 10 per group). 11βHSD2 (C) and 11βHSD1 (D) activity (% conversion 11DHC→Cort) in SCAT of 24-week-old transgenic and WT mice on chow and HFD (n = 10 per group). Data are expressed as means ± SE. ***P < 0.001 between genotypes on a given diet; ###P < 0.001 between diet groups for a given genotype.
Likewise, 11βHSD2 activity was detected in all adipose tissue depots of transgenic mice (Fig. 1B). 11βHSD2 activity in transgenic mice expressed as percent activity in murine kidney was as follows: transgenic-chow: SCAT 137.73%, BAT 111.35%, PGAT 139.16%, ISWAT 152.72%, and MAT 52.74%; transgenic-HFD: SCAT 246.12%, BAT 87.18%, PGAT 155.36%, ISWAT 168.44%, and MAT 49.74%. 11βHSD2 activity was significantly higher in transgenic compared with WT mice (Fig. 1C). While endogenous 11βHSD2 has not been reported in adipocytes per se, low levels have been reported in vascular endothelium (31). Expression of endogenous m11βHSD2, in contrast to the h11βHSD2 transgene, was extremely low relative to murine kidney and was not different between genotypes on a given diet (m11βHSD2/18S for WT-chow 0.007 ± 0.002, transgenic-chow 0.009 ± 0.002, WT-HFD 0.015 ± 0.002, and transgenic-HFD 0.014 ± 0.002), suggesting that the difference in 11βHSD2 activity can be attributed to the h11βHSD2 transgene rather than to endogenous m11βHSD2.
Other determinants of GC action and systemic indexes of GC exposure do not differ between transgenic and WT mice
11βHSD1 expression (Table 1) and activity (Fig. 1D) in SCAT were similar between WT and transgenic mice. For both genotypes, 11βHSD1 expression and activity were decreased in mice on HFD compared with chow diet, consistent with previously published data showing downregulation of 11βHSD1 by HFD in mice (32). GRα expression (Table 1) in SCAT was not significantly affected by genotype or diet. In addition, serum corticosterone and various indexes of systemic GC exposure, including thymic weight, adrenal weight, bone density, lean body mass, and naso-anal length, were also similar between WT and transgenic mice (Table 2).
TABLE 1.
Gene expression in SCAT
| WT-chow | Transgenic-chow | WT-HFD | Transgenic-HFD | |
|---|---|---|---|---|
| m11βHSD1 | 100 ± 11 | 110 ± 8 | 42 ± 7† | 46 ± 8‡ |
| GRα | 100 ± 7 | 140 ± 23 | 79 ± 13 | 130 ± 28 |
| Soluble phosphenolpyruvate carboxykinase | 100 ± 8 | 97 ± 13 | 59 ± 4†* | 105 ± 20 |
| Adiponectin | 100 ± 10 | 83 ± 14 | 72 ± 7#* | 144 ± 6 |
| PPARγ | 100 ± 7 | 101 ± 17 | 82 ± 6* | 134 ± 23 |
| Uncoupling protein 2 | 100 ± 13 | 122 ± 16 | 209 ± 14‡* | 325 ± 47# |
| Leptin | 100 ± 19 | 118 ± 27 | 389 ± 29‡** | 225 ± 40† |
| Resistin | 100 ± 10 | 114 ± 20 | 217 ± 21‡* | 150 ± 19 |
| Tumor necrosis factor-α | 100 ± 22 | 85 ± 19 | 124 ± 17 | 89 ± 16 |
Data are means ± SE. Gene expression in SCAT of 24-week-old mice (n = 10 per group) using quantitative real-time PCR. Data are normalized to 18S and presented as percentage of WT-chow control.
P < 0.05,
P < 0.01 between genotypes on a given diet;
P < 0.05,
P < 0.001,
P < 0.0001 between diet for a given genotype.
TABLE 2.
Systemic indices of glucocorticoid exposure
| WT-chow | Transgenic-chow | WT-HFD | Transgenic-HFD | |
|---|---|---|---|---|
| Serum corticosterone (ng/ml) (male, 10 weeks old, n = 8) |
17.0 ± 13.5 | 18.2 ± 23.3 | — | — |
| Thymic weight (mg) (male, 10 weeks old, n = 8) |
89.9 ± 2.0 | 88.5 ± 2.0 | — | — |
| Adrenal weight (mg) (male, 24 weeks old, n = 10) |
3.21 ± 0.18 | 3.23 ± 0.21 | 3.65 ± 0.15 | 3.47 ± 0.10 |
| Bone mineral density (mg/cm2) (male, 24 weeks old, n = 10) |
524. ± 3 | 530 ± 3 | 543 ± 2 | 535 ± 5 |
| Lean body mass (g) (male, 24 weeks old, n = 10) |
25.1 ± 0.3 | 25.7 ± 0.3 | 26.7 ± 0.3 | 26.3 ± 0.4 |
| Naso-anal length (cm) (male, 24 weeks old, n = 10) |
10.0 ± 0.04 | 9.9 ± 0.03 | 10.3 ± 0.10 | 10.2 ± 0.10 |
Data are means ± SE.
Transgenic aP2-h11βHSD2 mice resist weight gain on HFD
Mice of both genotypes fed HFD gained significantly more weight than mice fed chow diet (body weight at 24 weeks: WT-chow 32.87 ± 0.54 g, transgenic-chow 32.82 ± 0.52 g, WT-HFD 44.52 ± 0.66 g, and transgenic-HFD 40.80 ± 1.04 g). Body weight for WT and transgenic mice fed chow diet were similar throughout the 24-week study. In contrast, transgenic mice fed HFD gained significantly less weight than WT mice fed HFD (Fig. 2).
FIG. 2.
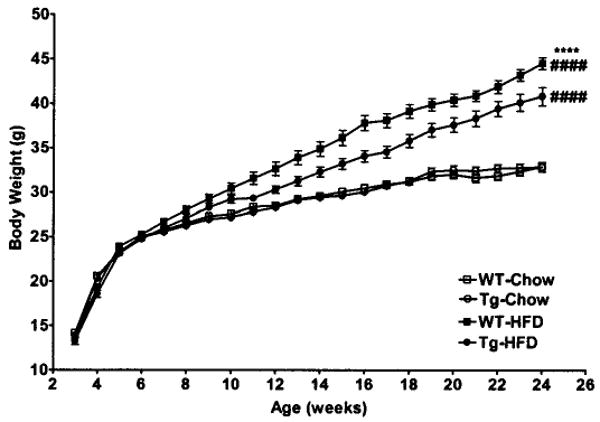
Body weight. Body weight in transgenic and WT mice fed chow or HFD from 3 to 24 weeks of age (n = 18–22 per group). □, WT-chow; ○, transgenic-chow; ■, WT-HFD; ●, transgenic-HFD. Data are expressed as means ± SE. ****P < 0.0001 between genotypes on a given diet; ####P < 0.0001 between diet groups for a given genotype by repeated-measures ANOVA.
Transgenic aP2-h11βHSD2 mice on HFD have reduced body fat
Percent fat mass by DEXA (Fig. 3A) was increased in mice of both genotypes fed HFD compared with mice fed chow diet (WT-chow 20.56 ± 0.57%, transgenic-chow 20.78 ± 0.82%, WT-HFD 37.78 ± 0.75%, and transgenic-HFD 34.11 ± 1.21%). Percent fat mass by DEXA was similar between WT and transgenic mice fed chow diet. In contrast, transgenic mice fed HFD had significantly lower percent fat mass than WT mice fed HFD. Lean body mass by DEXA and linear growth were not significantly different between genotypes on either diet (Table 2). Total fat pad weights (Fig. 3B) were increased in mice of both genotypes fed HFD compared with mice fed chow diet (total fat pad weight: WT-chow 2.70 ± 0.12 g, transgenic-chow 2.93 ± 0.28 g, WT-HFD 7.38 ± 0.43 g, and transgenic-HFD 5.51 ± 0.55 g). Total fat pad weights were similar in WT and transgenic mice fed chow diet. In contrast, transgenic mice fed HFD had significantly lower total fat pad weight than WT mice fed HFD. This difference in total fat pad weight was primarily due to decreased fat pad weight of the SCAT depot and, to a lesser extent, the perirenal plus retroperitoneal adipose tissue and ISWAT depots (Fig. 3C).
FIG. 3.
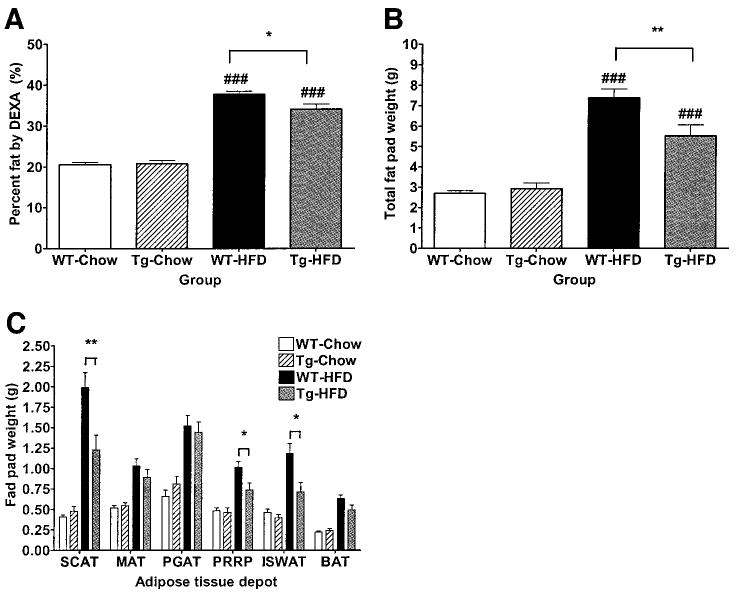
Fat mass. A: Percent fat mass by DEXA in 24-week-old transgenic and WT mice fed chow or HFD (n = 18–22 per group). Total (B) and individual (C) fat pad weights in 24-week-old transgenic and WT mice fed chow or HFD (n = 10 per group). For individual fat pad weights, significant differences between diet groups for a given genotype are omitted for clarity. Data are expressed as means ± SE. *P < 0.05 and **P < 0.01 between genotypes on a given diet; ###P < 0.001 between diet groups for a given genotype.
Transgenic aP2-h11βHSD2 mice have decreased food intake and increased energy expenditure
Average daily (Fig. 4A) and cumulative (Fig. 4B) food intake were lower in transgenic compared with WT mice on HFD (average daily food intake for WT-HFD 5.95 ± 0.25 g vs. transgenic-HFD 5.21 ± 0.23 g; cumulative food intake for WT-HFD 71.40 ± 2.95 g vs. transgenic-HFD 62.50 ± 2.80 g). In addition, oxygen consumption (Vo2) was significantly increased in transgenic compared with WT mice (WT-HFD 3,016 ± 34 mg · ml−1 · h−1 vs. transgenic-HFD 3,238 ± 34 mg · ml−1 · h−1) (Fig. 4C). This elevation in oxygen consumption in transgenic mice was present throughout the 24-h cycle for each day during the study period (Fig. 4D).
FIG. 4.
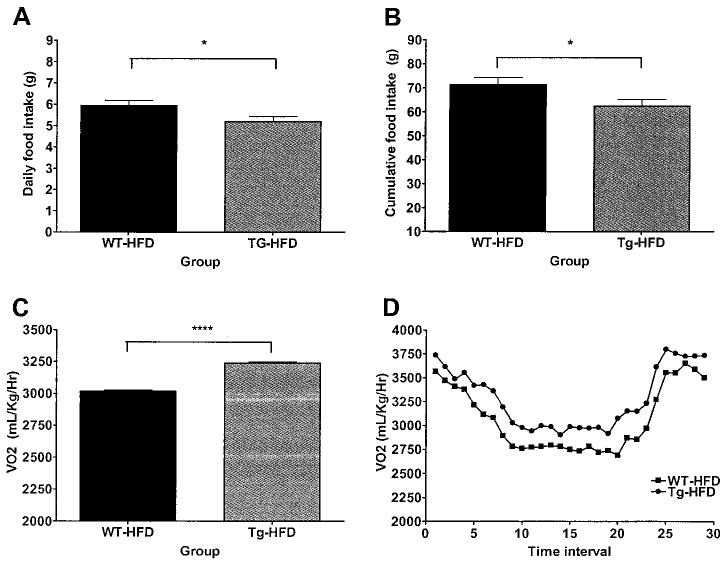
Food intake and energy expenditure. Daily food intake (A), cumulative food intake (B), oxygen consumption (Vo2) (C), and diurnal oxygen consumption profile (D) of 24-week-old transgenic and WT mice fed HFD (n = 8 per group) measured for 12 days in a comprehensive laboratory animal monitoring system. Data are expressed as means ± SE. ■, WT-HFD; ●, transgenic-HFD. For food intake, *P < 0.05 between genotypes. For oxygen consumption, ****P < 0.0001 between genotypes by mixed model analysis.
Transgenic aP2-h11βHSD2 mice have improved glucose tolerance and insulin sensitivity
Fasting plasma glucose (Fig. 5A) was higher in WT mice fed HFD compared with WT mice fed chow diet but was not different between transgenic mice fed HFD compared with transgenic mice fed chow diet (WT-chow 134 ± 7 mg/dl, WT-HFD 155 ± 5 mg/dl; transgenic-chow 137 ± 5 mg/dl, transgenic-HFD 148 ± 6 mg/dl). Fasting plasma glucose was not significantly different between WT and transgenic mice on either diet. Fasting serum insulin (Fig. 5B) was higher in WT mice fed HFD compared with WT mice fed chow diet but was not different between transgenic mice fed HFD compared with transgenic mice fed chow diet (WT-chow 0.4190 ± 0.0336 ng/ml, WT-HFD 0.7331 ± 0.0886 ng/ml; transgenic-chow 0.2893 ± 0.0285 ng/ml, transgenic-HFD 0.4350 ± 0.0759 ng/ml). Fasting serum insulin was significantly lower in transgenic compared with WT mice on both diets.
FIG. 5.
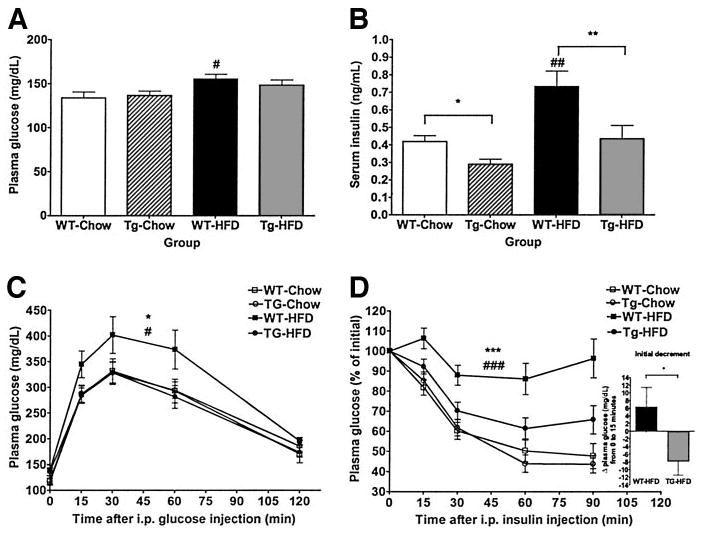
Glucose tolerance and insulin sensitivity. Plasma glucose (A) and serum insulin (B) after an overnight fast in 20-week-old transgenic and WT mice (n = 18–22 per group). Data are expressed as means ± SE. C: Glucose tolerance test. Glucose (1 g/kg) was administered intraperitoneally after an overnight fast to 16-week-old transgenic and WT mice fed chow or HFD (n = 10 per group). Data are expressed as means ± SE. D: Insulin tolerance test. Insulin (1.125 units/kg) was administered intraperitoneally after a 6-h fast to 21-week-old transgenic and WT mice fed chow or HFD (n = 18–22 per group). Values are expressed as means ± SE of percent initial value. The initial decrement from 0 to 15 min after insulin injection for WT-HFD and transgenic-HFD groups are indicated (inset). □, WT-chow; ○, transgenic-chow; ■, WT-HFD; ●, transgenic-HFD. *P < 0.05, **P < 0.01, and ***P < 0.001 between genotypes on a given diet; #P < 0.05, ##P < 0.01, and ###P < 0.001 between diet groups for a given genotype.
Glucose tolerance by glucose tolerance test (Fig. 5C) was impaired in WT mice fed HFD compared with WT mice fed chow diet but was similar in transgenic mice fed HFD versus transgenic mice fed chow diet. Glucose tolerance was similar between transgenic and WT mice fed chow diet. Glucose tolerance in transgenic mice fed HFD was improved compared with WT mice fed HFD and similar to glucose tolerance of mice of both genotypes fed chow diet. Insulin sensitivity by insulin tolerance test (Fig. 5D) was decreased in WT mice fed HFD compared with WT mice fed chow diet but was similar in transgenic mice fed HFD versus transgenic mice fed chow diet. Insulin sensitivity was similar between transgenic and WT mice fed chow diet. Insulin sensitivity in transgenic mice fed HFD was improved compared with WT mice fed HFD (initial decrement: WT-HFD +6.3 ± 5.1, transgenic-chow −7.8 ± 3.4 mg/dl) and similar to insulin sensitivity of mice of both genotypes fed chow diet.
Transgenic aP2-h11βHSD2 mice have a favorable adipose tissue gene expression profile
To evaluate the molecular mechanisms underlying this improved metabolic profile, expression of genes known to play important roles in adipose tissue metabolism and/or known to be regulated by GCs were evaluated (Table 1). In the SCAT of transgenic compared with WT mice fed HFD, expression of soluble phosphenolpyruvate carboxykinase, adiponectin, peroxisome proliferator-activated receptor γ, and uncoupling protein 2 were significantly increased, whereas expression of leptin and resistin were significantly decreased. Expression of tumor necrosis factor-α was also decreased in the SCAT of transgenic compared with WT mice fed HFD, albeit not significantly so. No significant differences in adipose tissue gene expression were observed for transgenic compared with WT mice fed chow diet.
DISCUSSION
We describe an animal model of adipocyte-specific GC inactivation through ectopic transgenic overexpression of h11βHSD2 under the control of the murine aP2 promoter. 11βHSD2 decreases GC action in vivo by catalyzing the unidirectional conversion of hormonally active 11β-hydroxylated GCs to their hormonally inactive 11β-keto metabolites. Since 11βHSD2 is not endogenously expressed in adipocytes, transgenic overexpression of 11βHSD2 under the control of the aP2 promoter provides a novel mechanism whereby GC action can be selectively reduced in adipose tissue, thus reversing the GC-amplifying effect of endogenous 11βHSD1. A similar approach using ectopic tissue-specific overexpression of 11βHSD2 has been used to generate an animal model of reduced GC action in hippocampal neurons for the study of neuronal stress response pathways (33).
Several lines of evidence support adipose tissue–specific GC inactivation in this model. First, in vitro 11βHSD2 activity data demonstrate increased NAD-dependent dehydrogenation of corticosterone to its inactive metabolite 11DHC in adipose tissue of transgenic but not WT mice. This activity is comparable to 11βHSD2 activity in kidney, a level known to be sufficient to inactivate GCs and protect mineralocorticoid receptors in the distal nephron where 11βHSD1 is also expressed. Second, endogenous m11βHSD2 is not contributing to this activity since expression of m11βHSD2 is extremely low in adipose tissue and is not significantly different between genotypes. Third, no significant differences between genotypes are identified for other determinants of GC action, including 11βHSD1 expression/activity or GRα expression. Fourth, expression of the human 11βHSD2 transgene is 2.55- and 12.74-fold greater than expression of endogenous 11βHSD1 in SCAT of transgenic mice on chow and HFD, respectively. Finally, a relative increase in GC-repressible genes (i.e., soluble phosphenolpyruvate carboxykinase) (34,35) and a decrease in GC-inducible genes (i.e., leptin) (36) is observed in transgenic compared with WT mice fed HFD. Taken together, these data support a net reduction in GC action in adipose tissue and indicate that the “adipokine” actions of GCs may be key.
Interestingly, 11βHSD2 expression and activity were not uniform among adipose tissue depots. This finding may suggest the presence of depot-specific regulatory factors that influence the activity of the aP2 promoter. While MAT has been implicated as the primary determinant of adverse metabolic consequences associated with the metabolic syndrome (3), most studies in humans have revealed significant correlations between 11βHSD1 expression/activity in SCAT and measures of body weight and insulin sensitivity (11–17). Although the greater 11βHSD2 transgene activity in SCAT relative to MAT supports a role for “peripheral” adipose tissue depots in determining metabolic phenotype in this model, it does not entirely exclude a substantial contribution by MAT. MAT has relatively higher GR density than other adipose depots, and such depot-specific differences in GR expression are also important determinants of local GC action. Furthermore, since endogenous 11βHSD2 is negligible in adipose tissue, transgenic 11βHSD2, even at very low levels, could have a substantial effect on GC action, particularly in MAT where GR expression is high. This question could be further explored by evaluating gene expression in MAT and/or by identifying and evaluating transgenic lines of aP2-11βHS2 mice with higher 11βHSD2 transgene expression in MAT.
Phenotypic characterization of transgenic aP2-h11βHSD2 mice reveal several similarities with 11βHSD1-deficient mice (23–25). Both models demonstrate an improved metabolic response to HFD characterized by resistance to diet-induced obesity, reduced fat mass, and improved glucose tolerance and insulin sensitivity (23–25). In addition, both models have reduced expression of leptin and resistin and increased expression of adiponectin, peroxisome proliferator-activated receptor γ, and uncoupling protein 2 in adipose tissue (25). Potential contributions of these genes to phenotype is discussed in detail elsewhere (25). Hence, these similarities suggest that altered GC metabolism in adipose tissue is a major contributor to these aspects of metabolic phenotype.
Nevertheless, there are several important differences between these two models. The major difference is that local GC inactivation is confined exclusively to adipose tissue in aP2-h11βHSD2 mice, whereas 11βHSD1-deficient mice have altered GC metabolism in all tissues expressing the enzyme, including the central nervous system, as well as strain-dependent modest increases in circulating GCs. In addition, aP2-h11βHSD2 mice have reduced food intake, while 11βHSD1-deficient mice have increased food intake (25). Leptin, a major adipocyte-derived hormonal determinant of energy intake, is unlikely to account for this difference, since adipose tissue leptin expression is similarly reduced in both aP2-h11βHSD2 and 11βHSD1-deficient mice. A more likely explanation is that nonadipocyte-mediated mechanisms contribute to altered energy intake in 11βHSD1-deficient mice. GCs are well known to influence appetite and feeding behavior (37,38). Both GRs (39–42) and 11βHSD1 (43,44) are expressed throughout the central nervous system, including several hypothalamic areas involved in the regulation of energy balance such as the arcuate nucleus (43,44). Whether primary central 11βHSD1 deficiency or secondary increases in circulating GCs is the main determinant of altered energy intake in 11βHSD1-deficient mice remains unclear. In contrast, the decrease in food intake in aP2-h11βHSD2 mice supports an adipocyte-mediated mechanism for altered energy intake in this model. This conclusion is further supported by the opposite observation, i.e., increased food intake, in aP2-h11βHSD1 mice (10). An as yet unidentified adipocyte-secreted factor may be contributing to the reduced food intake in aP2-h11βHSD2 mice.
Resistance to diet-induced obesity in aP2-h11βHSD2 mice results not only from decreased energy intake but also from increased energy expenditure, as indicated by an increase in oxygen consumption. A similar increase in metabolic rate has been suggested, albeit not proven, in 11βHSD1-deficient mice by an increase in core body temperature (25). For the reasons outlined above, however, it is difficult to ascertain the relative contribution of central versus peripheral mechanisms to this altered energy expenditure in 11βHSD1-deficient mice. Increased oxygen consumption in aP2-h11βHSD2 mice supports an adipocyte-mediated mechanism for altered energy expenditure. Leptin, which increases energy expenditure, is not the major determinant of energy expenditure in this model, since leptin expression in adipose tissue is decreased. GCs decrease thermogenesis (45,46) and inhibit adrenergic stimulation of uncoupling proteins (47). GCs also inhibit β3 adrenergic receptor signaling and expression (48,49), a major mechanism promoting thermogenesis in BAT (50). Thus, aP2-h11βHSD2 mice, via decreased GC action in adipose tissue, would be protected from this GC-mediated inhibition of β3 adrenergic receptors and energy expenditure. Whether the observed level of expression of the h11βHSD2 transgene in BAT of aP2-h11βHSD2 mice is sufficient to alter β3 adrenergic receptors gene expression and/or adrenergic stimulation of uncoupling proteins in BAT remains to be determined. However, data in both aP2-h11βHSD2 and 11βHSD1-deficient mice suggest that GC metabolism by endogenous 11βHSD1 may play an important role in regulating GC action in BAT. This question could be further explored by generated BAT-specific models of increased and decreased GC action via transgenic overexpression of 11βHSD1 and 11βHSD2, respectively
In summary, adipocyte-specific transgenic overexpression of h11βHSD2 has been used as a strategy to determine the role of adipocyte-specific GC action in systemic energy balance. Transgenic aP2-h11βHSD2 mice fed HFD exhibit resistance to diet-induced obesity, reduced fat mass, decreased food intake, increased energy expenditure, and improved glucose tolerance and insulin sensitivity. This metabolic phenotype is associated with reduced expression of leptin and resistin, and increased expression of adiponectin, peroxisome proliferator–activated receptor γ, and uncoupling protein 2 in adipose tissue. These data provide the first in vivo evidence that reduction of active GCs exclusively in adipose tissue is an important determinant of a favorable metabolic phenotype with respect to energy homeostasis and the metabolic syndrome. Further investigation of this model may help to identify novel adipocyte mediators of systemic energy balance.
Acknowledgments
This research is supported by National Instiutes of Health Grants K08KK65833 (to E.E.K.) and DK R37 28082 (to J.S.F.), a Takeda Chemical Industrustries grant (to J.S.F.), a Wellcome Trust Programme grant (to J.R.S.), and a Wellcome Trust Intermediate Fellowship (to N.M.M.).
- 11βHSD
11β-hydroxysteroid dehydrogenase
- 11DHC
11-dehydrocorticosterone
- aP2
adipocyte fatty acid binding protein
- BAT
brown adipose tissue
- DEXA
dual-energy X-ray absorptiometry
- GC
glucocorticoid
- GR
GC receptor
- HFD
high-fat diet
- ISWAT
intrascapular white adipose tissue
- MAT
mesenteric adipose tissue
- PGAT
perigonadal adipose tissue
- SCAT
subcutaneous adipose tissue
- WT
wild-type
References
- 1.Friedman TC, Mastorakos G, Newman TD, Mullen NM, Horton EG, Costello R, Papadopoulos NM, Chrousos GP. Carbohydrate and lipid metabolism in endogenous hypercortisolism: shared features with metabolic syndrome X and NIDDM. Endocr J. 1996;43:645–655. doi: 10.1507/endocrj.43.645. [DOI] [PubMed] [Google Scholar]
- 2.Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among US adults: findings from the Third National Health and Nutrition Examination Survey. JAMA. 2002;287:356–359. doi: 10.1001/jama.287.3.356. [DOI] [PubMed] [Google Scholar]
- 3.Wajchenberg BL. Subcutaneous and visceral adipose tissue: their relation to the metabolic syndrome. Endocr Rev. 2000;21:697–738. doi: 10.1210/edrv.21.6.0415. [DOI] [PubMed] [Google Scholar]
- 4.Nobel S, Abrahmsen L, Oppermann U. Metabolic conversion as a prereceptor control mechanism for lipophilic hormones. Eur J Biochem. 2001;268:4113–4125. doi: 10.1046/j.1432-1327.2001.02359.x. [DOI] [PubMed] [Google Scholar]
- 5.Stewart PM, Krozowski ZS. 11 beta-hydroxysteroid dehydrogenase. Vitam Horm. 1999;57:249–324. [PubMed] [Google Scholar]
- 6.Edwards CR, Stewart PM, Burt D, Brett L, McIntyre MA, Sutanto WS, de Kloet ER, Monder C. Localisation of 11 beta-hydroxysteroid dehydrogenase: tissue specific protector of the mineralocorticoid receptor. Lancet. 1988;2:986–989. doi: 10.1016/s0140-6736(88)90742-8. [DOI] [PubMed] [Google Scholar]
- 7.Funder JW, Pearce PT, Smith R, Smith AI. Mineralocorticoid action: target tissue specificity is enzyme, not receptor, mediated. Science. 1988;242:583–585. doi: 10.1126/science.2845584. [DOI] [PubMed] [Google Scholar]
- 8.Livingstone DE, Kenyon CJ, Walker BR. Mechanisms of dysregulation of 11 beta-hydroxysteroid dehydrogenase type 1 in obese Zucker rats. J Endocrinol. 2000;167:533–539. doi: 10.1677/joe.0.1670533. [DOI] [PubMed] [Google Scholar]
- 9.Livingstone DE, Jones GC, Smith K, Jamieson PM, Andrew R, Kenyon CJ, Walker BR. Understanding the role of glucocorticoids in obesity: tissue-specific alterations of corticosterone metabolism in obese Zucker rats. Endocrinology. 2000;141:560–563. doi: 10.1210/endo.141.2.7297. [DOI] [PubMed] [Google Scholar]
- 10.Masuzaki H, Paterson J, Shinyama H, Morton NM, Mullins JJ, Seckl JR, Flier JS. A transgenic model of visceral obesity and the metabolic syndrome. Science. 2001;294:2166–2170. doi: 10.1126/science.1066285. [DOI] [PubMed] [Google Scholar]
- 11.Rask E, Olsson T, Soderberg S, Andrew R, Livingstone DE, Johnson O, Walker BR. Tissue-specific dysregulation of cortisol metabolism in human obesity. J Clin Endocrinol Metab. 2001;86:1418–1421. doi: 10.1210/jcem.86.3.7453. [DOI] [PubMed] [Google Scholar]
- 12.Wake DJ, Rask E, Livingstone DE, Soderberg S, Olsson T, Walker BR. Local and systemic impact of transcriptional up-regulation of 11beta-hydroxysteroid dehydrogenase type 1 in adipose tissue in human obesity. J Clin Endocrinol Metab. 2003;88:3983–3988. doi: 10.1210/jc.2003-030286. [DOI] [PubMed] [Google Scholar]
- 13.Lindsay RS, Wake DJ, Nair S, Bunt J, Livingstone DE, Permana PA, Tataranni PA, Walker BR. Subcutaneous adipose 11 beta-hydroxysteroid dehydrogenase type 1 activity and messenger ribonucleic acid levels are associated with adiposity and insulinemia in Pima Indians and Caucasians. J Clin Endocrinol Metab. 2003;88:2738–2744. doi: 10.1210/jc.2002-030017. [DOI] [PubMed] [Google Scholar]
- 14.Paulmyer-Lacroix O, Boullu S, Oliver C, Alessi MC, Grino M. Expression of the mRNA coding for 11beta-hydroxysteroid dehydrogenase type 1 in adipose tissue from obese patients: an in situ hybridization study. J Clin Endocrinol Metab. 2002;87:2701–2705. doi: 10.1210/jcem.87.6.8614. [DOI] [PubMed] [Google Scholar]
- 15.Rask E, Walker BR, Soderberg S, Livingstone DE, Eliasson M, Johnson O, Andrew R, Olsson T. Tissue-specific changes in peripheral cortisol metabolism in obese women: increased adipose 11beta-hydroxysteroid dehydrogenase type 1 activity. J Clin Endocrinol Metab. 2002;87:3330–3336. doi: 10.1210/jcem.87.7.8661. [DOI] [PubMed] [Google Scholar]
- 16.Engeli S, Bohnke J, Feldpausch M, Gorzelniak K, Heintze U, Janke J, Luft FC, Sharma AM. Regulation of 11beta-HSD genes in human adipose tissue: influence of central obesity and weight loss. Obes Res. 2004;12:9–17. doi: 10.1038/oby.2004.3. [DOI] [PubMed] [Google Scholar]
- 17.Kannisto K, Pietilainen KH, Ehrenborg E, Rissanen A, Kaprio J, Hamsten A, Yki-Jarvinen H. Overexpression of 11{beta}-hydroxysteroid dehydrogenase-1 in adipose tissue is associated with acquired obesity and features of insulin resistance: studies in young adult monozygotic twins. J Clin Endocrinol Metab. 2004;89:4414–4421. doi: 10.1210/jc.2004-0153. [DOI] [PubMed] [Google Scholar]
- 18.Draper N, Echwald SM, Lavery GG, Walker EA, Fraser R, Davies E, Sorensen TI, Astrup A, Adamski J, Hewison M, Connell JM, Pedersen O, Stewart PM. Association studies between microsatellite markers within the gene encoding human 11beta-hydroxysteroid dehydrogenase type 1 and body mass index, waist to hip ratio, and glucocorticoid metabolism. J Clin Endocrinol Metab. 2002;87:4984–4990. doi: 10.1210/jc.2001-011375. [DOI] [PubMed] [Google Scholar]
- 19.Gelernter-Yaniv L, Feng N, Sebring NG, Hochberg Z, Yanovski JA. Associations between a polymorphism in the 11 beta hydroxysteroid dehydrogenase type I gene and body composition. Int J Obes Relat Metab Disord. 2003;27:983–986. doi: 10.1038/sj.ijo.0802327. [DOI] [PubMed] [Google Scholar]
- 20.Alberts P, Nilsson C, Selen G, Engblom LO, Edling NH, Norling S, Klingstrom G, Larsson C, Forsgren M, Ashkzari M, Nilsson CE, Fiedler M, Bergqvist E, Ohman B, Bjorkstrand E, Abrahmsen LB. Selective inhibition of 11 beta-hydroxysteroid dehydrogenase type 1 improves hepatic insulin sensitivity in hyperglycemic mice strains. Endocrinology. 2003;144:4755–4762. doi: 10.1210/en.2003-0344. [DOI] [PubMed] [Google Scholar]
- 21.Alberts P, Engblom L, Edling N, Forsgren M, Klingstrom G, Larsson C, Ronquist-Nii Y, Ohman B, Abrahmsen L. Selective inhibition of 11beta-hydroxysteroid dehydrogenase type 1 decreases blood glucose concentrations in hyperglycaemic mice. Diabetologia. 2002;45:1528–1532. doi: 10.1007/s00125-002-0959-6. [DOI] [PubMed] [Google Scholar]
- 22.Barf T, Vallgarda J, Emond R, Haggstrom C, Kurz G, Nygren A, Larwood V, Mosialou E, Axelsson K, Olsson R, Engblom L, Edling N, Ronquist-Nii Y, Ohman B, Alberts P, Abrahmsen L. Arylsulfonamidothiazoles as a new class of potential antidiabetic drugs: discovery of potent and selective inhibitors of the 11beta-hydroxysteroid dehydrogenase type 1. J Med Chem. 2002;45:3813–3815. doi: 10.1021/jm025530f. [DOI] [PubMed] [Google Scholar]
- 23.Kotelevtsev Y, Holmes MC, Burchell A, Houston PM, Schmoll D, Jamieson P, Best R, Brown R, Edwards CR, Seckl JR, Mullins JJ. 11beta-hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proc Natl Acad Sci U S A. 1997;94:14924–14929. doi: 10.1073/pnas.94.26.14924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morton NM, Holmes MC, Fievet C, Staels B, Tailleux A, Mullins JJ, Seckl JR. Improved lipid and lipoprotein profile, hepatic insulin sensitivity, and glucose tolerance in 11beta-hydroxysteroid dehydrogenase type 1 null mice. J Biol Chem. 2001;276:41293–41300. doi: 10.1074/jbc.M103676200. [DOI] [PubMed] [Google Scholar]
- 25.Morton NM, Paterson JM, Masuzaki H, Holmes MC, Staels B, Fievet C, Walker BR, Flier JS, Mullins JJ, Seckl JR. Novel adipose tissue-mediated resistance to diet-induced visceral obesity in 11β-hydroxysteroid dehydrogenase type 1–deficient mice. Diabetes. 2004;53:931–938. doi: 10.2337/diabetes.53.4.931. [DOI] [PubMed] [Google Scholar]
- 26.Masuzaki H, Yamamoto H, Kenyon CJ, Elmquist JK, Morton NM, Paterson JM, Shinyama H, Sharp MG, Fleming S, Mullins JJ, Seckl JR, Flier JS. Transgenic amplification of glucocorticoid action in adipose tissue causes high blood pressure in mice. J Clin Invest. 2003;112:83–90. doi: 10.1172/JCI17845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paterson JM, Morton NM, Fievet C, Kenyon CJ, Holmes MC, Staels B, Seckl JR, Mullins JJ. Metabolic syndrome without obesity: hepatic overexpression of 11beta-hydroxysteroid dehydrogenase type 1 in transgenic mice. Proc Natl Acad Sci U S A. 2004;101:7088–7093. doi: 10.1073/pnas.0305524101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brown RW, Chapman KE, Murad P, Edwards CR, Seckl JR. Purification of 11 beta-hydroxysteroid dehydrogenase type 2 from human placenta utilizing a novel affinity labelling technique. Biochem J. 1996;313:997–1005. doi: 10.1042/bj3130997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ross SR, Graves RA, Greenstein A, Platt KA, Shyu HL, Mellovitz B, Spiegelman BM. A fat-specific enhancer is the primary determinant of gene expression for adipocyte P2 in vivo. Proc Natl Acad Sci U S A. 1990;87:9590–9594. doi: 10.1073/pnas.87.24.9590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jamieson PM, Chapman KE, Edwards CR, Seckl JR. 11 beta-hydroxysteroid dehydrogenase is an exclusive 11 beta- reductase in primary cultures of rat hepatocytes: effect of physicochemical and hormonal manipulations. Endocrinology. 1995;136:4754–4761. doi: 10.1210/endo.136.11.7588203. [DOI] [PubMed] [Google Scholar]
- 31.Christy C, Hadoke PW, Paterson JM, Mullins JJ, Seckl JR, Walker BR. 11beta-hydroxysteroid dehydrogenase type 2 in mouse aorta: localization and influence on response to glucocorticoids. Hypertension. 2003;42:580–587. doi: 10.1161/01.HYP.0000088855.06598.5B. [DOI] [PubMed] [Google Scholar]
- 32.Morton NM, Ramage L, Seckl JR. Down-regulation of adipose 11beta-hydroxysteroid dehydrogenase type 1 by high-fat feeding in mice: a potential adaptive mechanism counteracting metabolic disease. Endocrinology. 2004;145:2707–2712. doi: 10.1210/en.2003-1674. [DOI] [PubMed] [Google Scholar]
- 33.Kaufer D, Ogle WO, Pincus ZS, Clark KL, Nicholas AC, Dinkel KM, Dumas TC, Ferguson D, Lee AL, Winters MA, Sapolsky RM. Restructuring the neuronal stress response with anti-glucocorticoid gene delivery. Nat Neurosci. 2004;7:947–953. doi: 10.1038/nn1296. [DOI] [PubMed] [Google Scholar]
- 34.Meisner H, Loose DS, Hanson RW. Effect of hormones on transcription of the gene for cytosolic phosphoenolpyruvate carboxykinase (GTP) in rat kidney. Biochemistry. 1985;24:421–425. doi: 10.1021/bi00323a027. [DOI] [PubMed] [Google Scholar]
- 35.Sasaki K, Cripe TP, Koch SR, Andreone TL, Petersen DD, Beale EG, Granner DK. Multihormonal regulation of phosphoenolpyruvate carboxykinase gene transcription: the dominant role of insulin. J Biol Chem. 1984;259:15242–15251. [PubMed] [Google Scholar]
- 36.De Vos P, Saladin R, Auwerx J, Staels B. Induction of ob gene expression by corticosteroids is accompanied by body weight loss and reduced food intake. J Biol Chem. 1995;270:15958–15961. doi: 10.1074/jbc.270.27.15958. [DOI] [PubMed] [Google Scholar]
- 37.Dallman MF, la Fleur SE, Pecoraro NC, Gomez F, Houshyar H, Akana SF. Minireview: Glucocorticoids: food intake, abdominal obesity, and wealthy nations in 2004. Endocrinology. 2004;145:2633–2638. doi: 10.1210/en.2004-0037. [DOI] [PubMed] [Google Scholar]
- 38.Dallman MF, Pecoraro N, Akana SF, La Fleur SE, Gomez F, Houshyar H, Bell ME, Bhatnagar S, Laugero KD, Manalo S. Chronic stress and obesity: a new view of “comfort food”. Proc Natl Acad Sci U S A. 2003;100:11696–11701. doi: 10.1073/pnas.1934666100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morimoto M, Morita N, Ozawa H, Yokoyama K, Kawata M. Distribution of glucocorticoid receptor immunoreactivity and mRNA in the rat brain: an immunohistochemical and in situ hybridization study. Neurosci Res. 1996;26:235–269. doi: 10.1016/s0168-0102(96)01105-4. [DOI] [PubMed] [Google Scholar]
- 40.Ahima RS, Harlan RE. Charting of type II glucocorticoid receptor-like immunoreactivity in the rat central nervous system. Neuroscience. 1990;39:579–604. doi: 10.1016/0306-4522(90)90244-x. [DOI] [PubMed] [Google Scholar]
- 41.Sousa RJ, Tannery NH, Lafer EM. In situ hybridization mapping of glucocorticoid receptor messenger ribonucleic acid in rat brain. Mol Endocrinol. 1989;3:481–494. doi: 10.1210/mend-3-3-481. [DOI] [PubMed] [Google Scholar]
- 42.Cintra A, Zoli M, Rosen L, Agnati LF, Okret S, Wikstrom AC, Gustaffsson JA, Fuxe K. Mapping and computer assisted morphometry and microdensitometry of glucocorticoid receptor immunoreactive neurons and glial cells in the rat central nervous system. Neuroscience. 1994;62:843–897. doi: 10.1016/0306-4522(94)90481-2. [DOI] [PubMed] [Google Scholar]
- 43.Moisan MP, Edwards CR, Seckl JR. Ontogeny of 11 beta-hydroxysteroid dehydrogenase in rat brain and kidney. Endocrinology. 1992;130:400–404. doi: 10.1210/endo.130.1.1727713. [DOI] [PubMed] [Google Scholar]
- 44.Moisan MP, Seckl JR, Edwards CR. 11 beta-hydroxysteroid dehydrogenase bioactivity and messenger RNA expression in rat forebrain: localization in hypothalamus, hippocampus, and cortex. Endocrinology. 1990;127:1450–1455. doi: 10.1210/endo-127-3-1450. [DOI] [PubMed] [Google Scholar]
- 45.Strack AM, Bradbury MJ, Dallman MF. Corticosterone decreases nonshivering thermogenesis and increases lipid storage in brown adipose tissue. Am J Physiol. 1995;268:R183–R191. doi: 10.1152/ajpregu.1995.268.1.R183. [DOI] [PubMed] [Google Scholar]
- 46.Moriscot A, Rabelo R, Bianco AC. Corticosterone inhibits uncoupling protein gene expression in brown adipose tissue. Am J Physiol. 1993;265:E81–E87. doi: 10.1152/ajpendo.1993.265.1.E81. [DOI] [PubMed] [Google Scholar]
- 47.Soumano K, Desbiens S, Rabelo R, Bakopanos E, Camirand A, Silva JE. Glucocorticoids inhibit the transcriptional response of the uncoupling protein-1 gene to adrenergic stimulation in a brown adipose cell line. Mol Cell Endocrinol. 2000;165:7–15. doi: 10.1016/s0303-7207(00)00276-8. [DOI] [PubMed] [Google Scholar]
- 48.Feve B, Baude B, Krief S, Strosberg AD, Pairault J, Emorine LJ. Inhibition by dexamethasone of beta 3-adrenergic receptor responsiveness in 3T3–F442A adipocytes: evidence for a transcriptional mechanism. J Biol Chem. 1992;267:15909–15915. [PubMed] [Google Scholar]
- 49.Bakopanos E, Silva JE. Opposing effects of glucocorticoids on beta(3)-adrenergic receptor expression in HIB-1B brown adipocytes. Mol Cell Endocrinol. 2002;190:29–37. doi: 10.1016/s0303-7207(02)00027-8. [DOI] [PubMed] [Google Scholar]
- 50.Collins S, Surwit RS. The beta-adrenergic receptors and the control of adipose tissue metabolism and thermogenesis. Recent Prog Horm Res. 2001;56:309–328. doi: 10.1210/rp.56.1.309. [DOI] [PubMed] [Google Scholar]