

Abstract
A mechanistic study of the enantioselective asymmetric allylboration of ketones with allyldiisopropoxyborane catalyzed by chiral biphenols resulted in the development of improved reaction process. In a ligand exchange process involving the chiral biphenol and the boronate to liberate isopropanol as the key step, addition of isopropanol to the reaction was found to increase the overall rate and enantioselectivity. In the design of an improved reaction, a boronate possessing a tethered alcohol would more readily liberate catalyst at the end of a reaction. The use of allyldioxaborinane with 2 mol% (S)-3,3′-Br2-BINOL and 2 equivalents t-BuOH relative to ketone at room temperature results in high yields and enantioselectivities. Insight gathered from the mechanistic investigation resulted in the development of a reaction process that uses less catalyst (from 15 mol% to 2 mol%) at warmer temperatures (from -35 °C to room temperature).
The asymmetric allylboration reaction is an extensively studied synthetic method.1 The operational simplicity and chiral building blocks afforded readily account for the utility of the reaction.2 Studies aimed at the development of a catalytic enantioselective allylboration of ketones3 identified chiral biphenols as effective promoters of the reaction; however, the reaction required the use of relatively high catalyst loadings (0.15 equivalent, equation 1).4 Reducing catalyst loadings employed in organocatalytic reactions has proven challenging but recent advances in this area have been successful (2 mole % or lower),5 especially when coupled with experiments designed to provide insight into the reaction mechanism.6 We initiated a study of the reaction mechanism with the aim of indentifying a reaction that requires less catalyst and would also simultaneously address the scope and limitations of the method. Herein we report the results of our mechanistic experiments that led to the development of a reaction that uses 2 mole % of the catalyst under solvent free reaction conditions at room temperature to afford the products in ≥98:2 enantiomeric ratio (e.r.).
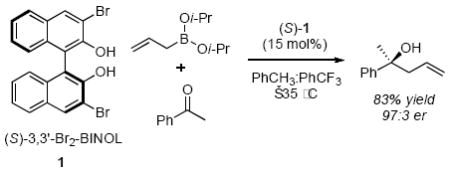 |
(1) |
Our study began by evaluating the role of the catalyst and isopropanol in the catalytic cycle. The key steps include a crucial ligand exchange processes at the beginning and end of the catalytic cycle; a process crucial for activation of the boronate (Scheme 1).4,7,8 Catalyst 1 reacts with allylboronate 2 to afford exchange product 3, a species that can be detected via direct ESI-MS analysis of the reaction mixture. We presumed this step is reversible. Spectroscopic analysis of the reaction mixture using 1H NMR indicates the formation of 3 along with free catalyst 1. Our previous studies determined the rate dependence on catalyst 1 to be first order.4 However, the order in isopropanol had not been established. If the overall rate of reaction was dependent on the initial exchange process, the addition of isopropanol to the reaction should inhibit the observed rate. This would indeed be the case for a single or double exchange process leading to 3 or a cyclic boronate.9 We performed experiments designed to ascertain the effect of isopropanol on rate and enantioselectivity in the allylboration of acetophenone (Figure 1).
Scheme 1. Proposed Catalytic Cycle for Asymmetric Allylboration Reaction Catalyzed by Chiral Biphenols.

Figure 1. Effect of isopropanol on rate and enantioselectivity[a].
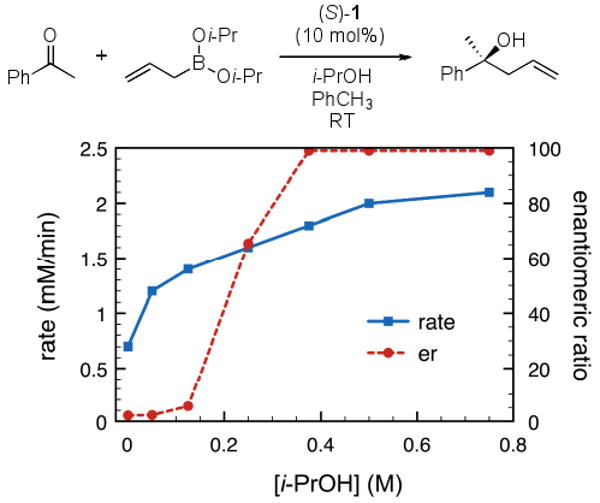
[a] Reactions were run with 0.075 mmol 1, 1.5 mmol boronate 2, and 0.75 mmol acetophenone in PhCH3 (0.25 M) for 15 h under Ar at RT, followed by flash chromatography on silica gel. Enantiomeric ratios were determined by chiral HPLC methods.
The formation of product was monitored using in situ ReactIR monitoring of the reaction as a function of isopropanol concentration ([i-PrOH]). For operational simplicity the reactions were performed at room temperature using 10 mol% 1. Under these conditions, the enantiomeric ratio (e.r.) of the product formed was 2.2:1 without i-PrOH. As a preliminary study, we chose to monitor the dependence of rate and e.r. on [i-PrOH] over a 2-fold concentration range relative to boronate. The observed rate increased almost 3-fold (Figure 1); coincident with the rate increase was a substantial increase in the observed enantioselectivity. With the addition of 1 equivalent i-PrOH relative to boronate the reaction rate doubled and the enantioselectivity increased to 65:1 e.r. At a concentration of i-PrOH 1.5 times [boronate], the reaction rate began to plateau at 3-fold greater than the parent rate and the enantioselectivity was determined to be 99:1 e.r. The inclusion of >1 equivalent i-PrOH resulted in a substantially improved reaction process exhibiting higher enantioselectivities and increased rates in comparison to the parent reaction at room temperature.
The improved reaction may be understood using the proposed catalytic cycle (Scheme 1). Our observations demonstrate that the rate determining exchange process is not the initial formation of active boronate species 3 but liberation of catalyst 1 from allylation product 6 (kex). Although the parent catalyzed reaction was nominally selective (e.r. = 2.2:1), there is a 4-fold increase over the reaction run in the absence of catalyst. The catalyst 1 serves to increase the overall rate of reaction; however, the effective catalyst concentration is reduced by the formation of 6 in addition to the other species in the reaction ([1] = [cattotal] − [3] − [5] − [6]). The inclusion of i-PrOH to the catalyzed reaction increases the overall catalyst concentration ([1]) thereby increasing the overall rate; consequently changing the rate-limiting step of the reaction process. The effective catalyst concentration approaches the actual catalyst concentration as the exchange rate increases by the addition of i-PrOH (kex[6][i-PrOH]). The apparent rate of reaction is the maximum rate possible for the allylboration bond formation process. Observations made from our investigations led us to consider further improvements of the reaction.
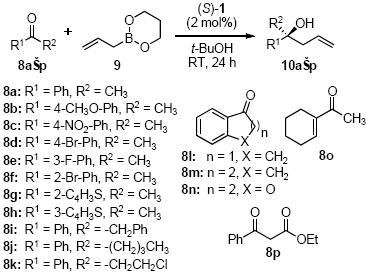
We focused our attention on the identity of the boronate utilized in the reaction and catalyst concentration as areas for improvement. The characteristic of allyldiisopropoxyboronate 2 that makes it ideal for use in the catalytic reaction is the same characteristic that makes it a difficult reagent to prepare and store; the lability of the isopropoxy groups result in a hydrolytically unstable boronate reagent. We sought to identify a boronate possessing a desirable balance of kinetic reactivity and thermodynamic stability. Cyclic boronates such as dioxaborolanes and dioxaborinanes are substantially more stable.10 They can be prepared and purified with greater ease and stored for longer times than acyclic boronates. In addition to the enhancement of stability, cyclic boronates would produce a tethered alcohol upon catalyst exchange that would more readily liberate the catalyst at the end of a reaction cycle (Scheme 1). Use of allyldioxaborinane 9 in the allylboration reaction with 15 mol% 1 with acetophenone 8a (0.1 M in PhCH3) at room temperature resulted in good yields of the product in 4:1 e.r. after 24 h. We postulated that the addition of another alcohol may facilitate the reaction. Isopropanol was added to the reaction of 8a and 9 to yield the product 10a with low yields (60% after 3 days) but high enantiopurity (99:1 e.r.). The addition of i-PrOH effectively reduced the background reaction while coincidentally inhibiting the catalytic reaction. This observation can be understood via a Lewis acid-base coordination of B-allyldioxaborinane and i-PrOH. We reasoned that a less coordinating alcohol such as t-BuOH would still accelerate the catalyzed reaction by facilitating ligand exchange while not inhibiting the overall rate of reaction. The addition of t-BuOH to the reaction in slight excess relative to boronate concentration ([9]) afforded the desired product in near quantitative yields with excellent enantioselectivity (e.r. > 99:1). While investigating the reaction of 8a and 9 we found that the reaction proceeded well in the absence of solvent11 with complete conversion and no loss in enantioselectivity. The optimized reaction conditions using allyldioxaborinane 9 required the use of 2 mol% 1 and 2 equivalents t-BuOH relative to ketone at room temperature. The catalyst concentration should be noted. The catalyst loading is calculated based on ketone concentration. Since the catalyst activates the boronate, relative to [boronate] the catalyst loading is 1.3 mol%. The use of lower catalyst loadings resulted in lower rates of reaction with no loss in enantioselectivity. Finally, the reaction of acetophenone could be scaled to 5 g achieving similar yields and enantioselectivities. The catalyst could be recovered in 90% yield from the reaction.
The reaction conditions proved general for a number of substrates (Table 1). Excellent yields and enantioselectivies were achieved for a broad range of ketones (>90% yield, >97:3 e.r.). In some examples, the reaction proved slow. For these substrates, additional catalyst was used to improve the rate (Table 1, Entries 6–8, 11, 13, 16). The reaction using 9 also exhibits a broader scope than the previous reaction. Phenyl acetophenone 8i was found to be a poor substrate under the previous reaction conditions (<15% yield, low enantioselectivities). However, the reaction of 8i with 9 afforded the allylboration product in 98% yield and 99:1 e.r. (Table 1, Entry 9). Boronate 9 was also reacted in high enantioselectivities with β-ketoester 8p (Entry 16), a particularly difficult substrate due to facile enolization. Crotylation reactions with 8a using E- and Z-crotyldioxaborinane, 11a and 11b respectively, provided products 12a and 12b in excellent yields with high enantio- and diastereoselectivities (Scheme 2).
Table 1.
Asymmetric allylboration of ketones[a]
 | ||||
|---|---|---|---|---|
| Entry | ketone | product | Yield [%][b] | e.r.[c] |
| 1 | 8a | 10a | 96 | 99:1 |
| 2 | 8b | 10b | 88 | 99:1 |
| 3 | 8c | 10c | 93 | 99:1 |
| 4 | 8d | 10d | 97 | 99:1 |
| 5 | 8e | 10e | 95 | 99:1 |
| 6[d] | 8f | 10f | 95 | 98:2 |
| 7[e] | 8g | 10g | 93 | >99:1 |
| 8[e] | 8h | 10h | 92 | 991 |
| 9 | 8i | 10i | 98 | >99:1 |
| 10 | 8j | 10j | 93 | 99:1 |
| 11[e] | 8k | 10k | 95 | >99:1 |
| 12 | 8l | 10l | 95 | 98:2 |
| 13[e] | 8m | 10m | 97 | 99:1 |
| 14 | 8n | 10n | 95 | 99:1 |
| 15 | 8o | 10o | 96 | 98:2 |
| 16[e] | 8p | 10p | 98 | 99:1 |
Reactions were run with 0.02 mmol (S)-1, 2.0 mmol t-BuOH, 1.0 mmol of ketone 8, and 1.5 mmol B-allyl-1,3,2-dioxaborinane 9 at RT for 24 h under Ar, followed by flash chromatography on silica gel.
Yield of isolated product.
Determined by chiral HPLC methods.
Reaction was run in PhCH3 (0.25 M) with 0.075 mmol (S)-1.
Reaction was run with 0.04 mmol (S)-1.
Scheme 2. Asymmetric crotylboration of acetophenone.

We performed mechanistic experiments to determine if the allylboration reaction of 9 follows the previously proposed reaction mechanism or via an alternative reaction process. The reaction exhibited a first-order rate dependence on [1], consistent with our previous findings. We also observed the formation of the corresponding exchange product complex of boronate 8 and 1 via ESI-MS and 1H NMR, similarly consistent with our proposed mechanism (Scheme 1).4,7c,8 The reaction rate and enantioselectivity was also monitored as a function of [t-BuOH] (Figure 2). Similar to the observations obtained using boronate 2 and i-PrOH, there was a coincident increase in rate and enantioselectivity with increasing concentrations of t-BuOH. However, at [t-BuOH]/[9] > 1 the rate became slower. Based on our previous observations using i-PrOH with 9, the rate may be inhibited by a weakly coordinating Lewis acid-base coordination of B-allyldioxaborinane.10 While cyclic boronate 9 appears to be more sensitive to Lewis base coordination, the mechanism by which 9 proceeds is consistent with our previous observations.
Figure 2. Effect of t-butanol on rate and enantioselectivity[a].
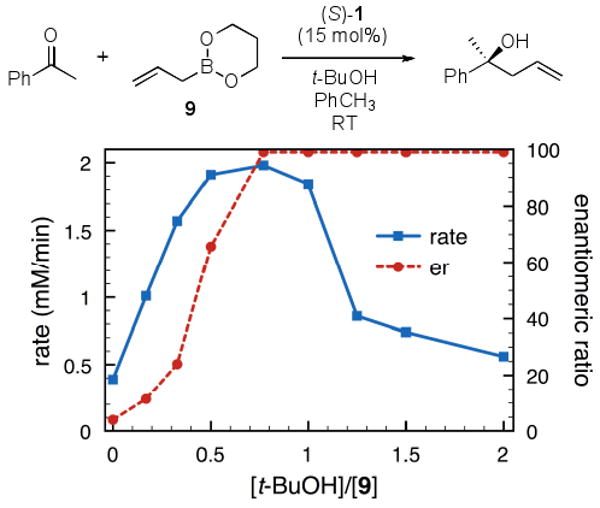
[a] Reactions were run with 0.15 mmol 1, 1.5 mmol boronate 9, and 0.75 mmol acetophenone in PhCH3 (0.3 M) for 15 h under Ar, followed by flash chromatography on silica gel. Enantiomeric ratios were determined by chiral HPLC methods.
In summary, a mechanistic investigation of the asymmetric allylboration reaction of ketones catalyzed by chiral biphenols has resulted in the development of a highly optimized reaction. Key observations about the ligand exchange process aided the design of a reaction that uses less catalyst (from 15 mol% to 2 mol%) at ambient temperatures (from -35 °C to room temperature). The new reaction exhibits similar mechanistic characteristics to the original with a first-order rate dependence on catalyst concentration and chiral biphenol-boronate complex formation. Insight afforded by this study will enable further reaction development, application of the method to asymmetric synthesis, and identification of novel catalytic processes.
Supplementary Material
Footnotes
This research was supported by the NIH (R01 GM078240)
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.Roush WR. In: Comprehensive Organic Synthesis. Trost BA, Fleming I, editors. Vol. 2. Permagon Press; Oxford, U.K.: 1991. [Google Scholar]; (b) Chemler SR, Roush WR. In: Modern Carbonyl Chemistry. Otera J, editor. Wiley-VCH; Weinheim: 2000. p. 403. Chapter 11. [Google Scholar]; (c) Denmark SE, Almstead NG. In: Modern Carbonyl Chemistry. Otera J, editor. Wiley-VCH; Weinheim: 2000. p. 299. Chapter 10. [Google Scholar]; (d) Kennedy JWJ, Hall DG. In: Boronic Acids. Hall DG, editor. Wiley-VCH; Weinheim: 2005. p. 241. Chapter 6. [Google Scholar]
- 2.For examples of asymmetric ketone allylations in total synthesis, see: Nielsen DK, Nielsen LL, Jones SB, Toll L, Asplund MC, Castle SL. J Org Chem. 2009;74:1187. doi: 10.1021/jo802370v.Li F, Tartakoff SS, Castle SL. J Am Chem Soc. 2009;131:6674. doi: 10.1021/ja9024403.
- 3.Asymmetric catalytic ketone allylations: Casolari S, D'Addario D, Tagliavini E. Org Lett. 1999;1:1061.Hanawa H, Kii S, Maruoka K. Adv Synth Catal. 2001;343:57.Cunningham A, Woodward S. Synlett. 2002:43.Cunningham A, Mokal-Parekh V, Wilson C, Woodward S. Org Biomol Chem. 2004;2:741. doi: 10.1039/b313384b.Wada R, Oisaki K, Kanai M, Shibasaki M. J Am Chem Soc. 2004;126:8910. doi: 10.1021/ja047200l.Teo YC, Goh JD, Loh TP. Org Lett. 2005;7:2743. doi: 10.1021/ol051018n.Lu J, Hong ML, Ji SJ, Teo YC, Loh TP. Chem Commun. 2005:4217. doi: 10.1039/b507768k.Wadamoto M, Yamamoto H. J Am Chem Soc. 2005;127:14556. doi: 10.1021/ja0553351.Zhang X, Chen D, Liu X, Feng X. J Org Chem. 2007;72:5227. doi: 10.1021/jo0706325.Miller JJ, Sigman MS. J Am Chem Soc. 2007;129:2752. doi: 10.1021/ja068915m.Schneider U, Ueno M, Kobayashi S. J Am Chem Soc. 2008;130:13824. doi: 10.1021/ja804182j.Itoh J, Han SB, Krische MJ. Angew Chem. 2009;121:6431. doi: 10.1002/anie.200902328.Angew Chem Int Ed. 2009;48:6313.Yazaki R, Kumagai N, Shibasaki M. J Am Chem Soc. 2009;131:3195. doi: 10.1021/ja900001u.
- 4.Lou S, Moquist PN, Schaus SE. J Am Chem Soc. 2006;128:12660. doi: 10.1021/ja0651308. [DOI] [PubMed] [Google Scholar]
- 5.(a) Saaby S, Bella M, Jørgensen KA. J Am Chem Soc. 2004;126:8120. doi: 10.1021/ja047704j. [DOI] [PubMed] [Google Scholar]; (b) Shirakawa S, Yamamoto K, Kitamura M, Ooi T, Maruoka K. Angew Chem. 2005;117:631. doi: 10.1002/anie.200462035. [DOI] [PubMed] [Google Scholar]; Angew Chem, Int Ed. 2005;44:625. doi: 10.1002/anie.200462035. [DOI] [PubMed] [Google Scholar]; (c) Kitamura M, Shirakawa S, Maruoka K. Angew Chem. 2005;117:1573. doi: 10.1002/anie.200462257. [DOI] [PubMed] [Google Scholar]; Angew Chem, Int Ed. 2005;44:1549. [Google Scholar]; (d) Terada M, Machioka K, Sorimachi K. Angew Chem. 2006;118:2312. doi: 10.1002/anie.200503477. [DOI] [PubMed] [Google Scholar]; Angew Chem, Int Ed. 2006;45:2254. [Google Scholar]; (e) Rueping M, Antonchick AP, Theissmann T. Angew Chem. 2006;118:6903. doi: 10.1002/anie.200601832. [DOI] [PubMed] [Google Scholar]; Angew Chem, Int Ed. 2006;45:6751. [Google Scholar]; (f) Pan SC, Zhou J, List B. Angew Chem. 2007;119:3028. doi: 10.1002/anie.200603630. [DOI] [PubMed] [Google Scholar]; Angew Chem, Int Ed. 2007;46:612. [Google Scholar]; (g) Liu TY, Cui HL, Long J, Li BJ, Wu Y, Ding LS, Chen YC. J Am Chem Soc. 2007;129:1878. doi: 10.1021/ja068703p. [DOI] [PubMed] [Google Scholar]; (h) Guo QX, Liu H, Guo C, Luo SW, Gong LZ. J Am Chem Soc. 2007;129:3790. doi: 10.1021/ja068236b. [DOI] [PubMed] [Google Scholar]; (i) Terada M, Machioka K, Sorimachi K. J Am Chem Soc. 2007;129:10336. doi: 10.1021/ja0739584. [DOI] [PubMed] [Google Scholar]; (j) Rueping M, Theissmann T, Kuenkel A, Koenigs RM. Angew Chem. 2008;120:6903. doi: 10.1002/anie.200802139. [DOI] [PubMed] [Google Scholar]; Angew Chem, Int Ed. 2008;47:6798. [Google Scholar]; (k) Cheon CH, Yamamoto H. J Am Chem Soc. 2008;130:9246. doi: 10.1021/ja8041542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vachal P, Jacobsen EN. J Am Chem Soc. 2002;124:10012. doi: 10.1021/ja027246j. [DOI] [PubMed] [Google Scholar]; (b) Kotke M, Schreiner PR. Synthesis. 2007:779. [Google Scholar]
- 7.Wu TR, Chong JM. J Am Chem Soc. 2005;127:3244. doi: 10.1021/ja043001q. [DOI] [PubMed] [Google Scholar]; (b) Wu TR, Chong JM. J Am Chem Soc. 2007;129:4908. doi: 10.1021/ja0713734. [DOI] [PubMed] [Google Scholar]; (c) Lou S, Moquist PN, Schaus SE. J Am Chem Soc. 2007;129:15398. doi: 10.1021/ja075204v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Ishiyama T, Ahiko T, Miyaura N. J Am Chem Soc. 2002;124:12414. doi: 10.1021/ja0210345. [DOI] [PubMed] [Google Scholar]; (b) Rauniyar V, Hall DG. J Am Chem Soc. 2004;126:4518. doi: 10.1021/ja049446w. [DOI] [PubMed] [Google Scholar]; (c) Rauniyar V, Zhai H, Hall DG. J Am Chem Soc. 2008;130:8481. doi: 10.1021/ja8016076. [DOI] [PubMed] [Google Scholar]; (d) Sakata K, Fujimoto H. J Am Chem Soc. 2008;130:12519. doi: 10.1021/ja804168z. [DOI] [PubMed] [Google Scholar]
- 9.Paton RS, Goodman JM, Pellegrinet SC. Org Lett. 2009;11:37. doi: 10.1021/ol802270u. [DOI] [PubMed] [Google Scholar]
- 10.(a) Brown HC, Racherla US, Pellechia PJ. J Org Chem. 1990;55:1868. [Google Scholar]; (b) Roy CD, Brown HC. J Organomet Chem. 2007;692:784. [Google Scholar]
- 10.(a) Tokunaga M, Larrow JF, Kakuichi F, Jacobsen EN. Science. 1997;277:936. doi: 10.1126/science.277.5328.936. [DOI] [PubMed] [Google Scholar]; (b) Schaus SE, Brandes BD, Larrow JF, Tokunaga M, Hansen KB, Gould AE, Furrow ME, Jacobsen EN. J Am Chem Soc. 2002;124:1307. doi: 10.1021/ja016737l. [DOI] [PubMed] [Google Scholar]; (c) Long J, Hu J, Shen X, Ji B, Ding K. J Am Chem Soc. 2002;124:10. doi: 10.1021/ja0172518. [DOI] [PubMed] [Google Scholar]; (d) Dolman SJ, Sattely ES, Hoveyda AH, Schrock RR. J Am Chem Soc. 2002;124:6991. doi: 10.1021/ja012534l. [DOI] [PubMed] [Google Scholar]; (e) Yuan Y, Zhang X, Ding K. Angew Chem. 2003;115:5636. doi: 10.1002/anie.200352535. [DOI] [PubMed] [Google Scholar]; Angew Chem, Int Ed. 2003;42:5478. [Google Scholar]; (f) Jeon SJ, Li H, Walsh PJ. J Am Chem Soc. 2005;127:16416. doi: 10.1021/ja052200m. [DOI] [PubMed] [Google Scholar]; (g) Wooten AJ, Kim JG, Walsh PJ. Org Lett. 2007;9:381. doi: 10.1021/ol062264h. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.