

Abstract
Despite the widespread use of cardiac troponins as biomarkers for the diagnosis and quantitation of cardiac injury, the effect of troponin release and a possible autoimmune response to the troponins is unknown. Other investigators reported that programmed cell death – 1 (PD-1) – receptor deficient mice developed severe cardiomyopathy with autoantibodies to troponin I. We found that immunization of genetically susceptible mice with troponin I but not troponin T induced a robust autoimmune response leading to marked inflammation and fibrosis in the myocardium. At later times, antibodies to cardiac myosin were detected in troponin – immunized mice. The severity of inflammation correlated with expression of chemokines RANTES, MIP-2, IP-10 and MCD-1 in the myocardium. Prior immunization with troponin I increased the severity of experimental infarctions, indicating that an autoimmune response to troponin I aggravates acute cardiac damage. Cardiac inflammation, fibrosis and functional impairment were transferred from immunized to naive recipients by CD4+ T cells, and the cytokine profile suggested both a Th2 and Th17 profile in A/J mice. Finally we identified an 18-mer of troponin I containing an immuno-dominant epitope.
Introduction
In the field of cardiovascular disease, troponins have emerged as the most reliable clinical measure of myocyte injury [1-11]. Despite the widespread use of cardiac troponins for diagnosis of myocyte injury and risk stratification in acute cardiac disorders, little is known about the precise role of an autoimmune response to the troponins on cardiac function. Recently, investigators made the surprising discovery that mice treated with monoclonal anti-cTnI antibodies developed myocardial dysfunction [12]. Shortly after, it has been reported that autoantibodies to cTnI are also present in patients with acute coronary syndrome [13,14]. These findings indicate that induction of an autoimmune response to cTnI is not a rare event in patients. This review will summarize our investigations on the role of cardiac troponins in the pathogenesis of autoimmune myocarditis and of heart failure.
Heart failure is an increasingly prevalent disorder with considerable morbidity and mortality. While many causal mechanisms such as inherited cardiomyopathies, ischemic cardiomyopathy or muscular overload are easily identified in clinical practice, the events that determine the progression of cardiac injury to heart failure or ventricular remodelling are still unclear. Yet, there is compelling evidence that inflammatory mechanisms contribute to progressive heart failure [15] and that autoimmune responses are involved in the pathogenesis of many cardiovascular diseases [16-18]. Thus, myocardial infiltration of lymphocytes and mononuclear cells, increased expression of pro-inflammatory chemokines and cytokines and circulating autoantibodies are frequently observed in myocarditis, and in dilated cardiomyopathy (DCM) and subsequent heart failure [19-26].
Myocarditis is a clinically heterogeneous myocardial inflammatory condition that is most definitively diagnosed by endomyocardial biopsy [27]. It may be genetic, infectious, or autoimmune in etiology and may lead to DCM [28,29]. The pathogenetic progression leading from an infection such as initial viral myocarditis to postinflammatory DCM represent different stages of an organ-specific autoimmune process occurring in genetically predisposed individuals. The first stage is dominated by the viral infection itself, the second stage by the onset of multiple autoimmune reactions, and the third stage by fibrosis, dilatation and cardiac dysfunction [17] (Fig. 1).
Fig. 1.

Autoimmune Myocarditis and Dilated Cardiomyopathy: a triphasic disease process.
In animal models, cell-mediated or antibody-mediated autoimmune myocarditis/dilated cardiomyopathy can be initiated by a viral infection or by immunization with heart-specific autoantigens. In patients with a diagnosis of autoimmune myocarditis a myocardial biopsy may reveal the established histological signs (Dallas criteria) [29], characteristic immunohistological changes and cardiac-reactive autoantibodies [30-34]. The antibodies are directed against different cardiac antigens and may predict DCM development among relatives of patients with DCM years before disease onset. Some antibodies (such as autoantibodies to β1-adrenergic and M2 muscarinic receptors, to cardiac troponin I and to cardiac myosin) may produce effects on myocytes in animal models and possibly in some patients with DCM who are responsive to extracorporal immunoadsorption [25,33-41,12,42-47].
Because of the overlap of pathophysiological stages in inflammatory cardiomyopathy, design of rational therapy is difficult. Immunosuppressive treatments may be effective only in the absence of persistant virus. Immunosuppressive treatments should not be applied to patients with evidence of persistent viral genome in the myocardium. Clinical trials with antiviral agents, such as interferons, are in progress. Kühl et al. [48] investigated in a phase II study of patients with myocardial virus persistence whether interferon-β therapy is safe and achieves virus clearance, preventing deterioration of left ventricular function. They showed clearance of viral genomes in all 22 patients receiving antiviral therapy. Clearance of virus was paralleled by a significant increase in left ventricular ejection fraction. Other studies showed beneficial effects of extracorporal immunoadsorption and use of hyperimmune globulins [35,36,42,43,49]. However, the appropriate treatment of inflammatory cardiomyopathy still remains imperfect. Consequently, experimental studies are needed to devise novel approaches to therapy.
1.1 Experimental Models of Myocarditis in Mice
There is compelling evidence that cardiac myosin is one of the dominant autoantigens in virus–induced myocarditis in mice [50]. The disease can be reproduced by immunization of susceptible strains of mice with cardiac myosin [51]. Myosin-induced myocarditis can be adoptively transferred by CD4+ T lymphocytes [52]. In addition to T cells, passive administration of antimyosin monoclonal antibodies was found to induce myocarditis in DBA/2 but not in BALB/c mice because of the presence of myosin or a myosin like protein in the extracellular matrix of DBA/2 mice [53]. Therefore, both antibodies and T cells may contribute to the pathogenesis of inflammatory myocardial lesions. Gauntt et al. [54] and Cunningham [55] investigated the relationship between coxsackievirus and myosin, and suggested that molecular mimicry between myosin and coxsackieviruses may play a role in myocarditis. Anti-coxsackievirus-neutralizing antibodies produced myocardial inflammation in mice [54]. On the other hand, Horwitz et al. [56] presented evidence that virus-mediated damage to the heart is necessary for the induction of the autoimmune response.
Studies to explore the inductive and the effector mechanisms involved in the development of experimental autoimmune myocarditis (EAM) implicate both innate and adaptive immune responses. Important roles have been shown for autoreactive T cells [52], cardiac-specific autoantibodies [53,57], various cytokines and chemokines [58-66], natural killer cells [67], and the complement system [68,69].
1.1.1 Coxsackievirus B3–Induced Autoimmune Myocarditis
The murine model of autoimmune myocarditis is based on genetic differences among inbred mouse strains in their initial immune response to Coxsackievirus B3 infection. Currently, there are two models of autoimmune myocarditis. The first model, based on infection with heart-passaged Coxsackievirus B3, resolves in certain mouse strains into an early phase characterized by myocyte damage due to viral cytotoxicity and a late phase that is associated with the production of heart-muscle–specific autoantibodies [70]. In the second model the later phase of Coxsackievirus B3-induced heart disease can be mimicked by immunization of mice with purified murine cardiac myosin in the absence of viral infection. This experimental cardiac myosin–induced myocarditis has immunologic and histopathologic features that resemble postviral heart disease in genetically susceptible mice [51]. Thus, experimental myocarditis in the mouse offers a unique opportunity to study the factors contributing to the transition from a viral infection to an autoimmune disease.
1.1.2 Cardiac Myosin–Induced Autoimmune Myocarditis
Immunization of susceptible mice with cardiac myosin or with a myocardiotogenic peptide derived from the α-cardiac heavy chain emulsified in complete Freund's adjuvant (CFA) induces myocarditis in mice with a peak of inflammation in the heart around day 21 [59]. This inflammation is similar to that seen in the Coxsackievirus B3-induced autoimmune myocarditis during the chronic phase. The immunization with cardiac myosin is linked with production of cardiac myosin-specific autoantibodies and cardiac myosin-specific T cells [71,72].
1.2 Cardiac Troponins
Troponin participates in the regulatory complex of the myofibrillar thin filament that plays a critical role in regulating excitation-contraction coupling in the heart [73]. It is composed of three distinct gene products: troponin C, the 18-kD Ca2+-binding subunit, cardiac troponin I (cTnI), the 23-kD inhibitory subunit that prevents contraction in the absence of Ca2+ binding to troponin C; and cardiac troponin T (cTnT), the 35kD subunit that attaches troponin to tropomyosin and to the myofibrillar thin filament. The functional unit of the cardiac myocyte is the sarcomere. Sarcomere thin filament proteins are composed of actin and troponins C, T, and I. Sarcomere thick filament proteins include myosin heavy chain, myosin essential and regulatory light chains, myosin-binding protein-C and titin. Cardiac troponin I and T isoforms from the heart are structurally different from the corresponding forms found in skeletal muscle. After myocardial injury the troponins enter the circulation where they can be used for diagnosis of acute coronary syndromes (ACS) and for prognosis [1-11,13,14]. Because of this distribution, the measurement of cTnI and cTnT isoforms is superior to other serum biomarkers of cardiac lesions such as creatine kinase. In addition, the fact that the cTnI and cTnT are normally not found in the circulation means that the cardiac troponins provide a high level of clinical sensitivity and specificity even when cardiac lesions are small. Therefore, troponins in blood are now the preferred markers of myocardial damage [1,2,5,11,13]. Cardiac injury often results in the exposure of intracellular cardiac-specific proteins and, if recognized by the immune system, production of autoantibodies directed against these antigens. Despite the widespread use of cardiac troponins as biomarkers for diagnosis and risk stratification, the long term effect of troponin release and the possible pathogenic role of an autoimmune response to the troponins are not known.
2. Cardiac Troponins and Autoimmune Cardiomyopathy
2.1 Animal studies
Recently, Nishimura et al. [74] reported that PD-1 receptor deficient mice developed DCM with production of high-titered autoantibodies against a heart-specific, 30-kDa protein. Okazaki, et al. [12] purified this 30-kDa protein from heart extract and identified it as cardiac troponin I. They demonstrated that administration of monoclonal antibodies to cTnI induced dilatation and dysfunction of hearts in wild-type mice. Furthermore, they showed that monoclonal antibodies to cTnI stained the surface of cardiomyocytes and augmented the voltage-dependent L-type Ca2+ current of normal cardiomyocytes. Based on these findings the authors suggested that antibodies to cTnI induce heart dysfunction and dilatation by chronic stimulation of Ca2+ influx in cardiomyocytes [12]. In our studies [75] we induced a robust autoimmune reaction encompassing both humoral and cellular responses against murine (m) cTnI leading to severe inflammation and fibrosis in the myocardium of the mice. Persistent, prominent inflammation and fibrosis produce enlarged hearts (Fig. 3), increased endsystolic and enddiastolic diameters, reduced fractional shortening and reduced survival over 270 days compared to mice immunized with mcTnT or with control buffer. The inflammation in mcTnI immunized A/J mice was more severe than the inflammation observed in comparable BALB/c mice and females had more severe disease than male mice indicating that there is a genetically and sex-based predisposition to mcTnI induced inflammation in the myocardium comparable to other models of autoimmune disease [51].
Interestingly, when mcTnT was used for immunization, there was very little or no inflammation in the myocardium of both mouse strains used in our study, even though the immunization induced both strong humoral (antigen-specific autoantibody titers) and cellular (antigen-specific cytokine production) autoimmune response against mcTnT [75]. These results support the previously reported finding that cTnI is not strictly localized in intracellular compartments but also expressed on the surface of ventricular cardiomyocytes on normal heart sections [12]. In contrast, antibodies against cTnT stain the cytoplasm but not the surface of ventricular cardiomyocytes, indicating that the distribution of the two troponins is different [12]. Even though we could induce comparable levels of humoral and cellular immune responses by immunization with mcTnT or mcTnI only mcTnI immunized mice developed severe inflammation. We suggest that one of the mechanisms is because the mcTnI-specific autoantibodies could bind the target protein (troponin I) expressed on the surface of cardiomyocytes [12], whereas mcTnT-specific autoantibodies and T cells could not. In support of this concept, we could demonstrate mcTnI-reactive immunoglobulins in the heart extracts of mcTnI immunized mice, but no significant mcTnT-reactive immunglobulins could be found in the heart extracts of mcTnT immunized mice.
Cardiac myosin is known to be one of the main cardiac proteins reported by us and others to induce experimental autoimmune myocarditis [51,63,69.76]. Therefore, we measured antibodies against cardiac myosin in troponin-immunized mice. The mcTnI immunized mice later developed high titers of antibodies against cardiac myosin (days 90, 270), indicating that immunization with mcTnI caused myocardial damage with release of cardiac myosin and induction of an immune response to another cardiomyocyte-specific protein. This broadened autoimmune response may of course aggravate the inflammation and cardiac damage, since immunization with cardiac myosin alone is known to induce myocarditis and impair myocardial function [51,63,69.76].
Recently it has been shown that an adhesive, fibrotic pericarditis is associated with increased DCM and reduced survival in IFN-γ deficient mice following Coxsackievirus B3 infection [62]. The development of fibrosis is an important feature in a number of many pathological conditions including myocarditis, and is a key determinant in the clinical outcome of chronic heart disease [62]. Fibrosis involves proliferation of fibroblasts and deposition of extracellular matrix proteins like collagen. Fibroblasts are a major component of cardiac tissue and so it is not surprising that fibrosis is an important contributor to the development of DCM and congestive heart failure [77]. We observed increased fibrotic myocarditis in mcTnI immunized mice, but not in mcTnT or control buffer immunized mice. Continued myocardial inflammation was associated with the myocardial fibrosis.
Chemokines, such as MCP-1, MIP1-α and their major receptors, CCR2 and CCR5, play an important roles in the pathogenesis of many inflammatory diseases [78-80]. In our experiments, only the mcTnI immunized mice showed increased expression of these chemokines, correlating with increased myocardial inflammation in these mice. In our previous work we studied the expression of different CCRs during inflammation in the myocardium of mice and questioned if there is upregulation of other chemokine receptors in CCR2 or CCR5 deficient mice. We found that mainly the CCRs CCR1, CCR2 and CCR5 are expressed in the myocardium, but mRNA for the receptors CCR3 and CCR4 is not expressed at all or at very low levels. The CCR2- and CCR5- deficient mice did not show marked upregulation of the other CCR's tested compared to the wild type mice. Furthermore, we studied the mRNA expression levels of other chemokines in the myocardium of these mice on day 21 and found especially that the cytokines RANTES, MIP-2, IP-10, and MCP-1 were expressed in the myocardium in all mice tested. The mRNA expression levels of these chemokines correlated with the severity of myocarditis and were independent of CCR2- or CCR5- expression. The expression of these chemokines was continuously lower in CCR2- and CCR5- deficient mice (with overall mild to no inflammation), but with differences shown in the wild type (WT) mice by means of higher mRNA expression levels in mice with a higher myocarditis score, indicating that these chemokines were mostly released by the inflammatory cells infiltrating into the myocardium.
We also studied the effect of prior immunization with mcTnI on the infarct size and post-infarct fibrosis and inflammation in an acute cardiac damage model. Compared to control mice, animals pre-immunized with mcTnI showed significantly larger infarct size, more fibrosis and significantly more inflammation not only in the infarcted area but also in areas distant from the infarcted area [75]. These results indicate that an autoimmune response against troponin I aggravates the outcome of acute cardiac damage and may have a significant influence on post-infarct remodelling. This view is supported by the findings that tolerance induction by nasal vaccination with troponin reduced ischemia-reperfusion injury [81].
To investigate the role of cTnI-specific T cells, T cells were isolated from spleens of mice immunized with mcTnI and transferred to WT mice. Transfer of mcTnI-specific T cells led to severe inflammation and fibrosis in healthy recipient WT mice resulting in enlarged hearts, increased end-systolic and end-diastolic diameters, reduced fractional shortening, inflammation, fibrosis and heart failure [82]. Furthermore, CD4+ T cells are necessary for successful transfer of disease whereas CD8+ T cells do not seem to play a significant role. We could neither transfer disease with CD8+ T cells alone nor heighten the severity of disease when both CD4+ and CD8+ were transferred compared to transfer of CD4+ T cells alone [82]. In addition, we found significantly elevated levels of mcTnI-specific TNF-α, IL-1, IL-4, IL13, IL-17 production. This cytokine profile suggests that mcTnI induced myocarditis in A/J mice exhibits a Th2 (IL-4, IL13) and Th17 (IL-17) like phenotype. In the past, we demonstrated that experimental autoimmune myocarditis (EAM) in A/J mice induced by cardiac myosin has a Th2 phenotype [59]. Others described an important role for Th1 subsets in EAM, so that both of the CD4+ Th1 and Th2 subsets can contribute to disease [58,59,83]. Recently, Rangachari et al. [66] demonstrated a significant role for another subset of CD4+ T cells characterized by IL-17 production in EAM induced in Balb/c mice cardiac myosin peptide. We believe that the strain of mice used in the experiments is a crucial factor in deciding which subsets of CD4+ cells are involved in disease induction and progression.
To identify the antigenic determinants of troponin I responsible for the observed inflammation, fibrosis and heart failure, 16 overlapping 16-18mer peptides covering the entire amino acid sequence of mcTnI (211 residues) were synthesized (Table 1).
Table 1.
Sixteen overlapping (brown, cursive) 16-18 mer peptides covering the entire amino acid sequence of murine troponin I (211 residues) were synthesized followed by HPLC-purification.
| Peptide-Nr.: | Residues: | Amino Acid Sequences: | Histoscore: | Incidence: |
|---|---|---|---|---|
| 1 | 1-18 | MADES SDAAGEPQ 
|
0 (0-0-0-0-0) | 0 |
| 2 | 14-31 |
VRRRSSAN 
|
0 (0-0-0-0-0) | 0 |
| 3 | 27-44 |
TEPHAKKK 
|
0 (0-0-0-0-0) | 0 |
| 4 | 40-57 |
SRKLQLKT 
|
0 (0-0-0-0-0) | 0 |
| 5 | 53-70 |
AKQEMERE 
|
0 (0-0-0-0-0) | 0 |
| 6 | 66-83 |
GEKGRVLR 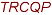
|
0 (0-0-0-0-0) | 0 |
| 7 | 79-96 |
LELDGLGF 
|
0 (0-0-0-0-0) | 0 |
| 8 | 92-109 |
LCRQLHAR 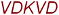
|
0 (0-0-0-0-0) | 0 |
| 9 | 105-122 |
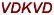 EERYDVEA EERYDVEA 
|
1,4 (3-1-0-2-1) | 80 % |
| 10 | 118-135 |
 ITEIADLT ITEIADLT 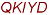
|
0 (0-0-0-0-0) | 0 |
| 11 | 131-148 |
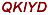 LRGKFKRP LRGKFKRP 
|
0,2 (1-0-0-0-0) | 20 % |
| 12 | 144-161 |
 RISADAMM RISADAMM 
|
0 (0-0-0-0-0) | 0 |
| 13 | 157-174 |
TRAKESLD 
|
0 (0-0-0-0-0) | 0 |
| 14 | 170-187 |
KQVKKEDI 
|
0 (0-0-0-0-0) | 0 |
| 15 | 183-200 |
EVGDWRKN 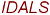
|
0 (0-0-0-0-0) | 0 |
| 16 | 196-211 |
GMEGRKKK FEG |
0 (0-0-0-0-0) | 0 |
| positive control (murine TnI) | 1,6 (3-2-0-1-2) | 80 | ||
| negative control (control buffer) | 0 (0-0-0-0-0) | 0 | ||
We identified two amino acid sequences of mcTnI that led to heart failure by inducing inflammation and fibrosis [82]. Peptide 9 (residues 105-122) of mcTnI was the strongest inducer of inflammation and fibrosis in the myocardium accompanied by chemokines RANTES, IP-10, MCP-1, MIP-1α, MIP-1β, MIP-2, TCA-3 and eotaxin, and of the chemokine receptors CCR1, CCR2, CCR5. McTnI residues 131-148 (peptide 11) contained a minor epitope inducing milder inflammation. The corresponding human cTnI (hcTnI) residues 104-121 differ in only one amino acid from the mcTnI residues 105-122; it induced milder inflammation in mice. In contrast none of the mice immunized with skeletal troponin I showed significant signs of inflammation. Furthermore, mice immunized with mcTnI also developed antibodies directed against the residues 105-122 of mcTnI (peptide 9). Conversely, immunization with the residues 105-122 of mcTnI (peptide 9) induced production of antibodies reacting with the complete protein mcTnI. Thus, the residues 105-122 of mcTnI represent an immunodominant epitope in the complex structure of the whole mcTnI protein. This epitope becomes more interesting when one realizes that it helps form a parallel helix, suggesting that a coiled coil in cTnI and cTnT has important physiological roles that are characteristic of troponin [84].
2.2 Clinical studies
Although the mechanisms of cardiomyocyte degeneration in patients with progressive heart failure are not fully understood, some studies provide evidence of myocardial damage in patients with dilated cardiomyopathy is accompanied by leakage of cTnT and cTnI into circulation. Eriksson et al. [85] reported the presence of autoantibodies against cTnI in patients with acute coronary syndrome (ACS) with the potential to interfere with diagnostic assays by impeding detection of cTnI. This points to an early induction of an autoimmune response to cTnI in these patients. Others [86,87] demonstrated the presence of circulating autoantibodies against cTnI in patients with heart failure. We screened patients with DCM or ischemic cardiomyopathy (ICM) for the presence of anti-cTnI and anti-cTnT antibodies [25]. Altogether, in 70% of patients with DCM and in 9.2% with ICM, an anti-cTnI IgG antibody titer ≥160 was measured. In contrast, only in 1.7% of patients with DCM and in 0.5% with ICM an anti-cTnT IgG antibody titer ≥160 was detected.
To elucidate the potential effect of these antibodies on cardiac function after acute myocardial infarction, we followed 108 patients with acute myocardial infarction, recording their heart function and the presence of anti-cardiac troponin antibody titers [25]. Evolution of cardiac remodeling was determined by magnetic resonance imaging that was performed after a median of 4 days after admission and after 6-9 months. Ten of 108 patients included in the follow-up study tested positive for cTnI-Ab with IgG Ab titers ≥160. Both groups had comparable demographic data and cardiovascular risk factor constellations. CTnI-Ab negative patients showed a significant increase in left ventricular ejection fraction (LVEF) and stroke volume 6-9 months after acute myocardial infarction. In contrast, there was no significant increase in LVEF and stroke volume in cTnI-Ab positive patients. Thus, antibodies in a titer ≥160 are associated with impaired recovery of the heart after myocardial injury. Patients with a lower titer or no detectable antibody showed amelioration in heart function after 6-9 months [25]. One might assume that smaller lesions in skeletal muscle can prompt immunization or cross-reactivity. We therefore analyzed blood samples from athletes running a 216 km ultra-endurance marathon. These well-trained athletes all had great experience and a history of marathon running before the time of the analysis. It is likely that their immune system had already been confronted with the release of cardiac proteins (and skeletal troponins). However, none of them showed elevated titers for antibodies against cardiac troponin I or T [25]. Also, the documented rhabdomyolysis in one of the runners did not induce cTnI- or cTnT antibodies.
These findings may suggest new opportunities for therapeutic approaches. Miettinen et al. [87] demonstrated the presence of autoantibodies against cTnI in patients with dilated cardiomyopathy. They reported that patients with high levels of cTnI autoantibodies remained strongly positive throughout the follow-up period, whereas patients with low levels showed cTnI autoantibody positivity usually only at 1 point during follow-up. Even though there were no significant differences between the groups, there was a trend for more dilated ventricle, more prominent cardiac sympathetic activation, and higher levels of circulating norepinephrine in patients with higher cTnI autoantibody levels. Although the patient population in both studies was well characterized, the number of patients and follow-up time remained rather limited. Thus, large-scale studies are necessary to confirm these preliminary observations.
3. Conclusion
Myocarditis is an inflammatory heart muscle disease, resulting from various etiologies, both noninfectious and infectious, which may be associated with cardiac dysfunction. Its course is unpredictable: it may resolve spontaneously or evolve into dilated cardiomyopathy and heart failure. A possible connection between myocarditis and dilated cardiomyopathy has long been postulated, but the underlying mechanisms linking these two conditions are still unknown. Understanding the mechanisms underlying the relationship between myocarditis and dilated cardiomyopathy will help in identifying effective strategies of treatment aimed to stop and prevent cardiac damage.
Despite the widespread use of cardiac troponin I for diagnosis of myocyte injury and risk stratification in acute cardiac disorders, little is known about the precise effect of an autoimmune response to the troponins on cardiac function. Recently, investigators made the surprising discovery that mice treated with monoclonal anti-cTnI antibodies developed myocardial dysfunction. Shortly after, it was reported that autoantibodies to cTnI are also present in patients with acute coronary syndrome (ACS). These findings indicate that induction of an autoimmune response to cTnI is not a rare event in patients. Lately, we demonstrated that the prevalence of cTnI antibodies in patients with ACS has an impact on the improvement of the LVEF. Furthermore, the role of autoantibodies in heart failure has been supported by clinical studies demonstrating that removing immunoglobulins by immunoadsorption can improve the ejection fraction in patients with dilated cardiomyopathy. Our findings suggest that the prevalence of cTnI antibodies in patients with ACS has an impact on the improvement of the left ventricular function.
To clarify the effect of inducing an autoimmune response to troponins we developed an animal model. We showed that induction of an immune response against cTnI but not cTnT induces severe inflammation and fibrosis in the myocardium of A/J mice leading to heart failure and reduced survival. Furthermore, we demonstrated that mice pre-immunized with cTnI prior to ligation of the left anterior descending artery showed greater infarct size, more fibrosis, higher inflammation score and reduced fractional shortening. Furthermore, we demonstrated the important role of troponin-specific T cells in inducing the autoimmune inflammation and identified one major and one minor epitope of troponin I that seem to be responsible for disease induction. Using these newly identified epitope sequences of troponin I instead of the whole troponin molecule, a more specific screening test of patients with high risk for progressive autoimmune inflammatory heart failure will be possible.
We believe that these findings affect our fundamental understanding of the etiology of inflammatory cardiovascular diseases and post-infarction remodelling. They may aid in developing new approaches to therapy, risk stratification and clinical studies initiated to investigate the role of autoimmunity against troponins in patients with acute coronary syndrome and dilated cardiomyopathy.
Fig. 2.
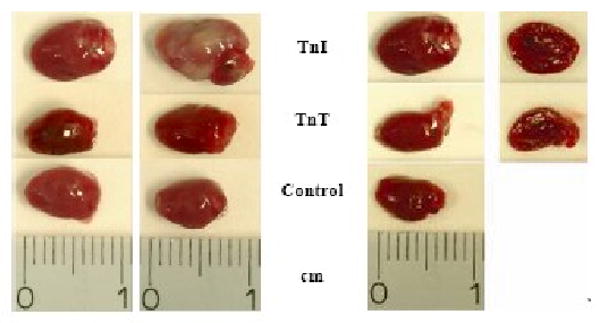
Representative on hearts removed from day 270 of mcTnl immunized mice with severe inflammation. Representative hearts of mcTnT or control buffer treated mice showed little or no inflammation.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft, research grants KA 1797/3-1 and KA 1974/4-1 (to Dr Kaya), by the Ernst und Berta Grimmke Stiftung (to Dr Kaya) and in part by National Institutes of Health research grants HL067290 and HL077611 (to Dr Rose).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Horwich TB, Patel J, MacLellan WR, Fonarow GC. Cardiac troponin I is associated with impaired hemodynamics, progressive left ventricular dysfunction, and increased mortality rates in advanced heart failure. Circulation. 2003;108:833–838. doi: 10.1161/01.CIR.0000084543.79097.34. [DOI] [PubMed] [Google Scholar]
- 2.La VL, Mezzena G, Zanolla L, Paccanaro M, Varotto L, Bonanno C, Ometto R. Cardiac troponin I as diagnostic and prognostic marker in severe heart failure. J Heart Lung Transplant. 2000;19:644–652. doi: 10.1016/s1053-2498(00)00120-0. [DOI] [PubMed] [Google Scholar]
- 3.Katus HA. Development of the cardiac troponin T immunoassay. Clin Chem. 2008;54:1576–1577. doi: 10.1373/clinchem.2008.104810. [DOI] [PubMed] [Google Scholar]
- 4.Ordonez-Llanos J, Santalo-Bel M, Merce-Muntanola J, Collinson PO, Gaze D, Haass M, Katus HA, Chwallek F, Hirschl MM, Derhaschnig U, Mueller-Bardorff M, Kellett J, Sylven C, Schulz I, Zerback R. Risk stratification of chest pain patients by point-of-care cardiac troponin T and myoglobin measured in the emergency department. Clin Chim Acta. 2006;365:93–97. doi: 10.1016/j.cca.2005.07.035. [DOI] [PubMed] [Google Scholar]
- 5.Hamm CW, Giannitsis E, Katus HA. Cardiac troponin elevations in patients without acute coronary syndrome. Circulation. 2002;106:2871–2872. doi: 10.1161/01.cir.0000044342.50593.63. [DOI] [PubMed] [Google Scholar]
- 6.Giannitsis E, Muller-Bardorff M, Lehrke S, Wiegand U, Tolg R, Weidtmann B, Hartmann F, Richardt G, Katus HA. Admission troponin T level predicts clinical outcomes, TIMI flow, and myocardial tissue perfusion after primary percutaneous intervention for acute ST-segment elevation myocardial infarction. Circulation. 2001;104:630–635. doi: 10.1161/hc3101.093863. [DOI] [PubMed] [Google Scholar]
- 7.Giannitsis E, Lehrke S, Wiegand UK, Kurowski V, Muller-Bardorff M, Weidtmann B, Richardt G, Katus HA. Risk stratification in patients with inferior acute myocardial infarction treated by percutaneous coronary interventions: the role of admission troponin T. Circulation. 2000;102:2038–2044. doi: 10.1161/01.cir.102.17.2038. [DOI] [PubMed] [Google Scholar]
- 8.Hartmann F, Giannitsis E, Kurowski V, Frey N, Kampmann M, Katus HA. Risk stratification and therapeutic decision making in patients with acute coronary syndrome--the role of cardiac troponin T. Clin Chem Lab Med. 1999;37:1107–1111. doi: 10.1515/CCLM.1999.161. [DOI] [PubMed] [Google Scholar]
- 9.Christenson RH, Duh SH, Newby LK, Ohman EM, Califf RM, Granger CB, Peck S, Pieper KS, Armstrong PW, Katus HA, Topol EJ. Cardiac troponin T and cardiac troponin I: relative values in short-term risk stratification of patients with acute coronary syndromes. GUSTO-IIa Investigators. Clin Chem. 1998;44:494–501. [PubMed] [Google Scholar]
- 10.Ohman EM, Armstrong PW, Christenson RH, Granger CB, Katus HA, Hamm CW, O'Hanesian MA, Wagner GS, Kleiman NS, Harrell FE, Jr, Califf RM, Topol EJ. Cardiac troponin T levels for risk stratification in acute myocardial ischemia. GUSTO IIA Investigators. N Engl J Med. 1996;335:1333–1341. doi: 10.1056/NEJM199610313351801. [DOI] [PubMed] [Google Scholar]
- 11.Katus HA, Remppis A, Neumann FJ, Scheffold T, Diederich KW, Vinar G, Noe A, Matern G, Kuebler W. Diagnostic efficiency of troponin T measurements in acute myocardial infarction. Circulation. 1991;83:902–912. doi: 10.1161/01.cir.83.3.902. [DOI] [PubMed] [Google Scholar]
- 12.Okazaki T, Tanaka Y, Nishio R, Mitsuiye T, Mizoguchi A, Wang J, Ishida M, Hiai H, Matsumori A, Minato N, Honjo T. Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD-1-deficient mice. Nat Med. 2003;9:1477–1483. doi: 10.1038/nm955. [DOI] [PubMed] [Google Scholar]
- 13.Katus HA, Remppis A, Looser S, Hallermeier K, Scheffold T, Kubler W. Enzyme linked immuno assay of cardiac troponin T for the detection of acute myocardial infarction in patients. J Mol Cell Cardiol. 1989;21:1349–1353. doi: 10.1016/0022-2828(89)90680-9. [DOI] [PubMed] [Google Scholar]
- 14.Hamm CW, Ravkilde J, Gerhardt W, Jorgensen P, Peheim E, Ljungdahl L, Goldmann B, Katus HA. The prognostic value of serum troponin T in unstable angina. N Engl J Med. 1992;327:146–150. doi: 10.1056/NEJM199207163270302. [DOI] [PubMed] [Google Scholar]
- 15.Kawai C. From myocarditis to cardiomyopathy: mechanisms of inflammation and cell death: learning from the past for the future. Circulation. 1999;99:1091–1100. doi: 10.1161/01.cir.99.8.1091. [DOI] [PubMed] [Google Scholar]
- 16.Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res. 2002;53:31–47. doi: 10.1016/s0008-6363(01)00434-5. [DOI] [PubMed] [Google Scholar]
- 17.Mason JW. Myocarditis and dilated cardiomyopathy: an inflammatory link. Cardiovasc Res. 2003;60:5–10. doi: 10.1016/s0008-6363(03)00437-1. [DOI] [PubMed] [Google Scholar]
- 18.Call JT, Deliargyris EN, Newby LK. Focusing on inflammation in the treatment of atherosclerosis. Cardiol Rev. 2004;12:194–200. doi: 10.1097/01.crd.0000111822.34362.71. [DOI] [PubMed] [Google Scholar]
- 19.Maisch B, Portig I, Ristic A, Hufnagel G, Pankuweit S. Definition of inflammatory cardiomyopathy (myocarditis): on the way to consensus. A status report. Herz. 2000;25:200–209. doi: 10.1007/s000590050007. [DOI] [PubMed] [Google Scholar]
- 20.Maisch B, Richter A, Sandmoller A, Portig I, Pankuweit S. Inflammatory dilated cardiomyopathy (DCMI) Herz. 2005;30:535–544. doi: 10.1007/s00059-005-2730-5. [DOI] [PubMed] [Google Scholar]
- 21.Landsberger M, Staudt A, Choudhury S, Trimpert C, Herda LR, Klingel K, Kandolf R, Schultheiss HP, Kroemer HK, Volker U, Felix SB. Potential role of antibodies against cardiac Kv channel-interacting protein 2 in dilated cardiomyopathy. Am Heart J. 2008;156:92–99. doi: 10.1016/j.ahj.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 22.Kallwellis-Opara A, Dorner A, Poller WC, Noutsias M, Kuhl U, Schultheiss HP, Pauschinger M. Autoimmunological features in inflammatory cardiomyopathy. Clin Res Cardiol. 2007;96:469–480. doi: 10.1007/s00392-007-0524-x. [DOI] [PubMed] [Google Scholar]
- 23.Schultheiss HP, Kuhl U. Overview on chronic viral cardiomyopathy/chronic myocarditis. Ernst Schering Res Found Workshop. 2006:3–18. doi: 10.1007/3-540-30822-9_1. [DOI] [PubMed] [Google Scholar]
- 24.Dorner A, Kallwellis-Opara A, Pauschinger M, Kuhl U, Schultheiss HP. Cardiac autoantibodies in viral myocarditis. Heart Fail Clin. 2005;1:333–343. doi: 10.1016/j.hfc.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 25.Leuschner F, Li J, Goser S, Reinhardt L, Ottl R, Bride P, Zehelein J, Pfitzer G, Remppis A, Giannitsis E, Katus HA, Kaya Z. Absence of auto-antibodies against cardiac troponin I predicts improvement of left ventricular function after acute myocardial infarction. Eur Heart J. 2008;29:1949–1955. doi: 10.1093/eurheartj/ehn268. [DOI] [PubMed] [Google Scholar]
- 26.Schulz-Menger J, Maisch B, bdel-Aty H, Pankuweit S. Integrated biomarkers in cardiomyopathies: cardiovascular magnetic resonance imaging combined with molecular and immunologic markers--a stepwise approach for diagnosis and treatment. Herz. 2007;32:458–472. doi: 10.1007/s00059-007-3046-4. [DOI] [PubMed] [Google Scholar]
- 27.Pauschinger M, Noutsias M, Lassner D, Schultheiss HP, Kuehl U. Inflammation, ECG changes and pericardial effusion: whom to biopsy in suspected myocarditis? Clin Res Cardiol. 2006;95:569–583. doi: 10.1007/s00392-006-0427-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, O'Connell J, Olsen E, Thiene G, Goodwin J, Gyarfas I, Martin I, Nordet P. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation. 1996;93:841–842. doi: 10.1161/01.cir.93.5.841. [DOI] [PubMed] [Google Scholar]
- 29.Aretz HT, Billingham ME, Edwards WD, Factor SM, Fallon JT, Fenoglio JJ, Jr, Olsen EG, Schoen FJ. Myocarditis. A histopathologic definition and classification. Am J Cardiovasc Pathol. 1987;1:3–14. [PubMed] [Google Scholar]
- 30.Herskowitz A, Ahmed-Ansari A, Neumann DA, Beschorner WE, Rose NR, Soule LM, Burek CL, Sell KW, Baughman KL. Induction of major histocompatibility complex antigens within the myocardium of patients with active myocarditis: a nonhistologic marker of myocarditis. J Am Coll Cardiol. 1990;15:624–632. doi: 10.1016/0735-1097(90)90637-5. [DOI] [PubMed] [Google Scholar]
- 31.Kuhl U, Seeberg B, Schultheiss HP, Strauer BE. Immunohistological characterization of infiltrating lymphocytes in biopsies of patients with clinically suspected dilated cardiomyopathy. Eur Heart J. 1994;15 C:62–67. doi: 10.1093/eurheartj/15.suppl_c.62. [DOI] [PubMed] [Google Scholar]
- 32.Kuhl U, Noutsias M, Schultheiss HP. Immunohistochemistry in dilated cardiomyopathy. Eur Heart J. 1995;16 O:100–106. doi: 10.1093/eurheartj/16.suppl_o.100. [DOI] [PubMed] [Google Scholar]
- 33.Wallukat G, Morwinski M, Kowal K, Forster A, Boewer V, Wollenberger A. Autoantibodies against the beta-adrenergic receptor in human myocarditis and dilated cardiomyopathy: beta-adrenergic agonism without desensitization. Eur Heart J. 1991;12 D:178–181. doi: 10.1093/eurheartj/12.suppl_d.178. [DOI] [PubMed] [Google Scholar]
- 34.Wallukat G, Wollenberger A, Morwinski R, Pitschner HF. Anti-beta 1-adrenoceptor autoantibodies with chronotropic activity from the serum of patients with dilated cardiomyopathy: mapping of epitopes in the first and second extracellular loops. J Mol Cell Cardiol. 1995;27:397–406. doi: 10.1016/s0022-2828(08)80036-3. [DOI] [PubMed] [Google Scholar]
- 35.Wallukat G, Reinke P, Dorffel WV, Luther HP, Bestvater K, Felix SB, Baumann G. Removal of autoantibodies in dilated cardiomyopathy by immunoadsorption. Int J Cardiol. 1996;54:191–195. doi: 10.1016/0167-5273(96)02598-3. [DOI] [PubMed] [Google Scholar]
- 36.Mobini R, Staudt A, Felix SB, Baumann G, Wallukat G, Deinum J, Svensson H, Hjalmarson A, Fu M. Hemodynamic improvement and removal of autoantibodies against beta1-adrenergic receptor by immunoadsorption therapy in dilated cardiomyopathy. J Autoimmun. 2003;20:345–350. doi: 10.1016/s0896-8411(03)00042-8. [DOI] [PubMed] [Google Scholar]
- 37.Staudt A, Mobini R, Fu M, Grosse Y, Stangl V, Stangl K, Thiele A, Baumann G, Felix SB. beta(1)-Adrenoceptor antibodies induce positive inotropic response in isolated cardiomyocytes. Eur J Pharmacol. 2001;423:115–119. doi: 10.1016/s0014-2999(01)01113-x. [DOI] [PubMed] [Google Scholar]
- 38.Limas CJ, Limas C. Beta-adrenoceptor antibodies and genetics in dilated cardiomyopathy--an overview and review. Eur Heart J. 1991;12 D:175–177. doi: 10.1093/eurheartj/12.suppl_d.175. [DOI] [PubMed] [Google Scholar]
- 39.Magnusson Y, Wallukat G, Waagstein F, Hjalmarson A, Hoebeke J. Autoimmunity in idiopathic dilated cardiomyopathy. Characterization of antibodies against the beta 1-adrenoceptor with positive chronotropic effect. Circulation. 1994;89:2760–2767. doi: 10.1161/01.cir.89.6.2760. [DOI] [PubMed] [Google Scholar]
- 40.Fu LX, Magnusson Y, Bergh CH, Liljeqvist JA, Waagstein F, Hjalmarson A, Hoebeke J. Localization of a functional autoimmune epitope on the muscarinic acetylcholine receptor-2 in patients with idiopathic dilated cardiomyopathy. J Clin Invest. 1993;91:1964–1968. doi: 10.1172/JCI116416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goldman JH, Keeling PJ, Warraich RS, Baig MK, Redwood SR, Dalla LL, Sanderson JE, Caforio AL, McKenna WJ. Autoimmunity to alpha myosin in a subset of patients with idiopathic dilated cardiomyopathy. Br Heart J. 1995;74:598–603. doi: 10.1136/hrt.74.6.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Felix SB, Staudt A, Landsberger M, Grosse Y, Stangl V, Spielhagen T, Wallukat G, Wernecke KD, Baumann G, Stangl K. Removal of cardiodepressant antibodies in dilated cardiomyopathy by immunoadsorption. J Am Coll Cardiol. 2002;39:646–652. doi: 10.1016/s0735-1097(01)01794-6. [DOI] [PubMed] [Google Scholar]
- 43.Felix SB, Staudt A, Dorffel WV, Stangl V, Merkel K, Pohl M, Docke WD, Morgera S, Neumayer HH, Wernecke KD, Wallukat G, Stangl K, Baumann G. Hemodynamic effects of immunoadsorption and subsequent immunoglobulin substitution in dilated cardiomyopathy: three-month results from a randomized study. J Am Coll Cardiol. 2000;35:1590–1598. doi: 10.1016/s0735-1097(00)00568-4. [DOI] [PubMed] [Google Scholar]
- 44.Jahns R, Boivin V, Schwarzbach V, Ertl G, Lohse MJ. Pathological autoantibodies in cardiomyopathy. Autoimmunity. 2008;41:454–461. doi: 10.1080/08916930802031603. [DOI] [PubMed] [Google Scholar]
- 45.Jahns R, Boivin V, Lohse MJ. Beta 1-adrenergic receptor-directed autoimmunity as a cause of dilated cardiomyopathy in rats. Int J Cardiol. 2006;112:7–14. doi: 10.1016/j.ijcard.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 46.Jahns R, Boivin V, Lohse MJ. beta(1)-Adrenergic receptor function, autoimmunity, and pathogenesis of dilated cardiomyopathy. Trends Cardiovasc Med. 2006;16:20–24. doi: 10.1016/j.tcm.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 47.Jahns R, Boivin V, Siegmund C, Inselmann G, Lohse MJ, Boege F. Autoantibodies activating human beta1-adrenergic receptors are associated with reduced cardiac function in chronic heart failure. Circulation. 1999;99:649–654. doi: 10.1161/01.cir.99.5.649. [DOI] [PubMed] [Google Scholar]
- 48.Kuhl U, Pauschinger M, Schwimmbeck PL, Seeberg B, Lober C, Noutsias M, Poller W, Schultheiss HP. Interferon-beta treatment eliminates cardiotropic viruses and improves left ventricular function in patients with myocardial persistence of viral genomes and left ventricular dysfunction. Circulation. 2003;107:2793–2798. doi: 10.1161/01.CIR.0000072766.67150.51. [DOI] [PubMed] [Google Scholar]
- 49.Dorffel WV, Wallukat G, Dorffel Y, Felix SB, Baumann G. Immunoadsorption in idiopathic dilated cardiomyopathy, a 3-year follow-up. Int J Cardiol. 2004;97:529–534. doi: 10.1016/j.ijcard.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 50.Neu N, Beisel KW, Traystman MD, Rose NR, Craig SW. Autoantibodies specific for the cardiac myosin isoform are found in mice susceptible to Coxsackievirus B3-induced myocarditis. J Immunol. 1987;138:2488–2492. [PubMed] [Google Scholar]
- 51.Neu N, Rose NR, Beisel KW, Herskowitz A, Gurri-Glass G, Craig SW. Cardiac myosin induces myocarditis in genetically predisposed mice. J Immunol. 1987;139:3630–3636. [PubMed] [Google Scholar]
- 52.Smith SC, Allen PM. Myosin-induced acute myocarditis is a T cell-mediated disease. J Immunol. 1991;147:2141–2147. [PubMed] [Google Scholar]
- 53.Liao L, Sindhwani R, Rojkind M, Factor S, Leinwand L, Diamond B. Antibody-mediated autoimmune myocarditis depends on genetically determined target organ sensitivity. J Exp Med. 1995;181:1123–1131. doi: 10.1084/jem.181.3.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gauntt CJ, Arizpe HM, Higdon AL, Wood HJ, Bowers DF, Rozek MM, Crawley R. Molecular mimicry, anti-coxsackievirus B3 neutralizing monoclonal antibodies, and myocarditis. J Immunol. 1995;154:2983–2995. [PubMed] [Google Scholar]
- 55.Cunningham MW. T cell mimicry in inflammatory heart disease. Mol Immunol. 2004;40:1121–1127. doi: 10.1016/j.molimm.2003.11.023. [DOI] [PubMed] [Google Scholar]
- 56.Horwitz MS, La Cava A, Fine C, Rodriguez E, Ilic A, Sarvetnick N. Pancreatic expression of interferon-gamma protects mice from lethal coxsackievirus B3 infection and subsequent myocarditis. Nat Med. 2000;6:693–697. doi: 10.1038/76277. [DOI] [PubMed] [Google Scholar]
- 57.Neumann DA, Lane JR, Lafond-Walker A, Allen GS, Wulff SM, Herskowitz A, Rose NR. Heart-specific autoantibodies can be eluted from the hearts of Coxsackievirus B3-infected mice. Clin Exp Immunol. 1991;86:405–412. doi: 10.1111/j.1365-2249.1991.tb02945.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Afanasyeva M, Wang Y, Kaya Z, Stafford EA, Dohmen KM, Sadighi Akha AA, Rose NR. Interleukin-12 receptor/STAT4 signaling is required for the development of autoimmune myocarditis in mice by an interferon-gamma-independent pathway. Circulation. 2001;104:3145–3151. doi: 10.1161/hc5001.100629. [DOI] [PubMed] [Google Scholar]
- 59.Afanasyeva M, Wang Y, Kaya Z, Park S, Zilliox MJ, Schofield BH, Hill SL, Rose NR. Experimental autoimmune myocarditis in A/J mice is an interleukin-4-dependent disease with a Th2 phenotype. Am J Pathol. 2001;159:193–203. doi: 10.1016/S0002-9440(10)61685-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cihakova D, Barin JG, Afanasyeva M, Kimura M, Fairweather D, Berg M, Talor MV, Baldeviano GC, Frisancho S, Gabrielson K, Bedja D, Rose NR. Interleukin-13 protects against experimental autoimmune myocarditis by regulating macrophage differentiation. Am J Pathol. 2008;172:1195–1208. doi: 10.2353/ajpath.2008.070207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fairweather D, Yusung S, Frisancho S, Barrett M, Gatewood S, Steele R, Rose NR. IL-12 receptor beta 1 and Toll-like receptor 4 increase IL-1 beta- and IL-18-associated myocarditis and coxsackievirus replication. J Immunol. 2003;170:4731–4737. doi: 10.4049/jimmunol.170.9.4731. [DOI] [PubMed] [Google Scholar]
- 62.Fairweather D, Frisancho-Kiss S, Yusung SA, Barrett MA, Davis SE, Steele RA, Gatewood SJ, Rose NR. IL-12 protects against coxsackievirus B3-induced myocarditis by increasing IFN-gamma and macrophage and neutrophil populations in the heart. J Immunol. 2005;174:261–269. doi: 10.4049/jimmunol.174.1.261. [DOI] [PubMed] [Google Scholar]
- 63.Kaya Z, Dohmen KM, Wang Y, Schlichting J, Afanasyeva M, Leuschner F, Rose NR. Cutting edge: a critical role for IL-10 in induction of nasal tolerance in experimental autoimmune myocarditis. J Immunol. 2002;168:1552–1556. doi: 10.4049/jimmunol.168.4.1552. [DOI] [PubMed] [Google Scholar]
- 64.Eriksson U, Kurrer MO, Sonderegger I, Iezzi G, Tafuri A, Hunziker L, Suzuki S, Bachmaier K, Bingisser RM, Penninger JM, Kopf M. Activation of dendritic cells through the interleukin 1 receptor 1 is critical for the induction of autoimmune myocarditis. J Exp Med. 2003;197:323–331. doi: 10.1084/jem.20021788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Eriksson U, Kurrer MO, Schmitz N, Marsch SC, Fontana A, Eugster HP, Kopf M. Interleukin-6-deficient mice resist development of autoimmune myocarditis associated with impaired upregulation of complement C3. Circulation. 2003;107:320–325. doi: 10.1161/01.cir.0000043802.38699.66. [DOI] [PubMed] [Google Scholar]
- 66.Rangachari M, Mauermann N, Marty RR, Dirnhofer S, Kurrer MO, Komnenovic V, Penninger JM, Eriksson U. T-bet negatively regulates autoimmune myocarditis by suppressing local production of interleukin 17. J Exp Med. 2006;203:2009–2019. doi: 10.1084/jem.20052222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fairweather D, Kaya Z, Shellam GR, Lawson CM, Rose NR. From infection to autoimmunity. J Autoimmun. 2001;16:175–186. doi: 10.1006/jaut.2000.0492. [DOI] [PubMed] [Google Scholar]
- 68.Fairweather D, Frisancho-Kiss S, Njoku DB, Nyland JF, Kaya Z, Yusung SA, Davis SE, Frisancho JA, Barrett MA, Rose NR. Complement receptor 1 and 2 deficiency increases coxsackievirus B3-induced myocarditis, dilated cardiomyopathy, and heart failure by increasing macrophages, IL-1beta, and immune complex deposition in the heart. J Immunol. 2006;176:3516–3524. doi: 10.4049/jimmunol.176.6.3516. [DOI] [PubMed] [Google Scholar]
- 69.Kaya Z, Afanasyeva M, Wang Y, Dohmen KM, Schlichting J, Tretter T, Fairweather D, Holers VM, Rose NR. Contribution of the innate immune system to autoimmune myocarditis: a role for complement. Nat Immunol. 2001;2:739–745. doi: 10.1038/90686. [DOI] [PubMed] [Google Scholar]
- 70.Rose NR, Herskowitz A, Neumann DA, Neu N. Autoimmune myocarditis: a paradigm of post-infection autoimmune disease. Immunol Today. 1988;9:117–120. doi: 10.1016/0167-5699(88)91282-0. [DOI] [PubMed] [Google Scholar]
- 71.Afanasyeva M, Georgakopoulos D, Belardi DF, Bedja D, Fairweather D, Wang Y, Kaya Z, Gabrielson KL, Rodriguez ER, Caturegli P, Kass DA, Rose NR. Impaired up-regulation of CD25 on CD4+ T cells in IFN-gamma knockout mice is associated with progression of myocarditis to heart failure. Proc Natl Acad Sci U S A. 2005;102:180–185. doi: 10.1073/pnas.0408241102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Afanasyeva M, Georgakopoulos D, Belardi DF, Ramsundar AC, Barin JG, Kass DA, Rose NR. Quantitative analysis of myocardial inflammation by flow cytometry in murine autoimmune myocarditis: correlation with cardiac function. Am J Pathol. 2004;164:807–815. doi: 10.1016/S0002-9440(10)63169-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Morita H, Seidman J, Seidman CE. Genetic causes of human heart failure. J Clin Invest. 2005;115:518–526. doi: 10.1172/JCI200524351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, Sasayama S, Mizoguchi A, Hiai H, Minato N, Honjo T. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001;291:319–322. doi: 10.1126/science.291.5502.319. [DOI] [PubMed] [Google Scholar]
- 75.Goser S, Andrassy M, Buss SJ, Leuschner F, Volz CH, Ottl R, Zittrich S, Blaudeck N, Hardt SE, Pfitzer G, Rose NR, Katus HA, Kaya Z. Cardiac troponin I but not cardiac troponin T induces severe autoimmune inflammation in the myocardium. Circulation. 2006;114:1693–1702. doi: 10.1161/CIRCULATIONAHA.106.635664. [DOI] [PubMed] [Google Scholar]
- 76.Goser S, Ottl R, Brodner A, Dengler TJ, Torzewski J, Egashira K, Rose NR, Katus HA, Kaya Z. Critical role for monocyte chemoattractant protein-1 and macrophage inflammatory protein-1alpha in induction of experimental autoimmune myocarditis and effective anti-monocyte chemoattractant protein-1 gene therapy. Circulation. 2005;112:3400–3407. doi: 10.1161/CIRCULATIONAHA.105.572396. [DOI] [PubMed] [Google Scholar]
- 77.Manabe I, Shindo T, Nagai R. Gene expression in fibroblasts and fibrosis: involvement in cardiac hypertrophy. Circ Res. 2002;91:1103–1113. doi: 10.1161/01.res.0000046452.67724.b8. [DOI] [PubMed] [Google Scholar]
- 78.Rao AR, Quinones MP, Garavito E, Kalkonde Y, Jimenez F, Gibbons C, Perez J, Melby P, Kuziel W, Reddick RL, Ahuja SK, Ahuja SS. CC chemokine receptor 2 expression in donor cells serves an essential role in graft-versus-host-disease. J Immunol. 2003;171:4875–4885. doi: 10.4049/jimmunol.171.9.4875. [DOI] [PubMed] [Google Scholar]
- 79.Ni W, Kitamoto S, Ishibashi M, Usui M, Inoue S, Hiasa K, Zhao Q, Nishida K, Takeshita A, Egashira K. Monocyte chemoattractant protein-1 is an essential inflammatory mediator in angiotensin II-induced progression of established atherosclerosis in hypercholesterolemic mice. Arterioscler Thromb Vasc Biol. 2004;24:534–539. doi: 10.1161/01.ATV.0000118275.60121.2b. [DOI] [PubMed] [Google Scholar]
- 80.Bruhl H, Cihak J, Schneider MA, Plachy J, Rupp T, Wenzel I, Shakarami M, Milz S, Ellwart JW, Stangassinger M, Schlondorff D, Mack M. Dual role of CCR2 during initiation and progression of collagen-induced arthritis: evidence for regulatory activity of CCR2+ T cells. J Immunol. 2004;172:890–898. doi: 10.4049/jimmunol.172.2.890. [DOI] [PubMed] [Google Scholar]
- 81.Frenkel D, Pachori A, Zhang L, Vaknin AD, Stoijkovic SP, Dzau V, Weiner H. Nasal Vaccination with Troponin Reduces Myocardial Ischemia-Reperfusion Injury By Inducing IL-10 Secreting CD4+ T-Cells. Clin Immunol. 2006;119(Supplement):OR.104. [Google Scholar]
- 82.Kaya Z, Goser S, Buss SJ, Leuschner F, Ottl R, Li J, Volkers M, Zittrich S, Pfitzer G, Rose NR, Katus HA. Identification of cardiac troponin I sequence motifs leading to heart failure by induction of myocardial inflammation and fibrosis. Circulation. 2008;118:2063–2072. doi: 10.1161/CIRCULATIONAHA.108.788711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cunningham MW. Cardiac myosin and the TH1/TH2 paradigm in autoimmune myocarditis. Am J Pathol. 2001;159:5–12. doi: 10.1016/S0002-9440(10)61665-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Takeda S, Yamashita A, Maeda K, Maeda Y. Structure of the core domain of human cardiac troponin in the Ca(2+)-saturated form. Nature. 2003;424:35–41. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
- 85.Eriksson S, Hellman J, Pettersson K. Autoantibodies against cardiac troponins. N Engl J Med. 2005;352:98–100. doi: 10.1056/NEJM200501063520123. [DOI] [PubMed] [Google Scholar]
- 86.Shmilovich H, Danon A, Binah O, Roth A, Chen G, Wexler D, Keren G, George J. Autoantibodies to cardiac troponin I in patients with idiopathic dilated and ischemic cardiomyopathy. Int J Cardiol. 2007;117:198–203. doi: 10.1016/j.ijcard.2006.04.077. [DOI] [PubMed] [Google Scholar]
- 87.Miettinen KH, Eriksson S, Magga J, Tuomainen P, Kuusisto J, Vanninen EJ, Turpeinen A, Punnonen KR, Pettersson K, Peuhkurinen KJ. Clinical significance of troponin I efflux and troponin autoantibodies in patients with dilated cardiomyopathy. J Card Fail. 2008;14:481–488. doi: 10.1016/j.cardfail.2008.02.009. [DOI] [PubMed] [Google Scholar]