

Conspectus
Synthetic methods to form the carbon-nitrogen bonds in aromatic amines are fundamental enough to be considered part of introductory organic courses. Arylamines are important because they are common precursors to or substructures within active pharmaceutical ingredients and herbicides produced on ton scales, as well as conducting polymers and layers of organic light-emitting diodes produced on small scale. For many years, this class of compound was prepared from classical methods, such as nitration, reduction and reductive alkylation, copper-mediated chemistry at high temperatures, addition to benzyne intermediates, or direct nucleophilic substitution on particularly electron-poor aromatic or heteroaromatic halides. During the past decade, these methods to form aromatic amines have been largely supplanted by palladium-catalyzed coupling reactions of amines with aryl halides. The scope and efficiency of the palladium-catalyzed processes has gradually improved with successive generations of catalysts to the point of being useful for the synthesis of both milligrams and kilograms of product.
This Account describes the conceptual basis and utility of our latest, “fourth-generation” catalyst for the coupling of amines and related reagents with aryl halides. The introductory sections of this account describe the progression of catalyst development from the first-generation to current systems and the motivation for selection of the components of the fourth-generation catalyst. This progression began with catalysts containing palladium and sterically hindered monodentate aromatic phosphines used initially for coupling of tin amides with haloarenes in the first work on C-N coupling. A second generation of catalysts was then developed based on the combination of palladium and aromatic bisphosphines. These systems were then followed by third-generation systems catalysts on the combination of palladium and a sterically hindered alkylmonophosphine or N-heterocyclic carbene.
During the past five years, we have studied a fourth-generation catalyst for these reactions containing ligands that combine the chelating properties of the second-generation systems with the steric hindrance and strong electron donation of the third-generation systems. This combination has created a catalyst that couples aryl chlorides, bromides and iodides with primary amines, N-H imines, and hydrazones in high yield, with broad scope, high functional group tolerance, nearly perfect selectivity for monoarylation, and the lowest levels of palladium that have been used for C-N coupling. This catalyst is based on palladium and a sterically hindered version of the Josiphos family of ligands that possesses a ferrocenyl-1-ethylbackbone, a hindered di-tert-butylphosphino group, and a hindered dicyclohexylphosphino group. This latest generation of catalyst not only improves the coupling of primary amines and related nucleophiles, but it has dramatically improved the coupling of thiols with haloarenes to form C-S bonds. This catalyst system couples both aliphatic and aromatic thiols with chloroarenes with much greater scope, functional group tolerance, and turnover numbers than had been observed previously.
The effects of structural features of the Josiphos ligand on catalyst activity have been revealed by examining the reactivity of catalysts generated from ligands lacking one or more of the structural elements of the most active catalyst. These modified ligands lack the relative stereochemistry of the ferrocenyl-1-ethyl backbone, the strong electron donation of the dialkylphosphino groups, the steric demands of the alkylphosphine groups, or the stability of the ferrocenyl unit. This set of studies showed that each one of these structural features contributed to the high reactivity and selectivity of the catalyst containing the hindered, bidentate Josiphos ligand.
Finally, a series of studies on the effect of electronic properties on the rates of reductive elimination have recently distinguished between the effect of the properties of the M-N σ-bond and the nitrogen electron pair on the rate of reductive elimination. These studies have shown that the effect of substituents attached to the metal-bound nitrogen or carbon atoms on the rate of reductive elimination are similar. Because the amido ligands contain an electron pair, while the alkyl ligands do not, we have concluded that the major electronic effect is transmitted through the σ-bond. In other words, we have concluded that the electronic effect on the metal-nitrogen σ bond dominates an electronic effect on the nitrogen electron pair.

Introduction
For many years, the development of cross-coupling reactions focused on C-C bond formation.1 My group has sought to extend the reach of cross-coupling chemistry to reactions of aromatic electrophiles with heteroatom nucleophiles2,3,4,5 and later with enolate nucleophiles to form sp2 - sp3 carbon-carbon bonds.6,7,8,9 These efforts, along with those of other groups contributing to this issue of Accounts have led to practical synthetic methods to generate aryl amines, ethers, and sulfides, as well as α-aryl carbonyl compounds and α-aryl nitriles. This chemistry has been widely utilized in medicinal chemistry groups as well as the synthesis of clinic candidates on kilogram scale. Several review articles have appeared recently on cross-couplings that form amines, ethers, and sulfides.10–12 An Account from our group13 has summarized the coupling of enolates with aryl halides. Therefore, this account will focus on our recent development of a new class of catalyst for the coupling of aryl halides with amines and thiols. This catalyst couples primary amines, N-H imines, hydrazones, and thiols with aryl chlorides, bromides, and iodides with broad scope and with remarkably low loadings of palladium.
While developing this synthetic methodology, we sought to address several fundamental questions about the factors that control carbon-heteroatom bond cleavage and formation. When we began to develop the coupling of amines with aryl halides, reductive eliminations that form the C-N bonds in amines were rare.14 Since that time, this reaction has proven to occur with a variety of anionic nitrogen ligands and a variety of ancillary dative ligands on palladium,15 and a portion of our mechanistic work has focused on understanding the role of the electronic properties of the heteroatom on the rates of these reductive eliminations. A comparison of the electronic effects on reductive elimination to form C-N bonds from amido complexes and C-C bonds from alkyl complexes has helped to dissect the role of bond polarity and the presence of an electron pair on nitrogen. These electronic effects will be addressed at the conclusion of this account.
Background on Palladium-Catalyzed Amination of Aryl Halides
The first modern studies of palladium catalyzed coupling chemistry to form amines3 were stimulated by a report by Kosugi, Kameyama and Migita16 on the coupling of tin amides with aryl halides catalyzed by palladium and the hindered tri-ortho-tolylphosphine (eq 1). Practical protocols for the coupling of amines with aryl halides in the presence of a base using the same “first generation” catalyst as in the previous work (eq 2) were published in 1995 by Buchwald’s laboratory17 and the authors’ laboratory18. This new procedure eliminated the need for toxic and relatively unstable aminostannane reagents. This chemistry began to encompass reactions of primary amines through work by Buchwald’s laboratory and the author’s laboratory with “second generation” catalysts containing aromatic bisphosphines (eq 3).19–21 Buchwald’s laboratory focused on reactions catalyzed by palladium complexes of BINAP,19, 21 and our group focused on the fundamental organometallic chemistry and coupling reactions catalyzed by palladium complexes of DPPF.20
 |
(1) |
 |
(2) |
 |
(3) |
 |
(4) |
More recent work on this process has focused on developing systems that couple aryl chlorides under mild conditions. Mild aminations of aryl chlorides were conducted in our laboratory by a postdoc Blake Hamann,22 using a member of the Josiphos class of ligands originally developed at Ciba-Geigy for asymmetric hydrogenation (eq 4).23 With a Josiphos ligand containing one diphenylphosphino and one di-tert-butylphosphino group (PPFtBu), reactions of primary amines with several chloroarenes occurred at 85 °C (eq 4). More than five years later, we returned to studying this class of ligand, and this more recent work on C-N couplings using Josiphos ligands is the focus of the majority of this account.
 |
(5) |
In the mean time, our efforts and those of many other groups began to focus on reactions catalyzed by “third-generation” systems containing hindered monodentate ligands.12, 24 Researchers at Tosoh Company reported the coupling of aryl bromides with secondary amines catalyzed by Pd(OAc)2 and tri-tert-butylphosphine with 800–900 turnovers, the highest obtained at that time.25 The Tosoh group also reported the coupling of phenyl chloride with piperazine using 0.5 mol % palladium at 140 °C to form the coupled product with 88% conversion.
Our work with hindered alkylphosphines began with mechanistic studies on these catalysts containing PtBu3. Our studies showed that catalysts generated from a 1:1 ratio of this phosphine to palladium coupled aryl bromides and chlorides at room temperature or slightly above.26 This finding later led us to initiate reactions with the palladium(I) dimers [Pd(PtBu3)Br]2 and {Pd[PtBu2(1-Ad)]Br}2 containing one PtBu3 or PtBu2(1-Ad) (1-Ad = 1-adamantyl) ligand on the palladium catalyst precursor.27 The rates of these reactions (eq 5) were spectacularly fast for the coupling of secondary alkyl amines with aryl chlorides.
 |
(6) |
In parallel, through a series of mechanistic studies on a catalyst generated from Pd(dba)2 and 1,1’-bis(di-tert-butylphosphino)ferrocene, we discovered that this bisphosphine underwent P-C bond cleavage and arylation of one cyclopentadienyl group to generate the sterically-hindered pentaphenylferrocenyl di-tert-butylphosphine ligand.28 The palladium catalyst containing this modified ligand was remarkably active for C-N coupling. Both secondary amines (cyclic and acyclic) and primary amines coupled under mild conditions with aryl bromides and chlorides (eq 6).29 Reactions of primary amines occurred with high selectivity for formation of the monoarylation product. In separate work, we have used catalysts containing a 1:1 ratio of palladium to P(tBu)3 and catalysts containing the Q-phos ligand to develop the coupling of enolate nucleophiles with aryl halides into a process that occurs with broad scope, with enolates containing different main group metals, and with much higher turnover numbers than had been achieved previously.13, 30–34
In parallel, other groups have developed particularly valuable catalysts for the coupling of heteroatom nucleophiles with aryl halides based on monophosphines or N-heterocyclic carbenes that are strongly electron donating and sterically hindered. Buchwald and coworkers have developed a family of biaryl dialkylphosphine ligands that has expanded the scope of the C-N coupling of aryl chlorides with weak nitrogen nucleophiles, such as amides, sulfonamides, and electron-poor arylamines.12 This class of catalyst has also increased the ability of palladium complexes to catalyze the etherification of aryl halides with alcohol and phenol nucleophiles. Beller and coworkers developed heteroaryl dialkylphosphines that are related to the biaryldialkylphosphine ligands and can be prepared by simple syntheses.35 Finally, Nolan and coworkers developed catalysts based on hindered N-heterocyclic carbene ligands, including single-site versions of these catalysts, that catalyze the coupling of amines with aryl bromides and chlorides.36
A Fourth-Generation Catalyst for the Amination of Aryl Halides
Although the catalysts we developed containing P(tBu)3 and the Q-phos ligand, as well as the hindered alkyl monophosphine and carbene catalysts described in the previous section developed by other groups, are highly reactive toward the coupling of secondary amines with halooarenes, they are less reactive toward coupling of primary amines, often generate mixtures of monoarylation and diarylation products, and typically require high loadings of palladium for the coupling of heteroaromatic halides. Thus, our recent work on the coupling of amines with aryl halides has focused most intensively on developing a fourth generation system that would improve the activity and selectivity for couplings of primary amines and thiols, as well as the coupling of heteroaromatic halides.
To do so, we returned to studies on catalysts derived from the Josiphos class of ligands.22 Because these ligands were developed for enantioselective catalysis, principally asymmetric hydrogenation, they have typically been used in combination with iridium, rhodium, and ruthenium catalyst precursors.37,38 Moreover, the use of a ligand developed for enantioselective catalysis to prepare achiral products was unconventional. However, an exceptionally hindered and electron-donating version of the Josiphos ligands containing one dicyclohexylphosphino group and one di-tert-butylphosphino group on the ferrocenyl-1-ethyl backbone (CyPFtBu)23 became commercially available (Figure 1), and we envisioned that the steric and electronic properties of this ligand were particularly well suited to address some of the current limitations on catalyst scope and activity.
Figure 1.

Josiphos ligand CyPFtBu in the fourth-generation catalyst.
For example, we envisioned that the steric property of this ligand could improve the selectivity for monoarylation of primary amines.27, 29 We also considered that its chelation could create a catalyst that would be more stable toward displacement of the ancillary ligand by primary amines and improve turnover numbers. Finally, we considered that chelation would prevent displacement of the phosphine by basic heterocycles39, 40 and, thereby, improve the coupling of halopyridines. If the bidentate ligand possessed a backbone that pre-organized the two phosphines toward metal binding, then it might stay bound to palladium despite severe steric demands; the ferrocenyl-1-ethyl backbone of the Josiphos ligands creates this pre-organization.
With these ligands, we have sought to improve four aspects of the scope of the aminations of aryl halides. First, we sought to conduct the coupling of aryl chlorides with high turnover numbers. By doing so, we would take advantage of the lower cost of these haloarenes without offsetting their cost advantage by using large amounts of catalyst. Second, we sought to couple primary amines with high turnover numbers and high selectivity for monoarylation. Third, we sought to conduct the coupling of heteroaromatic halides with high turnover numbers. Finally, we sought to exploit the chelation and steric hindrance of the ligand for the coupling aryl halides with ammonia to form monoarylamines. Each of these goals was met by using the combination of a palladium precursor and the Josiphos ligand CyPFtBu. As a side note, this Josiphos ligand is air stable in solution and as a solid.
Our first studies confronted three of these challenges simultaneously. We investigated the coupling of primary amines with heteroaromatic chlorides containing a basic nitrogen atom.41, 42 As shown in Chart 1, the reaction of 2-chloropyridine with octylamine occurred to completion and in high yield using only 10 ppm of palladium. Although this reaction can be conducted at higher temperatures in more polar solvents without a catalyst, only a few percent of the heteroarylamine product formed under the mild conditions of the catalytic reactions. Perhaps more striking, the reaction of the unactivated 3-chloropyridine occurred in high yield with only 50 ppm of the catalyst. The analogous reaction of 4-chloropyridine occurred to completion with only 0.01 mol % of the catalyst. Other examples of the coupling of primary amines with heteroaryl halides in Chart 1 show that the reaction occurs with halopyridines containing a substituent ortho to the halogen despite the steric hindrance of the ligand. Moreover, reactions of primary amines that are branched α to nitrogen, as well as benzyl amines, occurred in high yield with low loadings of catalyst. Because the cost of the ligands used for most palladium-catalyzed aminations of aryl halides is similar or greater to the cost of palladium, the 1:1 ratio of metal to ligand in this catalyst system is noteworthy.
Chart 1.

If the poisoning of the catalyst by pyridine is avoided by using a chelating ligand, then the coupling of halopyridines is most similar to the coupling of electron-poor chloroarenes, which are typically more reactive than electron-rich chloroarenes. Therefore, we were particularly pleased that the same conditions that we used for the coupling of chloropyridines led to the coupling of electron-rich or electron-poor chloroarenes using quantities of palladium that were orders of magnitude lower than required previously for the coupling of these substrates with primary amines.29, 43 As shown by the selected examples in Chart 2, several of these reactions occurred with 50–100 ppm of palladium, and the particularly challenging reaction of an α-branched, acyclic primary amine21 occurred in quantitative yields.
Chart 2.

The tolerance of coupling chemistry for ancillary functionality is a hallmark of palladium-catalyzed coupling. We showed that the coupling of aryl and heteroaryl chlorides possessing protic functionality occurs when the reactions are conducted with two equivalents of lithium hexamethyldisilazide as base,42,44 the first to deprotonate the protic functionality and the second to be used during the actual coupling process. By this method, reactions of primary amines with aryl halides possessing hydroxyalkyl groups, phenolic hydroxyl groups, enolizable keto groups, primary and secondary amides, and even a carboxylate occurred in high yield (Chart 3). Examples of couplings of these types of aryl halides with primary amines had not been demonstrated previously.
Chart 3.

The combination of palladium and the hindered Josiphos ligand also efficiently catalyzed couplings of other primary nitrogen nucleophiles (Chart 4). For example, reactions of chloropyridines and chloroarenes with benzophenone imine occurred with loadings as low as 0.005 mol % of the combination of palladium and the Josiphos ligand,41, 42 and reactions of several aryl halides occurred with 50 ppm to 0.1 mol %. Reactions of benzophenone hydrazone also occurred with loadings of this catalyst ranging from 0.1 % to 1.0 %.
Chart 4.

A comparison of several catalysts for two prototypical couplings of primary amines revealed the unusual reactivity of the combination of palladium and the Josiphos ligand. Table 1 summarizes reactions of 3-chloropyridine with octylamine catalyzed by complexes of CyPFtBu, the biaryl dialkylphosphine X-phos,45 and the hindered N-heterocyclic carbene SIPr.46 Reactions conducted with low loadings of the monophosphine and the carbene occurred to low conversion. Even reactions with 100 times more catalyst than is needed with CyPFtBu did not occur to completion.42 Moreover, when run with more catalyst, these reactions generated mixtures of mono- and diarylation products. Likewise, the reaction of 4-chlorotoluene with octylamine occurred to low conversion when using low loadings of the catalysts derived from the monodentate ligands, and reactions conducted with more catalyst formed mixtures of mono- and di-arylation products.42
Table 1.
Comparison of the Activity of CyPFtiBu, XPhos, and SIPr for the Reactions of a Heteroaryl Chloride with a Primary Alkylamine.
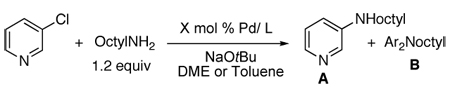 | ||||||
|---|---|---|---|---|---|---|
| Entry | Pd/L | X mol % Pd | T [°C] | t [h] | Conversion (yield, %) |
A/B |
| 1 | Pd(OAc)2/1 | 0.005 | 90 | 24 | 100(92) | 100:0 |
| 2 | Pd(OAc)2/2 | 0.05 | 90 | 24 | 40 | 5.4:1 |
| 3 | Pd(OAc)2/2 | 0.1 | 90 | 24 | 85(80) | 2.4:1 |
| 4 | Pd(OAc)2/2 | 0.5 | 90 | 12 | 88(81) | 2.6:1 |
| 5 | 3 | 0.005 | 110 | 24 | <10 | - |
| 6 | 3 | 0.05 | 110 | 48 | 58 | >25:1 |
| 7 | 3 | 0.5 | 110 | 12 | 100(71) | 7.6:1 |
A similar comparison of catalysts for the coupling of the particularly electron-rich, deprotonated 4-chlorophenol was published by Buchwald and coworkers.47 In this case, the reactions with the X-phos ligand occurred under milder conditions (40 °C) than with CyPFtBu,47 although a closely related reaction at 100 °C occurred in 72% isolated yield.34 These comparisons illustrate that some classes of coupling reactions benefit from the properties of the Josiphos ligands, while others benefit from the properties of the biaryl dialkylphosphines.
Couplings of Aryl Bromides and Iodides Catalyzed by the Pd-Josiphos System
Aryl bromides tend to react faster than aryl chlorides in palladium-catalyzed amination processes. Consistent with this trend, aryl bromides reacted with primary amines when using extremely low quantities of the palladium-Josiphos catalyst (Chart 5).42 Reactions of electron-poor bromoarenes occurred with 5–50 ppm of palladium, and the reactions of electron-neutral to electron-rich bromoarenes occurred with 0.005–0.1 mol % of palladium. These reactions occurred with linear and α-branched primary amines, as well as benzylic amines. Reactions of ortho-substituted bromoarenes also occurred using relatively low loadings of palladium. These bromoarenes included those containing electron-donating substituents in the ortho position, those containing a large ortho-isopropyl group, and those containing 2,6-disubstitution.
Chart 5.

Although iodoarenes are more reactive than bromoarenes for many cross-coupling processes, the coupling of amines with iodoarenes have occurred more slowly and in lower yields than coupling of bromoarenes.48, 49 Thus, we conducted studies to determine whether the Josiphos ligand would improve the scope and efficiency of the coupling of iodoarenes.42 These data are summarized in Chart 6. The reaction of the unactivated 3-iodopyridine occurred in good yield with a primary amine using just 0.05 mol % of palladium. Likewise the reaction of 4-iodotoluene with an α-branched primary amine occurred in good yield with the same loading of palladium. Although these reactions require a larger amount of palladium to run to completion than do the reactions of chloro- and bromoarenes, the amount of catalyst required is orders of magnitude lower than was needed for the coupling of iodoarenes using other catalysts, particularly for the coupling of primary amines.48, 49 Studies on reactions of bromoarenes and iodoarenes together have shown that the presence of the iodoarene retards the rate of the reaction. Although speculative at this point, we have proposed that the alkali metal iodide byproduct deactivates the catalyst.
Chart 6.

The First Palladium-Catalyzed Couplings of Ammonia with Aryl Halides to form Primary Arylamines
The combination of palladium and the hindered Josiphos ligand also led to the first coupling of ammonia with aryl halides to form primary aromatic amines (Chart 7).50 The reaction of the sterically unbiased bromoarene with an excess of ammonia in DME formed the primary aromatic amine in 86% yield with a 17:1 selectivity for the primary amine over the secondary amine. For reasons we do not yet fully understand, the selectivity of reactions with unhindered haloarenes was higher at lower concentrations (0.05 M) than at higher concentrations. However reactions of ortho-substituted bromoarenes occurred selectively at higher concentrations. For example, the reaction of 2-bromo-1-isopropylbenzene and 1-bromonaphthalene occurred at 0.25 M in high yield with greater than 50:1 selectivity for the primary amine. Heteroaryl bromides, such as 4-bromo and 5-bromoisoquinoline also reacted with ammonia to form the aminoisoquinolines in high yield, and reactions of chloro and iodoarenes occurred in good yield.
Chart 7.

In parallel, we reported similar reactions of haloarenes with lithium amide to form primary aromatic and heteroaromatic amines. The selectivity of these reactions with unhindered aryl halides was not as high as for reactions of ammonia, but was still acceptable. For example, the reaction of the sterically unbiased 1-bromo-4-tert-butylbenzene occurred in 72% isolated yield with 9.5:1 selectivity for formation of the primary amine. Like reactions of ammonia, the reactions of lithium amide with ortho-substituted haloarenes, bromonaphthalenes, and isoquinolines also occurred with essentially complete selectivity for the primary amine. Reactions of ammonia with haloarenes using catalysts derived from biaryl dialkylphosphines were recently reported by Buchwald.51
Coupling of Aryl Halides with Thiols Catalyzed by the Fourth-Generation Catalyst
Many catalysts are poisoned by thiols. Although the formation of palladium thiolates and the reductive elimination from arylpalladium thiolate complexes is fast,52, 53 the scope and efficiency of catalytic coupling of aryl halides with thiols has been limited.11, 54 Until recently, no catalyst had been developed that couples aryl halides with thiols possessing a significant range of ancillary functional groups, that couples arene thiols with high selectivity for the mixed diaryl sulfide, that couples aryl chlorides with thiols, or that couples any aryl halide with a thiol with high turnover numbers. We considered that the chelating property of the Josiphos ligand would minimize catalyst deactivation by thiols. Indeed, the palladium-Josiphos catalyst coupled a wide range of aryl chlorides and thiols with turnover numbers far exceeding those of previous systems.55
Chart 8 shows selected examples of the coupling of chloroarenes with alkane thiols catalyzed by palladium and CyPFtBu.56, 57 The coupling of chlorobenzene with 1-octanethiol using just 0.01 mol % of this catalyst formed the thioether in high yield. A range of more substituted chloroarenes reacted with similar efficiencies. Even the electron-rich 4-chloroanisole coupled when using just 0.1 mol % of palladium. Aliphatic thiols with branching α or β to sulfur also coupled efficiently. Some of these reactions were conducted with Pd(OAc)2 as precursor, and others with Pd2(dba)3. The effect of the pre-catalyst on the rates of these reactions is currently under investigation.
Chart 8.

The reactions of chloroarenes with arenethiols can be more challenging because of competing formation of symmetrical thioethers58 from reversible C-S bond formation. However, with the palladium-Josphos system, high yields of the unsymmetrical diarylsulfides were obtained without recourse to special solvents.59,55 These reactions were somewhat slower than those of aliphatic thiols, but the product was formed in high yield with 0.05–1 mol % of palladium and CyPFtBu. Like the C-N coupling, the C-S coupling of chloroarenes occurred with high functional group tolerance. For the C-S coupling, it was unnecessary to conduct reactions with silylamide base. Reactions conducted with NaOtBu or KOtBu as base occurred in high yield for haloarenes containing a variety of protic and electrophilic functional groups (Chart 9).
Chart 9.

As part of this work, we demonstrated a method to form diaryl sulfides without the need to use aromatic thiols as reagents or to isolate aromatic thiol intermediates.56 The reaction of triisopropylsilane thiol60 with a haloarene generated the TIPS-protected arenethiol. Reaction of this intermediate with a second haloarene in the presence of CsF and the palladium-CyPFtBu catalyst formed the unsymmetrical biaryl thioether in high yield.56
Ligand Structure-Reactivity Relationships
Having shown that the Josiphos ligand CyPFtBu generates a palladium catalyst that is highly active, but long lived, for a wide range of C-N and C-S couplings, we sought to determine which features of this ligand led to this favorable reactivity. To do so, we compared the reactivity (Table 3) of the catalyst generated from CyPFtBu to catalysts containing the ligand lacking the methyl group on the benzylic position of the backbone (A), ligands with propyl groups on the backbone (B, C), a ligand containing a phosphinoethylbenzene backbone (D), a ligand possessing an arene-chromium carbonyl unit in place of the ferrocene unit (E), a ligand based on a Xanthene backbone (F), a ligand (PPF-t-Bu) possessing a diphenylphosphino group in place of the dicyclohexylphosphino group, and ligands containing less sterically demanding groups on phosphorus.42
Table 3.
Effect of bidentate ligand structural properties of on C-N coupling with a heteroaryl chloride.
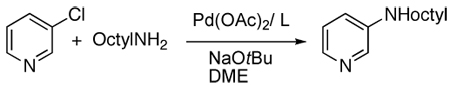 | ||||||
|---|---|---|---|---|---|---|
| Entry | Ligand | Loading | Temp | Time (h) | Yield | |
| 1 | CyPF-t-Bu | 0.005 | 90 °C | 24 | 93 | |
| 2 | PPF-t-Bu | 1.0 | 90 °C | 24 | 67 | |
| 3 | MePF-t-Bu | 0.005 | 90 °C | 24 | < 5% | |
| 4 | EtPF-t-Bu | 0.005 | 90 °C | 24 | < 5% | |
| 5 | CyPF-Cy | 1.0 | 90 °C | 24 | 46 | |
| 6 | CyPF-Ph | 1.0 | 90 °C | 24 | 48 | |
| 7 | tBuPF-Cy | 0.005 | 90 °C | 24 | 62 | |
| 8 | A | 0.005 | 90 °C | 24 | 50 | |
| 9 | B | 0.001 | 100 °C | 48 | 16 | |
| 10 | B | 0.005 | 90 °C | 24 | 93 | |
| 11 | C | 0.005 | 90 °C | 24 | < 5% | |
| 12 | D | 0.005 | 90 °C | 24 | < 5% | |
| 13 | E | 0.005 | 90 °C | 24 | < 5% | |
| 14 | F | 0.005 | 90 °C | 24 | < 5% | |
The lifetime of the desmethyl catalyst is long, but not as long as that for the catalyst containing CyPFtBu. However, ligands based on the xanthene and arene-chromium tri-carbonyl backbones generated much less reactive catalysts. We assume that the metal-ligand bonds in the complex generated from the hindered Xantphos analog is unstable under the conditions of the catalytic process and that the ligand containing the chromium carbonyl fragment is deactivated by nucleophilic attack onto the carbonyl units. The catalysts generated from PPFtBu were also less reactive than that generated from CyPFtBu, and the less hindered analogs of CyPFtBu containing dimethyl and diethylphosphino groups in place of the dicyclohexylphosphino group of CyPFtBu were much less reactive. These data imply that the preorganization imparted by the relative stereochemistry of the methyl group and the ferrocenyl unit, the steric properties of CyPFtBu, the strong electron donation of the alkylphosphino groups, and the high stability of the ferrocenylalkyl backbone each contribute to creating a catalyst that reacts with such high turnover numbers.
Mechanistic Considerations
The basic steps of the palladium-catalyzed amination and thioetherification have been discussed in review articles previously.10, 61, 62 The reaction is initiated by oxidative addition of a haloarene to an unsaturated palladium(0) fragment. Early studies with Pd(0) complexes of CyPFtBu showed that {Pd(CyPFtBu)[P(o-tol)3]} undergoes remarkably fast oxidative addition of typically unreactive aryl electrophiles.63 The resulting aryl halide complex then reacts with amine or thiol and base to generate an aryl palladium amide or thiolate intermediate. This intermediate then undergoes reductive elimination to form the carbon-nitrogen or -sulfur bond in the final product and regenerates the palladium(0) species.15, 61
A series of studies from our laboratory and others have shown that reductive elimination to form a carbon-heteroatom bond occurs more rapidly from complexes containing the more basic heteroatom.15 Reductive elimination from an alkylamido complex is faster than reductive elimination from an arylamido complex, which is faster than reductive elimination from a diarylamido complex.64 Recent studies on aryl palladium amidate complexes65 have shown that they undergo reductive elimination even more slowly than the diarylamido complexes. We have also shown recently that reductive elimination from an unsaturated, three-coordinate arylpalladium(II) diarylamido complex66 occurs more rapidly than reductive elimination from related arylpalladium alkoxo complex.67
One could attribute the faster rate of reductive elimination from the complex with the more basic heteroatom to more favorable attack of the electron pair on the ipso carbon of the aryl group in the transition state.68 Alternatively, one could attribute this faster rate to a decrease in the polarity, and thereby strength, of the metal-heteroatom bond. (Increasing polarity of a metal-heteroatom bond tends to lead to increasing its strength, in the absence of steric or mesomeric effects.) A comparison between the electronic effects on the rate of reductive elimination from arylpalladium amido and amidate complexes with those on the rate of reductive elimination from arylpalladium alkyl complexes69 helps dissect these potential contributions to the electronic effect.
Studies on the rates of reductive eliminations from arylpalladium alkyl complexes containing varying functional groups on the α carbon have shown that the C-C bond-forming reductive elimination is influenced by the electronic properties of the alkyl group in a fashion similar to the C-X bond-forming reductive elimination from amido and alkoxo complexes. Reductive elimination from the arylpalladium alkyl complexes occurs more slowly from the alkyl complexes containing an electron-withdrawing group on the α-carbon. Because the arylpalladium alkyl complexes lack an electron pair on the atom bound to the metal, we have concluded that the electronic effects are imparted more strongly through the metal-ligand sigma bond than through the lone electron pair on the anionic heteroatom ligand.
Summary
During the past several years, we have demonstrated that palladium catalysts containing sterically hindered alkylmonophosphines and catalysts containing sterically hindered alkylbisphosphines can significantly improve both the amination and thioetherification of aryl halides. In particular, the reactions of primary alkylamines, benzophenone imine, benzophenone hydrazone and even ammonia occur with high selectivities for monoarylation, and reactions of primary nitrogen nucleophiles occur with high turnover numbers using a very sterically hindered member of the Josiphos family of ligands. In parallel, we have shown that the reactions of thiols with haloarenes can now be conducted with high turnover numbers, including reactions to form unsymmetrical diaryl sulfides. Although no catalyst is optimal for all combinations of substrates, we anticipate that the high selectivity and efficiency of these catalysts for reactions of these classes of nucleophiles, in parallel with the high tolerance of the system for ancillary functional groups, will enable the efficient synthesis of alkyl arylamines, primary arylamines, N-aryl hydrazine derivatives, and aryl thioethers on both small and large scale.
Supplementary Material
Figure 2.

Electronic effects on C-X and C-C bond-forming reductive eliminations
Table 2.
Comparison of the Activity of CyPFtBu, XPhos, and SIPr for the Reactions of 4-Chlorotoluene with a Primary Alkylamine.
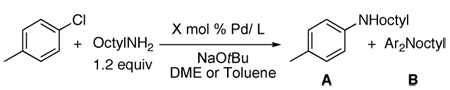 | ||||||
|---|---|---|---|---|---|---|
| Entry | Ligand | X mol % Pd/L | T [°C] | t [h] | Conversion (yield, %) | A:B |
| 1 | Pd(OAc)2/1 | 0.01 | 100 | 48 | 100(95) | 100:0 |
| 2 | Pd(OAc)2/2 | 0.05 | 100 | 48 | 13 | 18:1 |
| 3 | Pd(dba)2/2 | 0.1 | 100 | 48 | 25 | 10:1 |
| 4 | Pd(dba)2/2 | 0.5 | 100 | 12 | 100(93) | 2.3:1 |
| 5 | 3 | 0.005 | 110 | 48 | 40 | >50:1 |
| 6 | 3 | 0.05 | 110 | 48 | 76 | 15.3:1 |
| 7 | 3 | 0.5 | 110 | 12 | 100 | 4.3:1 |
Acknowledgements
We thank the NIH (GM-55382) for support of this work, Johnson-Matthey for palladium complexes, and Solvias for Josiphos ligands. I thank the many coworkers who have provided the experimental work and many of the conceptual insights into this chemistry. I particularly thank Blake Hamann, Amy Roy, Qilong Shen, and Manuel Fernandez-Rodriguez for their studies with the palladium-Josiphos catalysts.
Biography
John Hartwig was raised in outside Schenectady New York, received his A.B. from Princeton University conducting research with Maitland Jones, obtained his Ph.D. from U.C. Berkeley with Bob Bergman and Richard Andersen, and conducted an American Cancer Society postdoctoral fellowship at MIT with Stephen Lippard. In 1992 he began his independent career at Yale University and became the Irenée P. DuPont Professor in 2004. In 2006, he moved to the University of Illinois and is the Kenneth L. Rinehart Jr. Professor of Chemistry. In addition to coupling chemistry, his group has contributed to the discovery and mechanistic analysis of C-H bond functionalization, olefin hydroamination, and asymmetric allylic substitution. His textbook on organometallic chemistry and catalysis based on the classic 1987 book by Collman, Hegedus, Norton and Finke titled “Principles and Applications of Organotransition Metal Chemistry,” is currently under production by University Science Press. Outside of the office and lab, he cooks and enjoys time with his young daughters Amelia and Pauline.
References
- 1.de Meijere A, Diederich F. Metal-catalyzed cross-coupling reactions. Vol. 1. Weinheim: Wiley-VCH; 2004. [Google Scholar]
- 2.Four our first paper on this topic, see the following reference.
- 3.Paul F, Patt J, Hartwig JF. Palladium-Catalyzed Formation of Carbon-Nitrogen Bonds. Reaction Intermediates and Catalyst Improvements in the Hetero Cross-Coupling of Arylhalides and Tin Amides. J. Am. Chem. Soc. 1994;116:5969–5970. [Google Scholar]
- 4.For contemporaneous work see the following reference.
- 5.Guram AS, Buchwald SL. Palladium-Catalyzed Aromatic Aminations with In Situ Generated Aminostannanes. J. Am. Chem. Soc. 1994;116:7901–7902. [Google Scholar]
- 6.Hamann BC, Hartwig JF. Palladium-Catalyzed Direct Arylation of Ketones. Sterically Hindered Phosphines Enhance Reaction Yields. J. Am. Chem. Soc. 1997;119:12382–12383. [Google Scholar]
- 7.For contemporaneous work on this reaction, see the following two references.
- 8.Satoh T, Kawamura Y, Miura M, Nomura M. Palladium-Catalyzed Regioselective Mono-and Diarylation Reactions of 2-Phenylphenols and Naphthols with Aryl Halides. Angew. Chem. Int. Ed. Engl. 1997;36:1740–1742. [Google Scholar]
- 9.Palucki M, Buchwald SL. Palladium-Catalyzed Alpha-Arylation of Ketones. J. Am. Chem. Soc. 1997;119:11108–11109. [Google Scholar]
- 10.Shekhar S, Shen Q, Barrios-Landeros F, Hartwig JF. Synthesis of Anilines. In: Rapoport Z, editor. The Chemistry of Anilines. New York: Wiley Interscience; 2007. pp. 455–536. [Google Scholar]
- 11.Hartwig JF. Palladium-Catalyzed Synthesis of Aryl Ethers and Related Compounds Containing S and Se. In: Negishi EI, editor. Handbook of Organopalladium Chemistry for Organic Synthesis. Vol. 1. New York: Wiley-Interscience; 2002. pp. 1097–1106. [Google Scholar]
- 12.Muci AR, Buchwald SL. Practical palladium catalysts for C-N and C-O bond formation. Top. Curr. Chem. 2002;219:131–209. [Google Scholar]
- 13.Culkin DA, Hartwig JF. Palladium-Catalyzed α-Arylation of Carbonyl Compounds and Nitriles. Acc. Chem. Res. 2003;36:234–245. doi: 10.1021/ar0201106. [DOI] [PubMed] [Google Scholar]
- 14.Villanueva LA, Abboud KA, Boncella JM. Synthesis, Characterization, and Crystal Structures of Monomeric and Dimeric Palladium(II) Amide Complexes. Organometallics. 1994;13:3921–3931. [Google Scholar]
- 15.Hartwig JF. Electronic effects on reductive elimination to form carbon-carbon and carbon-heteroatom bonds from palladium(II) complexes. Inorg. Chem. 2007;46:1936–1947. doi: 10.1021/ic061926w. [DOI] [PubMed] [Google Scholar]
- 16.Kosugi M, Kameyama M, Migita T. Palladium-catalyzed aromatic amination of aryl bromides with diethylamino-tributyltin. Chem. Lett. 1983:927–928. [Google Scholar]
- 17.Guram AS, Rennels RA, Buchwald SL. A Simple Catalytic Method for the Conversion of Aryl Bromides to Arylamines. Angew. Chem. Int. Ed. Engl. 1995;34:1348–1350. [Google Scholar]
- 18.Louie J, Hartwig JF. Palladium-Catalyzed Synthesis of Arylamines from Aryl Halides. Mechanistic Studies Lead to Coupling in the Absence of Tin Reagents. Tetrahedron Lett. 1995;36:3609–3612. [Google Scholar]
- 19.Wolfe JP, Wagaw S, Buchwald SL. An Improved Catalyst System for Aromatic Carbon-Nitrogen Bond Formation: The Possible Involvement of Bis (Phosphine) Palladium Complexes as Key Intermediates. J. Am. Chem. Soc. 1996;118:7215–7216. [Google Scholar]
- 20.Driver MS, Hartwig JF. A Second Generation Catalyst for Aryl Halide Amination: Mixed Secondary Amines from Aryl Halides and Primary Amines Catalyzed by (DPPF)PdCl2. J. Am. Chem. Soc. 1996;118:7217–7218. [Google Scholar]
- 21.Wolfe JP, Buchwald SL. Scope and Limitation of the Pd/BINAP-catalyzed amination of aryl bromides. J. Org. Chem. 2000;65:1144–1157. doi: 10.1021/jo9916986. [DOI] [PubMed] [Google Scholar]
- 22.Hamann BC, Hartwig JF. Sterically Hindered Alkyl Phosphine Ligands Greately Accelerate the Palladium-Catalyzed Amination of Aryl Iodides, Bromides, Chlorides, and Tosylates. J. Am. Chem. Soc. 1998;120:7369–7370. [Google Scholar]
- 23.Spindler F, Wirth-Tijani A, Landert H. Preparation of ferrocenyldiphosphines as ligands for homogeneous catalysis. 1994 EP 612758 A1 19940831. [Google Scholar]
- 24.Wolfe JP, Wagaw S, Marcoux J-F, Buchwald SL. Rational Development of Practical Catalysts for Aromatic Carbon-Nitrogen Bond Formation. Acc. Chem. Res. 1998;31:805–818. [Google Scholar]
- 25.Nishiyama M, Yamamoto T, Koie Y. Synthesis of N-Arylpiperazines from Aryl Halides and Piperazine under a Palladium Tri-tert-butylphosphine Catalyst. Tetrahedron Lett. 1998;39:617–620. [Google Scholar]
- 26.Hartwig JF, Kawatsura M, Hauck SI, Shaughnessy KH, Alcazar-Roman LM. Room-temperature palladium-catalyzed amination of aryl bromides and chlorides and extended scope of aromatic C-N bond formation with a commercial ligand. J. Org. Chem. 1999;64:5575–5580. doi: 10.1021/jo990408i. [DOI] [PubMed] [Google Scholar]
- 27.Stambuli JP, Kuwano R, Hartwig JF. Unparalleled Rates for the Activation of Aryl Chlorides. Coupling with Amines and Boronic Acids in Minutes at Room Temperature. Angew. Chem. Int. Ed. Engl. 2002;41:4746–4747. doi: 10.1002/anie.200290036. [DOI] [PubMed] [Google Scholar]
- 28.Shelby Q, Kataoka N, Mann G, Hartwig JF. Unusual in situ Ligand Modification to Generate a Catalyst for Room Temperature Aromatic C-O Bond Formation. J. Am. Chem. Soc. 2000;122:10718–10719. [Google Scholar]
- 29.Kataoka N, Shelby Q, Stambuli JP, Hartwig JF. Air Stable, Sterically Hindered Ferrocenyl Dialkylphosphines for Palladium-Catalyzed C-C, C-N and C-O Bond-Forming Cross-Couplings. J. Org. Chem. 2002;67:5553–5566. doi: 10.1021/jo025732j. [DOI] [PubMed] [Google Scholar]
- 30.Liu X, Hartwig JF. Palladium-Catalyzed Arylation of Trimethylsilyl Enolates of Esters and Imides. High Functional Group Tolerance and Stereoselective Synthesis of -Aryl Carboxylic Acid Derivatives. J. Am. Chem. Soc. 2004;126:5182. doi: 10.1021/ja031544e. [DOI] [PubMed] [Google Scholar]
- 31.Hama T, Culkin DA, Hartwig JF. Palladium-catalyzed intermolecular alpha-arylation of zinc amide enolates under mild conditions. J. Am. Chem. Soc. 2006;128:4976–4985. doi: 10.1021/ja056076i. [DOI] [PubMed] [Google Scholar]
- 32.Hama T, Hartwig JF. α-Arylation of Esters Catalyzed by the Pd(I) Dimer {[P(t-Bu)3]PdBr}2. Org. Lett. 2008;10 doi: 10.1021/ol8002578. ASAP. [DOI] [PubMed] [Google Scholar]
- 33.Hama T, Hartwig JF. Org. Lett. 2008;10 doi: 10.1021/ol8002578. ASAP. [DOI] [PubMed] [Google Scholar]
- 34.Su W, Raders S, Verkade JG, Liao X, Hartwig JF. Palladium-Catalyzed alpha-Arylation of Trimethylsilyl Enol Ethers with Aryl Bromides and Chlorides. A Synergistic Effect of Two Metal Fluorides as Additives. Angew. Chem. Int. Ed. 2006;45:5852–5855. doi: 10.1002/anie.200601887. [DOI] [PubMed] [Google Scholar]
- 35.Harkal S, Rataboul F, Zapf A, Fuhrmann C, Riermeier T, Monsees A, Beller M. Dialkylphosphinoimidazoles as new ligands for palladium-catalyzed coupling reactions of aryl chlorides. Adv. Synth. Catal. 2004;346:1742–1748. [Google Scholar]
- 36.Nolan SP. Acc. Chem. Res. 2008;(this issue) [Google Scholar]
- 37.Blaser HU, Schmidt E. Asymmetric catalysis on industrial scale: challenges, approaches and solutions. Weinheim: Wiley-VCH; 2004. [Google Scholar]
- 38.For an exception, see: Magerlein W, Indolese AF, Beller M. Development of new palladium catalysts for the alkoxycarbonylation of aryl chlorides. J. Organomet. Chem. 2002;641:30–40.
- 39.Paul F, Patt J, Hartwig JF. Structural Characterization and Simple Synthesis of bis-o-tolylphopshine palladium(0). Characterization of the Dimeric Palladium(II) Complexes Obtained by Oxidative Addition of Aryl Bromides and their Reactivity with Amines. Organometallics. 1995;14:3030–3039. [Google Scholar]
- 40.Wagaw S, Buchwald SL. The Synthesis of Aminopyridines: A Method Employing Palladium-Catalyzed Carbon-Nitrogen Bond Formation. J. Org. Chem. 1996;61:7240–7241. doi: 10.1021/jo9612739. [DOI] [PubMed] [Google Scholar]
- 41.Shen Q, Shekhar S, Stambuli JP, Hartwig JF. Highly Reactive, General, and Long-Lived Catalysts for Coupling Heteroaryl and Aryl Chlorides with Primary Nitrogen Nucleophiles. Angew. Chem. Int. Ed. 2004;44:1371–1375. doi: 10.1002/anie.200462629. [DOI] [PubMed] [Google Scholar]
- 42.Shen Q, Ogata T, Hartwig JF. Highly Reactive, General and Long-Lived Catalysts for Palladium-Catalyzed Amination of Heteroaryl and Aryl Chlorides, Bromides and Iodides: Scope and Structure-Activity Relationships. J. Am. Chem. Soc. 2008;130:6586–6596. doi: 10.1021/ja077074w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wolfe JP, Tomori H, Sadighi JP, Yin JJ, Buchwald SL. Simple, efficient catalyst system for the palladium-catalyzed amination of aryl chlorides, bromides, and triflates. J. Org. Chem. 2000;65:1158–1174. doi: 10.1021/jo991699y. [DOI] [PubMed] [Google Scholar]
- 44.Harris MC, Huang X, Buchwald SL. Improved functional group compatibility in the palladium-catalyzed synthesis of aryl amines. Org. Lett. 2002;4:2885–2888. doi: 10.1021/ol0262688. [DOI] [PubMed] [Google Scholar]
- 45.Huang XH, Anderson KW, Zim D, Jiang L, Klapars A, Buchwald SL. Expanding Pd-catalyzed C-N bond-forming processes: The first amidation of aryl sulfonates, aqueous amination, and complementarity with Cu-catalyzed reactions. J. Am. Chem. Soc. 2003;125:6653–6655. doi: 10.1021/ja035483w. [DOI] [PubMed] [Google Scholar]
- 46.Marion N, Navarro O, Mei J, Stevens ED, Scott NM, Nolan SP. Modified (NHC)Pd(allyl)Cl (NHC = N-Heterocyclic Carbene) Complexes for Room-Temperature Suzuki-Miyaura and Buchwald-Hartwig Reactions. J. Am. Chem. Soc. 2006;128:4101–4111. doi: 10.1021/ja057704z. [DOI] [PubMed] [Google Scholar]
- 47.Anderson KW, Tundel RE, Ikawa T, Altman RA, Buchwald SL. Monodentate phosphines provide highly active catalysts for Pd-catalyzed C-N bond-forming reactions of heteroaromatic halides/amines and (H)N-heterocycles. Angew. Chem. Int. Ed. 2006;45:6523–6527. doi: 10.1002/anie.200601612. [DOI] [PubMed] [Google Scholar]
- 48.Wolfe JP, Buchwald SL. Palladium-Catalyzed Amination of Aryl Iodides. J. Org. Chem. 1996;61:1133–1135. [Google Scholar]
- 49.Urgaonkar S, Nagarajan M, Verkade JG. P(i-BuNCH2CH2)3N: An effective ligand in the palladium-catalyzed amination of aryl bromides and iodides. J. Org. Chem. 2003;68:452–459. doi: 10.1021/jo0205309. [DOI] [PubMed] [Google Scholar]
- 50.Shen Q, Hartwig JF. Palladium-Catalyzed Coupling of Ammonia and Lithium Amide. J. Am. Chem. Soc. 2006;128:10028–10029. doi: 10.1021/ja064005t. [DOI] [PubMed] [Google Scholar]
- 51.Surry DS, Buchwald SL. Selective Palladium-Catalyzed Arylation of Ammonia: Synthesis of Anilines as Well as Symmetrical and Unsymmetrical Di- and Triarylamines. J. Am. Chem. Soc. 2007;129:10354–10355. doi: 10.1021/ja074681a. [DOI] [PubMed] [Google Scholar]
- 52.Barañano D, Hartwig JF. Carbon-Heteroatom Bond Forming Reductive Elimination. Importance of Trapping Reagents and Unusual Electronic Effects in the Formation of Arylsulfides. J. Am. Chem. Soc. 1995;117:2937–2938. [Google Scholar]
- 53.Mann G, Barañano D, Hartwig JF, Rheingold AL, Guzei IA. Carbon-Sulfur Bond-Forming Reductive Elimination Involving sp-, sp2, and sp3-Hybridized Carbon. Mechanism, Steric Effects, and Electronic Effects on Sulfide Formation. J. Am. Chem. Soc. 1998;120:9205–9219. [Google Scholar]
- 54.Kosugi M, Shimizu T, Migita T. Reactions of aryl halides with thiolate anions in the presence of catalytic amounts of tetrakis(triphenylphosphine) palladium. Chem. Lett. 1978:13–14. [Google Scholar]
- 55.Murata M, Buchwald SL. A general and efficient method for the palladium-catalyzed cross-coupling of thiols and secondary phosphines. Tetrahedron. 2004;60:7397–7403. [Google Scholar]
- 56.Fernandez-Rodriguez MA, Shen QL, Hartwig JF. A general and long-lived catalyst for the palladium-catalyzed coupling of aryl halides with thiols. J. Am. Chem. Soc. 2006;128:2180–2181. doi: 10.1021/ja0580340. [DOI] [PubMed] [Google Scholar]
- 57.Fernandez-Rodriguez MA, Shen QL, Hartwig JF. Highly efficient and functional-group-tolerant catalysts for the palladium-catalyzed coupling of aryl chlorides with thiols. Chem. Eur. J. 2006;12:7782–7796. doi: 10.1002/chem.200600949. [DOI] [PubMed] [Google Scholar]
- 58.Yamamoto T, Sekine Y. Preparation of Diarylthiolatonickel(II) Complexes, Ni(SAr)2L2, by Oxidative Addition of Diaryl Disulfides to Ni(0) Complexes and Ni-Catalyzed Coupling Reactions between Thiophenols and Aryl Halides. Inorg. Chim. Acta. 1984;83:47–53. [Google Scholar]
- 59.In the following reference, this scrambling was alleviated by conducting the coupling in NEt3 as solvent.
- 60.Kreis M, Bräse S. A General and Efficient Method for the Synthesis of Silyl-Protected Arenethiols from Aryl Halides or Triflates. Adv. Synth. Catal. 2004;347:313–319. [Google Scholar]
- 61.Hartwig JF. Carbon-Heteroatom Bond-Forming Reductive Elimination of Amines, Ethers, and Sulfides. Acc. Chem. Res. 1998;31:852–860. [Google Scholar]
- 62.Hartwig JF. Transition Metal-Catalyzed Synthesis of Arylamines and Arylethers from Aryl Halides or Triflates. Angew. Chem. Int. Ed. Engl. 1998;37:2046–2067. doi: 10.1002/(SICI)1521-3773(19980817)37:15<2046::AID-ANIE2046>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 63.Roy AH, Hartwig JF. Oxidative Addition of Aryl Tosylates to Palladium(0) and Coupling of Unactivated Aryl Tosylates at Room Temperature. J. Am. Chem. Soc. 2003;125:8704–8705. doi: 10.1021/ja035835z. [DOI] [PubMed] [Google Scholar]
- 64.Driver MS, Hartwig JF. Carbon-Nitrogen Bond-Forming Reductive Elimination of Arylamines from Pd(II) J. Am. Chem. Soc. 1997;119:8232–8245. [Google Scholar]
- 65.Fujita KI, Yamashita M, Puschmann F, Alvarez-Falcon MM, Incarvito CD, Hartwig JF. Organometallic Chemistry from Amidate Complexes. Reductive elimination of N-Aryl Amidates from Palladium(II) J. Am. Chem. Soc. 2006;128:9044–9045. doi: 10.1021/ja062333n. [DOI] [PubMed] [Google Scholar]
- 66.Yamashita M, Hartwig JF. Synthesis, structure, and reductive elimination chemistry of three-coordinate arylpalladium amido complexes. J. Am. Chem. Soc. 2004;126:5344–5345. doi: 10.1021/ja0315107. [DOI] [PubMed] [Google Scholar]
- 67.Stambuli JR, Weng ZQ, Incarvito CD, Hartwig JF. Reductive elimination of ether from T-shaped, monomeric arylpalladium alkoxides. Angew. Chem. Int. Ed. 2007;46:7674–7677. doi: 10.1002/anie.200702809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Widenhoefer RA, Buchwald SL. Electronic dependence of C-O reductive elimination from palladium (aryl)neopentoxide complexes. J. Am. Chem. Soc. 1998;120:6504–6511. [Google Scholar]
- 69.Culkin DA, Hartwig JF. Carbon-Carbon Bond-Forming Reductive Elimination from Arylpalladium Complexes Containing Functionalized Alkyl Groups. Influence of Ligand Steric and Electronic Properties on Structure, Stability, and Reactivity. Organometallics. 2004;23:3398–3416. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.