

G-protein coupled receptors (GPCRs) are transmembrane proteins that play key roles in an array of diverse biological functions, such as vision, odor, taste, and memory, and have been the targets of ca. 50% drugs on the market. However, there is very limited information on the structures of GPCRs due to the difficulty of protein purification and stability requirement of the phospholipid membrane environment. To date, the bovine rhodopsin, a light receptor protein,1,2 is the only GPCR whose crystal structure has been determined. Rhodopsin consists of an opsin apoprotein and an 11-cis retinal chromophore, which is covalently bonded to Lys296 via a protonated Schiff base linkage. The photoactivation cycle of rhodopsin is triggered by the cis-to-trans isomerization of retinal and ends with the release of all-trans retinal. The crystal structures show a 7-helix bundle in the transmembrane segment and that the chromophore binds in a pocket enclosed by the transmembrane helices and the β-hairpin loop connecting helix 4 and helix 5 on the extracellular surface.
Despite the diverse functions, GPCRs are believed to share the 7-transmembrane helix bundle structure as in rhodopsin. Therefore, rhodopsin structure has been used as the template to model a large number of GPCR proteins, including those as drug targets.3 Because the binding pocket is buried inside the transmembrane domain, a very interesting question is how the chromophore enters and exits the protein. This question is of great interest because understanding ligand entrance routes may help to improve drug efficacy since many of the drugs bind to the same pocket. Although the uptake and release of retinal can be detected by monitoring the intrinsic protein fluorescence,4 direct evidence of the channeling routes is unavailable. The recent crystal structures showed no obvious channel through which the ligands can pass into the binding sites. Thus, the question regarding the paths for the uptake and release of retinal remains open.
In this work, we simulated the egress of all-trans retinal from the rhodopsin protein by applying the random acceleration molecular dynamics (RAMD) method,5 implemented by modification of the AMBER8 program.6 In the RAMD approach, a small randomly oriented force is added to the center of mass of retinal to enhance the dynamics, thus exploring the weakness of the binding pocket and allowing the retinal to exit in a computationally feasible time. The direction of the random force is kept for a certain period of time. If, during this time, retinal moves more than a certain distance, the direction of the force is maintained, otherwise a new direction is chosen randomly.
The rhodopsin molecule was embedded in a phospholipid bilayer of palmitoyloleoyl phosphatidylcholine (POPC) and solvated in water. From the X-ray structure (PDB entry 1U19),2 after the simulation of cis-to-trans isomerization followed by manual hydrolysis of retinal and 2.0 ns of standard molecular dynamics (MD) simulation with the free all-trans retinal aldehyde in the binding pocket (see Supporting Information), the protein structure remained similar to the dark state, consistent with the results of a much longer simulation.7 We then performed 38 RAMD simulations and obtained 36 egress trajectories as summarized in Table 1.
Table 1.
| pathway | no.b | acceleration (kcal/Å〉g) | trajectory length (ps) |
|---|---|---|---|
| A | 12 | 0.30–0.40 | 56–748 |
| B | 9 | 0.30–0.40 | 72–898 |
| C | 4 | 0.30–0.40 | 38–864 |
| D | 2 | 0.30, 0.40 | 110, 942 |
| E | 1 | 0.38 | 462 |
| othera | 8 | 0.30–0.40 | 272–1652 |
| no egress | 2 | 0.3, 0.35 | 5000, 1000 |
Retinal moved toward the cytoplasmic side and passed out of the protein there.
Number of trajectories.
The predominant egresses, in 28 out of 36, were near the extracellular side, which can be classified into five pathways according to the helices around the egresses (Figure 1). Retinal exited either from the clefts between helix 4 and helix 5 (pathway A) or between helix 5 and helix 6 (pathway B) in 21 out of 36 egress trajectories, with the β-ionone ring passing out first. The egress opening was made by transiently breaking the interhelical hydrophobic interactions, which were recovered immediately after retinal passed through (Figure 4 in Supporting information). In seven other simulations, retinal exited by first moving up toward the extracellular site and then passed out from the cleft between helix 1 and helix 7 (pathway C), the cleft between the β-hairpin and helices 6 and 7 (pathway D), and the cleft between helix 1 and helix 2 (pathway E), with the aldehyde group passing out first. Similar to pathways A and B, only hydrophobic interactions were involved in the channel opening. These pathways indicated that, although the rhodopsin protein structure is rather stable, the weakness in local interactions between the helices can be exploited and broken temporarily to allow ligands to pass through. Since most of the egress routes are near the extracellular side, they also suggest that ligands and drugs may enter the binding pocket without passing through the membrane.
Figure 1.
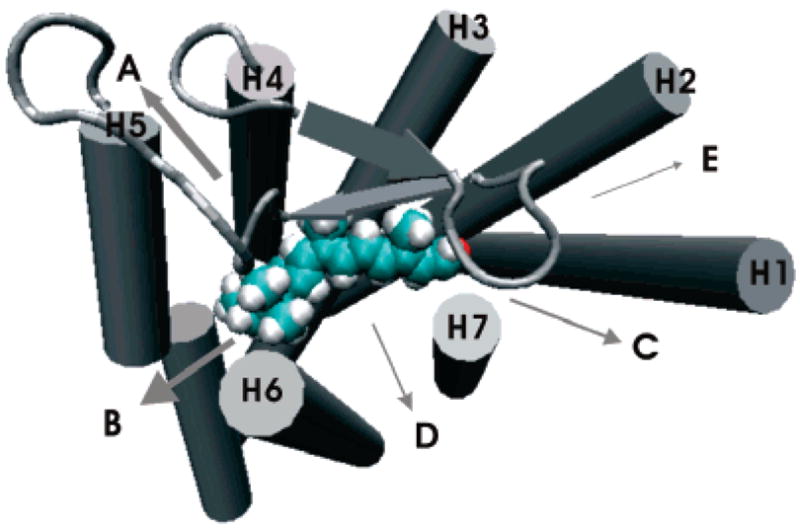
Retinal egress pathways near the extracellular side (out of the page toward the reader). They are classified into five (A, B, C, D, and E) pathways according to the helices around the egresses. The arrow widths represent the relative frequencies. See Figure 3 in Supporting Information for a stereoview.
In addition to the direct egress near the extracellular side, retinal was observed in two trajectories to make a long detour deep into the cytoplasmic side before its exit from the protein near the extracellular side. The path of retinal’s center of mass is shown in Figure 2 for one of the two trajectories. In eight other trajectories, retinal moved toward the cytoplasmic side by 5–10 Å and passed out from the interhelical clefts there (Figure 2). The egress opening was made by breaking the hydrophobic interactions between the residues in helices 5 and 6 or helices 6 and 7, located before the charged residues at the very end of the helices. These results suggest the possibility for retinal to egress near the cytoplasmic side through channeling in the transmembrane domain, although limited. Furthermore, if retinal is released by this way, it can be enzymatically converted to all-trans retinol in situ, as suggested by Schadel and co-workers.4 However, this may not be the preferred drug entrance route into GPCR proteins because it requires the passage of the membrane and long distance channeling to the putative binding site near the extracellular side. As our observations from the simulations indicate, this route is notably less preferred than egress pathways A–E from the extracellular side.
Figure 2.
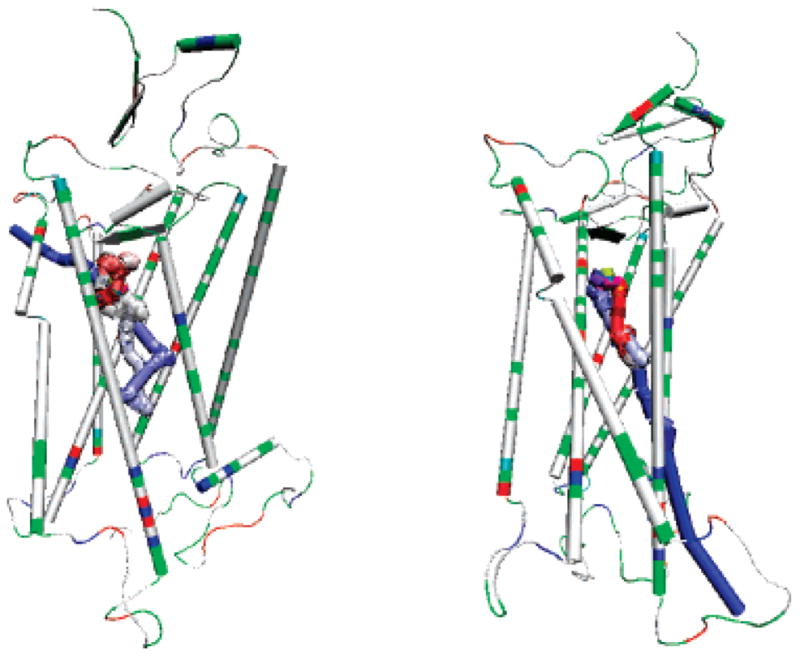
Paths of the center of mass of retinal in two of the trajectories. The center of mass of the retinal is indicated by the colored spheres, and the color scale is from yellow (start) to blue (end), indicating the trajectories of retinal. The protein is shown in cartoon and colored by residue types (charged residues are in red or blue and non-charged are in white or green). Left: Retinal made a long detour to the cytoplasmic side (the lower side) before it passed out of the protein from the cleft between helix 4 and helix 5 near the extracellular side (the upper side). Right: Retinal passed out of the protein from the cleft between helix 6 and helix 7 near the cytoplasmic side.
Although several different egress routes were identified in the simulations, none of the routes was through the interhelical clefts between helix 3 and other helices or the cleft between helix 3 and the extracellular β-hairpin loop. Unlike other helices, helix 3 inserts into the transmembrane domain and forms extensive contacts with other helices, including helices 2 and 4 on the extracellular side, and helices 4, 5, and 6 on the cytoplasmic side as well as the extracellular β-hairpin. Thus, the observation reinforces the high stability in the vicinity of helix 3 and the structural rigidity of the regions, as suggested in the thermal unfolding simulation study by Rader et al.8 The high stability is also consistent with the results of Martinez-Mayorga et al.7 who observed that rhodopsin remains stable until the completion of a 2000 ns simulation.
An important structural feature of rhodopsin is the highly conserved Cys110–Cys187 disulfide bond bridging helix 3 and the extracellular β-hairpin. Because of the disulfide bond, the β-hairpin is difficult to open and it effectively blocks a direct access channel to the binding pocket from the extracellular side. To investigate whether the Cys110–Cys187 disulfide bond plays a role in ligand egress, we carried out 10 RAMD simulations without the disulfide bond. In all 10 simulations, despite the removal of the disulfide bond, retinal did not exit from the β-hairpin route, instead it exited from the cleft between helix 5 and helix 6 (pathway B) similar to the exit paths when the disulfide bond was present. Therefore, irrespective to the lack of the disulfide bond, the main egress routes of retinal were the clefts between the helices rather than through the extracellular β-hairpin, suggesting that the highly conserved disulfide bond in GPCRs may serve to stabilize the tertiary structure of the protein9 and a stable binding cavity to facilitate high specificity binding.
It should be noted that the protein structure in our simulation was similar to the dark state of the rhodopsin and changes in helix clefts during the decay of the activated state to opsin may affect retinal egress pathways. However, this should not affect the implication of our results to drug entry routes because drugs initially populate on the extracellular side and typically access the GPCRs at their inactivated states.
Supplementary Material
Acknowledgments
We thank Dr. Rebecca Wade for providing the RAMD code, and UC Davis Genome Center for computer support. This work was supported by research grants from NIH (GM64458 and GM67168 to Y.D.). Usage of VMD and AMBER is gratefully acknowledged.
Footnotes
Supporting Information Available: Details of the system modeling and simulation. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 2.Okada T, Sugihara M, Bondar AN, Elstner M, Entel P, Buss V. J Mol Biol. 2004;342:571–583. doi: 10.1016/j.jmb.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 3.(a) Maeda K, Das D, Ogata-Aoki H, Nakata H, Miyakawa T, Tojo Y, Norman R, Takaoka Y, Ding J, Arnold GF, Arnold E, Mitsuya H. J Biol Chem. 2006;281:12688–12698. doi: 10.1074/jbc.M512688200. [DOI] [PubMed] [Google Scholar]; (b) Evers A, Klebe G. Angew Chem, Int Ed. 2004;43:248–251. doi: 10.1002/anie.200352776. [DOI] [PubMed] [Google Scholar]; (c) Hobrath JV, Wang S. J Med Chem. 2006;49:4470–4476. doi: 10.1021/jm0501634. [DOI] [PubMed] [Google Scholar]
- 4.Schadel SA, Heck M, Maretzki D, Filipek S, Teller DC, Palczewski K, Hofmann KP. J Biol Chem. 2003;278:24896–24903. doi: 10.1074/jbc.M302115200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Luedemann SK, Lounnas V, Wade RC. J Mol Biol. 2000;303:797–811. doi: 10.1006/jmbi.2000.4154. [DOI] [PubMed] [Google Scholar]; (b) Schleinkofer K, Sudarko, Winn PJ, Luedemann SK, Wade RC. EMBO Rep. 2005;6:584–589. doi: 10.1038/sj.embor.7400420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Case DA, Cheatham TE, III, Darden T, Gohlke H, Luo R, Merz KM, Jr, Onufriev A, Simmerling C, Wang B, Woods RJ. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]; (c) Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, Yang R, Cieplak P, Luo R, Lee T, Caldwell J, Wang J, Kollman P. J Comput Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]; (d) Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 7.Martinez-Mayorga K, Pitman MC, Grossfield A, Feller SE, Brown MF. J Am Chem Soc. 2006;128:16502–16503. doi: 10.1021/ja0671971. [DOI] [PubMed] [Google Scholar]
- 8.Rader AJ, Anderson G, Isin B, Khorana HG, Bahar I, Klein-Seetharaman J. Proc Natl Acad Sci USA. 2004;101:7246–7251. doi: 10.1073/pnas.0401429101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karnik SS, Sakmar TP, Chen HB, Khorana HG. Proc Natl Acad Sci USA. 1988;85:8459–8463. doi: 10.1073/pnas.85.22.8459. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.