

Abstract
Although the synaptogenic program for cholinergic synapses of the neuromuscular junction is well known, little is known of the identity or dynamic expression patterns of proteins involved in non-neuromuscular nicotinic synapse development. We have previously demonstrated abnormal presynaptic terminal morphology following loss of nicotinic acetylcholine receptor (nAChR) α9 subunit expression in adult cochleae. However, the molecular mechanisms underlying these changes have remained obscure. To better understand synapse formation and the role of cholinergic activity in the synaptogenesis of the inner ear, we exploit the nAChR α9 subunit null mouse. In this mouse, functional acetylcholine (ACh) neurotransmission to the hair cells is completely silenced. Results demonstrate a premature, effusive innervation to the synaptic pole of the outer hair cells in α9 null mice coinciding with delayed expression of cell adhesion proteins during the period of effusive contact. Collapse of the ectopic innervation coincides with an age-related hyperexpression pattern in the null mice. In addition, we document changes in expression of presynaptic vesicle recycling/trafficking machinery in the α9 null mice that suggests a bidirectional information flow between the target of the neural innervation (the hair cells) and the presynaptic terminal that is modified by hair cell nAChR activity. Loss of nAChR activity may alter transcriptional activity, as CREB binding protein expression is decreased coincident with the increased expression of N-Cadherin in the adult α9 null mice. Finally, by using mice expressing the nondesensitizing α9 L9′T point mutant nAChR subunit, we show that increased nAChR activity drives synaptic hyperinnervation.
Keywords: cochlea, acetylcholine receptors, synapse development, olivocochlear system, alpha9 nAChR
INTRODUCTION
The cochlea encodes auditory stimuli via activity of its primary sensory elements, the hair cells. There are two classes of hair cells—the inner hair cells (IHCs), responsible for the sense of hearing; and the outer hair cells (OHCs), motile cells largely responsible for modulating cochlear mechanics and generally regarded as the cells giving rise to the cochlear amplifier. Cochlear output is further modulated by descending efferent axons originating from cells located in the superior olivary complex, collectively making up the olivocochlear (OC) system. In rodents, OC synaptogenesis occurs during early postnatal ages, but prior to the onset of hearing, which occurs during the second postnatal week. The cellular interactions between OC fibers and their targets within the developing cochlea are crucial for the maturation of normal hearing. Surgical elimination of efferent connections to the cochlea during early postnatal life produces permanent reductions in both the sensitivity and selectivity of auditory nerve fibers to sound stimuli (Walsh et al., 1998). Therefore, understanding how the OC synapse forms and matures is an important link in understanding normal and pathological auditory processing.
Acetylcholine (ACh)-induced synaptic activity between efferent axons and both prehearing and post-onset hearing stage OHCs is mediated by a heteromeric receptor composed of α9 and α10 nicotinic acetylcholine receptor (nAChR) subunits, while IHCs express both subunits at prehearing stages, but only the α9 subunit following the onset of hearing (Elgoyhen et al., 1994, 2001). Neither subunit is expressed in the CNS. Therefore, data obtained from the inner ear following null ablation of the α9 and α10 subunits (Vetter et al., 1999, 2007) reflect local consequences occurring between hair cells and OC axons, and not consequences involving loss of the receptor from the CNS. The α9 subunit is responsible for many key functional properties of the hair cell nAChRs, including ligand binding, ion permeability, and pharmacological responses (Elgoyhen et al., 1994, 2001). Null ablation of the α9 gene results in total loss of nAChR-mediated activity (Vetter et al., 1999, 2007), while a point mutation to the α9 gene results in a hyperactive state of the receptor (Taranda et al., 2009). Using these transgenic mice, one may ascertain the role of cholinergic activity in coordinating synaptogenic programs within the inner ear.
The physiological role of α9 nAChRs at the olivocochlear synapse is well described (Vetter et al., 1999; Katz et al., 2000; Elgoyhen et al., 2001; Weisstaub et al., 2002). However, fundamental questions have remained regarding the molecular mechanisms invoked by the nAChR subunits during olivocochlear synapse formation that explain our previous findings of hypertrophic OC terminal structure and decreased density of OC innervation to OHCs following genetic ablation of α9 or α10 nAChR subunits (Vetter et al., 1999, 2007). To better define the role of OC nAChRs in synapse formation/stabilization with cochlear hair cells, we have examined the temporal aspects of postnatal synapse formation in the inner ears of mice harboring a null mutation for α9, and have probed numerous proteins known to be involved in synapse formation and/or maintenance to assess their expression levels at various important developmental ages. To further define whether the mechanisms underlying abnormal synaptogenesis occurring with loss of α9 gene expression are the result of loss of activity, or in some way the result of loss of protein interactions directly involving the nAChR complex, we have investigated the structural state of the OC synapse in an α9 L9′T point mutant (α9 knock-in mutant, α9KI) possessing abnormal ACh-mediated physiology, but normal α9 gene transcript expression (Taranda et al., 2009). Here, we report that the loss of α9 nAChR causes aberrant OC innervation during early stages of hair cell OC innervation, and misexpression of key synaptic proteins involved in the formation and maintenance of the OC synapse. We also demonstrate that heightened nAChR activity present in the α9KI mice results in hyperinnervation of outer hair cells, further suggesting that it is nAChR activity that is important for synapse formation in the inner ear.
METHODS
Animals
α9 null mice and α9 knock-in (α9KI) mice have been previously described (Vetter et al., 1999; Taranda et al., 2009). Homozygous null or knock-in male and female mice were bred to produce mice used for this study, while heterozygous males and females were bred to produce the wild-type mice that were then used as breeders to produce full litters of α9 wild-type mice matching the null background.
Immunostaining Radial Sections for Developmental Series Analysis
Immunostaining was performed on paraffin sections of cochleae from wild type and α9 null mice using antibodies selected because of their previously demonstrated usefulness in describing cochlear synaptic and hair cell development during postnatal ages. Cochleae from α9 null mice and wild-type mice at postnatal ages P3, P5, P7, P10, P13, P15, and P18 were perfused locally through the round and oval windows and isolated. At least three mice of each genotype, for each age group were used for the analysis (n = 6 cochleae). The isolated cochleae were immediately post-fixed in 4% paraformaldehyde in 0.1 M sodium phosphate buffer for 1 h at room temperature and decalcified (for ages P7 and above) in 8% EDTA in PBS overnight at room temperature on a rotator. The cochleae were then dehydrated and embedded in paraffin. Tissue was sectioned at 10 μm parallel to the modiolus. Before immunostaining, the cochlear radial sections were rehydrated and blocked with 5% normal donkey serum, 0.5% Triton X-100 for 1 h at room temperature. Cochlear hair cells were visualized using rabbit anti-Pep19 (1:2000, a gift from Dr. Jim Morgan), and efferent synaptic terminals were visualized using mouse anti-synaptophysin (1:1000, Millipore/Chemicon, Billerica, MA) overnight at room temperature. Other antibodies used included anti-pan Cadherin (1:1000, AbCam, Billerica, MA), and anti-cysteine string protein (CSP) (1:1000, Stress-Gen Biotechnologies Corp., Ann Arbor, MI). Fluorescent secondary antibodies included Texas red conjugated anti-rabbit (1:200) (Jackson Immunoresearch, West Grove, PA), and Oregon Green conjugated anti-mouse (1:200, InVitrogen, Carlsbad, CA). Sections were coverslipped using SlowFade Gold mounting media (InVitrogen).
Confocal Microscopy
Immunostained cochlear sections were analyzed using a Leica TCS SP2 confocal microscope equipped with an acoustico-optical beam splitter (AOBS) imaging system. Serial Z-stack images were collected.
Immunostaining on Whole Mount Tissue and Image Analysis for Efferent Innervation of OHC Region
Whole mount processing of adult (6- to 8-week old) cochlear turns was used for CSP immunostaining of α9 wild type and null cochleae (two mice each, n = 4 cochleae), as well as for synaptophysin immunostaining for α9KI and wild-type mice (three mice each, n = 6 cochleae). Cochleae were perfused through the round and oval windows with 4% paraformaldehyde in 0.1 M sodium phosphate buffer at room temperature. The cochleae were immediately isolated and postfixed in the same fixative for 1 h at room temperature. Following postfixation, the cochleae were decalcified in 8% EDTA in PBS overnight at room temperature on a rotator, or 2–3 days at 4°C. Cochleae were stripped of the bony outer capsule, and individual turns were cut and processed for whole mount immunostaining. Dissected turns were incubated in 5% normal donkey serum with 0.5% Triton X-100 in PBS for 1 h, followed by incubation with either an anti-synaptophysin or anti-CSP primary antibody overnight. Alexa Fluor-488 labeled goat anti-mouse secondary antibody (InVitrogen) at 1:500. Texas Red labeled anti-rabbit secondary (1:500) was used to visualize CSP. Sections were mounted onto slides and coverslipped with SlowFade Gold (In Vitrogen).
For quantifying terminal density of α9KI mice, mid-turns of the cochlea (the portion left over once the apical half turn and hook region are dissected off) were analyzed. For counting terminals under OHCs of α9KI mice, whole-mount preparations of cochleae from three adult mice each of wild type and homozygous α9KI (n = 6 cochleae for each genotype) were immunostained for synaptophysin and visualized using anti-mouse avidin-biotin-HRP complex linkage (ABC Kit, Vector laboratories, Burlingame, CA) and DAB/H2O2 chromogen reaction. Synaptophysin antibodies were also used for confocal microscopy to assess volume of α9KI terminals compared to wild type, and for presentation purposes. Sites of synaptophysin immunoreactivity were fluorescently labeled with Alexa-Fluor 488, and examined using a Leica TCS SP2 AOBS confocal microscope. For volume calculations of synaptic terminals, image projections in alternate planes were computed, and the volume of immunostained OC terminals from the image stacks determined using Amira three-dimensional visualization software (instruction, courtesy of Dr. M. Charles Liberman, Mass Eye and Ear Infirmary). Synaptophysin labeled efferent terminals were counted from 538 outer hair cells of adult wild type and 614 outer hair cells of adult α9KI mice. For volume measures of adult OC terminals, terminals innervating 95 OHCs of wild type and 119 OHCs of α9KI were examined.
Protein Expression Analysis: Lysate Production
Immunostaining procedures revealed that a number of proteins representing various cellular processes were differentially expressed over early postnatal ages in the α9 null mice. To better quantify the differential expression of proteins that changed in immunostaining experiments, and to further probe other proteins related to the cellular processes investigated via immunostaining procedures, quantitative western blot analyses were performed. Cochleae were obtained following standard gross dissection from P7, P10, and 6- to 8-week-old α9 null mice and wild-type mice. Each mouse underwent cervical dislocation, followed by rapid retrieval of the cochleae using a lateral approach to the bulla, which was removed to expose the cochlear capsule. The cochleae were rapidly removed (<1 min per animal) and immersed in ice-cold lysis buffer (T-Per supplemented with protease inhibitors, Thermo Scientific/Pierce, Rockford, IL). Lysates were generated from pooled samples. At least four mice (n = 8 cochleae) of each genotype, for each age group were used to produce lysate for analysis, and each analysis (i.e. on one pooled protein lysate) was performed in triplicate on a gel (technical replicate). At least two (and most often three) different lysates were produced from separate pools of mice and probed for each age, representing biological replicates. Care was taken during isolation of the cochleae to not include cerebellar tissue. Whole cochlear lysates were generated by manual homogenization in lysis buffer using a Kontes #21 ground glass tissue grinding pestle and tube. Samples were centrifuged at 13,000g for 20 min at 4°C to pellet cellular debris. Protein concentration was estimated using a micro-BCA kit (Thermo Scientific/Pierce). A 50 μg aliquot of each lysate was separated by 8–10% SDS-polyacrylamide gel electrophoresis and wet transferred to PVDF membrane (Bio-Rad, Hercules, CA).
Protein Expression Analysis: Immunoblotting
Proteins were detected with anti-pan-Neural Cell Adhesion Molecule (NCAM) (1:500, BD Pharmingen, San Jose, CA), anti-pan-cadherin (1:1000, AbCam, Cambridge, MA), anti-β-catenin (1:1000, InVitrogen/Zymed labs), anti-neuroligin-1 (1:5000, a gift from Dr. Nils Brose), anti-Munc interacting protein (MINT) (1:5000 Millipore/Chemicon), anti-Ca2+/calmodulin-activated Ser-Thr kinase (CASK) (1:1000, Millipore/Chemicon), anti-CSP (1:5000, Stress-Gen Biotechnologies Corp., Ann Arbor, MI), anti-synaptosomal associated protein-25 (SNAP-25) (1:5000, Sigma, St. Louis, MO), anti-Synapsin-1 (1:500, American Quale, San Clemente, CA), anti-Synaptophysin (1:1000, Millipore/Chemicon), anti-N-Cadherin (1:500, Sigma), anti-Presenillin (1:1000, Millipore/Chemicon), anti-CREB (1:1000, Millipore/Upstate, and Cell Signaling Technology, Danvers, MA), anti-CREB binding protein (CBP) (1:1000, Cell Signaling Technology), anti-high affinity choline transporter 1 (ChT-1) (1:1000, Millipore) and anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH, used as a lane loading control) (1:5000, Chemicon/Millipore). The primary antibodies were visualized using horseradish peroxidase conjugated anti-mouse or anti-rabbit secondary antibodies (1:2000, Jackson Immunoresearch), the SuperSignal West Dura enhanced chemiluminescence system (Thermo Scientific/Pierce) and a Kodak Image Station 440cf. Biotinylated molecular weight markers (Cell Signaling Technologies) and prestained protein standards (full range Rainbow recombinant protein, Amersham/GE HealthCare) were used for estimating the molecular weight of proteins. The blots were stripped and reprobed once, only for visualizing the GAPDH loading control after visualizing the protein of interest, and never for assessing a second protein of interest.
Protein Expression Analysis: Densitometric and Statistical Analyses
Protein expression levels were quantified using densitometric software (UN-SCAN-IT gel, Silk Software, Orem, UT). Density values of all bands were calculated by boxing the band of interest and calculating total pixel intensity after subtracting background signal using upper and lower band interpolation as recommended by the software manufacturer. In cases of multiple immunostained bands, the molecular weight of each band was calculated, and the band of the expected size (or within 10%) was used for quantification. In the case of a fuzzy band, Kodak ImageStation software was used to assess the band edges (defined as a two-thirds drop in intensity) prior to densitometry analysis to help establish consistent band borders. Data analysis was performed by first normalizing the raw expression levels of the protein of interest to GAPDH, used as a loading control. Next, the GAPDH normalized expression of proteins isolated from the null mice was represented as a percentage of the GAPDH normalized wild-type samples, yielding a value for the expression as percent of age matched wild-type control. Numbers for each analysis came from bands assayed from the same blots. The average of the technical replicates (i.e. from the same blot) was defined as the expression level for that lysate. Technical replicate measures across the biological replicates (i.e. across blots) were then used for the statistical analysis. Statistical analysis (either one-way ANOVA followed by a Tukey post-hoc test, or an unpaired Student’s t-test, as appropriate) was performed. All analyses were performed using Prism 5a (for Mac OSX), and statistical significance was defined at p ≤ 0.0500.
RESULTS
Efferent Innervation Defects During Postnatal Refinement of Synaptic Innervation in α9 Null Mice
Previous findings from the α9 (Vetter et al., 1999), α10 (Vetter et al., 2007), and SK2 (Murthy et al., 2009) null mice strongly suggested that nAChR activity as well as activity mediated by the functionally coupled, calcium activated SK2 potassium channel plays a significant role in shaping the structural aspects of synaptic innervation to OHCs of the cochlea. To gain further insight into the role of α9 nAChRs in olivocochlear (OC) efferent innervation and synaptogenesis, we investigated the nature and time course of OC innervation to the hair cells at various postnatal ages (P3, P5, P7, P10, P13, P15, and P18) in the α9 null mice.
Temporal expression of synaptophysin was consistent with previous axon tracing studies and synaptophysin immunostaining studies in wild-type mice showing that efferent axons initially accumulate below the IHCs prior to projection toward the first row of OHCs (Bruce et al., 2000; Simmons et al., 1996a,b, 2002). In P3 wild-type cochleae [Fig. 1(A)], synaptophysin-positive efferent terminals were observed contacting IHCs, but synaptophysin immunoreactive fibers were rarely detected under OHCs. By P5 [Fig. 1(C)], some efferent axons had crossed the tunnel of Corti to contact OHCs. Similarly, α9 nulls at P3 [Fig. 1(B)] and P5 [Fig. 1(D)] exhibited robust synaptophysin labeled puncta contacting IHCs. In contrast to wild-type mice, however, P3 α9 null mice already possessed a sparse synaptophysin-positive efferent terminal innervation below the first row of OHCs [Fig. 1(B), arrows], which increased by P5 [Fig. 1(D)].
Figure 1.

α9 nAChR is required for target recognition and navigation of efferent fibers during olivocochlear synaptogenesis. Mid-modiolar paraffin cross sections of cochlea from α9 null and wild-type littermate control mice were double stained with Pep19 (red; hair cell marker) and synaptophysin (green, vesiculated efferent fiber marker) at P3, P5, P7, P10, P13, and P18 postnatal stages. (A) P3 wild-type cochleae possessed synaptophysin-positive efferent terminals adjacent to inner hair cells (IHC), but synaptophysin immunoreactive terminals were not detected in the outer hair cell (OHC) region. (B) P3 α9 null mice already possessed small numbers of synaptophysin-positive efferent terminals below the first and second rows of OHCs (arrows). (C) No significant change in innervation to IHCs occurred at P5 in the wild-type mice. (D) Synaptophysin staining intensity increased at the IHCs of α9 null mice. A greater number of efferent fibers migrating across the tunnel of Corti towards the OHCs and an increase in number and intensity of synaptophysin-positive efferent terminals were also observed in the OHC region in the α9 nulls (arrow). (E) At P7 in wild-type mice, an increase in intensity of synaptophysin staining was observed at the IHCs, and the first clear indication of vesiculated synaptic endings (arrow) occurred under OHCs. (F) Similarly, there was an increase in synaptic terminal innervation to the IHCs of the α9 null mice at P7. At the OHCs, synaptophysin labeled efferent fibers (arrowhead) overshot the synaptic (basal) pole of the first row hair cell (asterisk) and enveloped the modiolar side lateral wall of the OHC. Large immunopositive profiles of accumulated efferent crossing fibers (arrow) were also located near the base of the first row OHC. (G) At P10, the robust innervation to the wild-type IHCs decreased, and innervation to OHCs took on a mature profile under all three rows. (H) In the α9 null mice, large numbers of distinct synaptophysin labeled terminals were present directly over supranuclear regions of the OHCs (arrows). Synaptophysin labeling at the IHCs remained greater than that observed in wild-type mice. (I) At P13, the efferent innervation in wild-type mice remained stable and adult-like. (J) However, in α9 null mice, synaptophysin labeled efferent fibers created a lattice-like network surrounding the OHC soma of all three rows in α9 null mice (arrowhead) while also hypertrophying at the base of the cells (arrows). (K) At P18, the innervation to the wild-type hair cells remained unchanged and adult-like. (L) In the α9 nulls, direct efferent fiber contacts with the IHCs decreased significantly, and fibers were present in a position below the IHCs. In the OHC region, the lattice-like innervation to these cells also disappeared, leaving only the hypertrophied basal terminal (arrow). All scale bars represent 10 μm.
Robust innervation of OHCs began at P7 in wild-type mice [Fig. 1(E)]. Innervation remained at the “synaptic” basal pole of OHCs. In contrast, at this stage in α9 null mice, efferent fibers did not remain at the synaptic pole of OHCs, but rather continued to meander toward the apex of the hair cell soma [Fig. 1(F), arrowhead]. Additionally, large synaptophysin-positive clumps of fibers were observed near the synaptic pole on the tunnel side of the first row of outer hair cells in α9 null mice at P7 [Fig. 1(F), arrow]. The ectopic efferent fibers present along the length of the OHC soma were still present at P10 in the α9 nulls, although their appearance changed, becoming thinner and creating a lattice-like network surrounding the soma [Fig. 1(H)]. These fibers remained through P13 in the α9 null mice [Fig. 1(J)], but retracted to the base of the OHC by P15 (data not shown).
At P15, a time normally coincident with the decrease in expression of α10 nAChR subunit in the IHCs during normal development and the time at which the majority of OC fibers have migrated from the IHCs to the OHC region in wild-type mice, dense synaptophysin labeling was still observed directly contacting the IHCs in the α9 null mice (data not shown). At P18, the OC fibers persisted near, but were no longer observed directly on IHCs in the α9 null mice as well as wild-type mice [Fig. 1(K,L)], and punctate hypertrophied bouton-like synapses were observed at the OHCs of the null mice [Fig. 1(L), arrow].
Altered Expression of Trans-Synaptic Adhesion Proteins in α9 Null Mice
Synaptic development and plasticity require the rearrangement of adhesion and cytoskeletal elements that otherwise bind the synaptic elements together (Arrikath and Reichardt, 2008; Muller et al., 2008). Given the abnormal structural aspects of synapse development observed over the period of early synaptogenesis in the nAChR nulls (Fig. 1), and the hypertrophied synaptic bouton observed in adults (Vetter et al., 1999), it is possible that alterations may occur in cell adhesion molecule expression in the α9 nulls that underlie the structural phenotype observed.
N-Cadherin immunostain was localized diffusely in IHC and OHC cell bodies and as a distinct punctate profile at the base of the OHCs in wild-type cochleae at P18 [Fig. 2(A,B)]. In the α9 null mice, however, N-Cadherin was only expressed by the IHCs at this age, and the OHC region was devoid of significant immunostaining [Fig. 2(C,D)]. However, by adulthood (2 months old), N-Cadherin expression was found as punctate immunopositive profiles at the base of the OHCs, within the region of synaptic contact between the OC fibers and the OHC soma. While by nature not quantitative due to the vagaries of fixation, antibody penetration, etc., it is nonetheless interesting to observe that the N-Cadherin immunostaining below OHCs in adult α9 nulls appears brighter than that of the wild-type mice [Fig. 2(E,F)].
Figure 2.
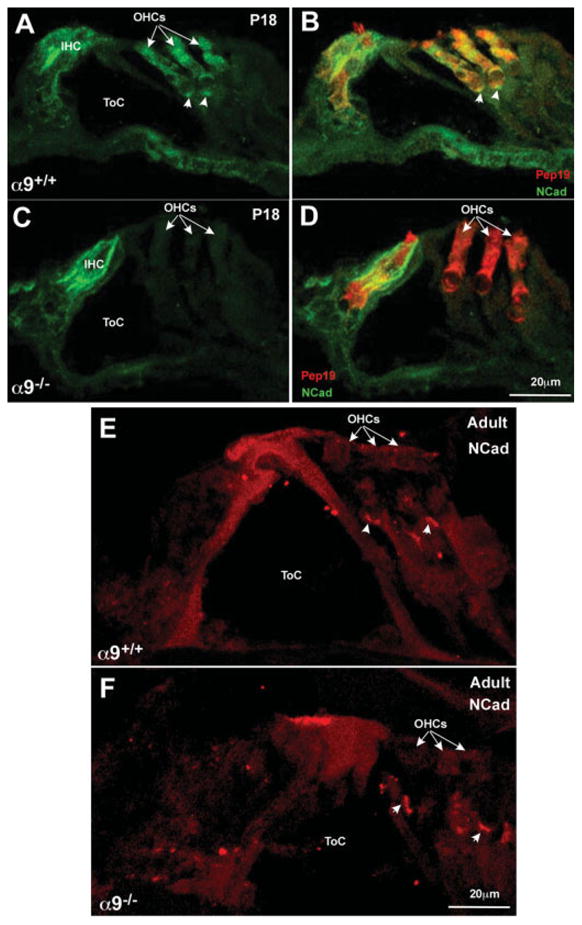
N-Cadherin immunostaining reveals loss of expression in OHCs in the α9 null mice. (A) Immunostaining for N-Cadherin (green) in wild-type P18 cochleae reveals expression primarily in the hair cells of the organ of Corti. (B) Double labeling the hair cells with Pep19 (red) reveals the extent of the hair cell boundary and that the hair cells do express N-Cadherin. (C) Immunostaining for N-Cadherin in P18 α9 null cochleae reveals expression in IHCs, but not in the OHC region (OHCs indicated by arrows). (D) Double labeling the section with Pep19 (red) reveals the coexistence of Pep19 and N-Cadherin in the IHC, but the OHCs do not express N-Cadherin, and remain fluorescent in red only. (E) In adults, N-Cadherin (red) is expressed predominantly in the OHC region, and denser staining can be observed at the basal (synaptic) pole of the cells (arrowheads). (F) In the α9 nulls, N-Cadherin staining is similar to that observed in the wild-type OHCs, but the staining near the synaptic pole of the cells is brighter and the immunopositive plaque is large than observed in the wild-type mice.
To more precisely determine whether the observed changes in synaptic structure and innervation in α9 null mice involves altered synaptic adhesion molecule expression, we examined the expression of a larger cadre of trans-synaptic signaling adhesion proteins, including N-Cadherin, Neuronal Cell Adhesion Molecule (NCAM), and Neuroligin 1 [Fig. 3(A)], all of which trigger synapse formation by mediating heterophilic and/or homophilic interactions. In addition, we examined the expression of the intracellular signaling molecule β-catenin, which is associated with trans-synaptic adhesion molecules such as cadherins. Changes in N-Cadherin and NCAM expression and distribution pattern are known to coincide with the appearance of efferent synapses (Whitlon et al., 1999).
Figure 3.

α9 nAChR regulates the expression of trans-synaptic adhesion proteins. (A) Total cochlear lysates from α9 null and wild-type control mice at P7, P10, and adult (2 months) ages were probed by Western blotting with antibodies against trans-synaptic and signaling proteins as indicated. In the case of the cadherins and the NCAM immunoblots, the antibodies detected previously recognized isoforms. Note that only the 140 kDa and 120 kDa form of NCAM in the cochlea, not the 180 kDa isoform, were detected. Similarly, the pan Cadherin antibody recognized multiple bands in the immunoblot. Based on mass, the upper band was identified as N-Cadherin, while the lower band was identified as E-Cadherin. Densitometric values were obtained only for the 140 kDa isoform of NCAM because of its expression over all the ages examined, and for N-Cadherin due to its role in synapse formation/maintenance. All data were normalized to densitometric values of loading control GAPDH. (B) The dynamic change in expression of the adhesion and adhesion-related proteins expressed by α9 null mice is displayed as percent change relative to wild-type levels in the time series bar graph. Quantification of the change in protein expression levels in α9 null relative to GAPDH was then normalized to the wild type expression GAPDH (and multiplied by 100) to better visualize changes in dynamics of protein expression. A value of 100 would therefore represent no change from the wild type expression values. One-way ANOVA and Tukey post-hoc multiple comparisons tests were used to assess changes in protein expression of α9 nulls normalized to wild type levels over time (*p < 0.05, **p < 0.005).
Quantitative analysis of the immunoblots revealed abnormal dynamics of protein expression in α9 null mice. Change in protein expression of null mice was assessed by first normalizing the band density of both wild type and α9 null mice to the GAPDH loading control, and then, to control for inter-blot variability, the null value was expressed as a percent of wild type expression level. Hence, a value of 100% represents equal expression between α9 null and wild-type cochlear samples.
NCAM, N-Cadherin, Neuroligin 1, and β-catenin exhibited significant variations in expression across the ages examined [Fig. 3(A,B)]. Cochlear samples from adult α9 null mice exhibited an upregulation of NCAM (37% ± 13%), N-Cadherin (47% ± 19%), Neuroligin-1 (45% ± 20%), and β-catenin (70% ± 16%), relative to expression levels found in wild-type mice [Fig. 3(B)]. Because of the structural changes observed developmentally following loss of α9 nAChRs, the expression level of the above proteins were examined at P7 and P10, ages that represent peak efferent synaptogenesis (P7) and an early stage of synaptic rearrangement prior to the onset of hearing (P10) (Katz et al., 2004). Except for NCAM, these adhesion proteins were downregulated at P7 and P10 in the α9 null mice compared to wild-type mice [Fig. 3(B)]. At P7 and P10, N-Cadherin was downregulated by 48% (±4%) and 54% (±6%), respectively. This downregulation of N-Cadherin expression mirrored the qualitative observations using immunohistochemical techniques [Fig. 2(A–D)]. Similarly, Neuroligin 1 was downregulated by 47% (±6%) and 33% (±6%) at P7 and P10, respectively, while β-catenin was downregulated by 34% (±9%) at P7, but comparably expressed at P10 relative to the wild-type control mice. At P7, NCAM expression was comparably expressed to wild-type mice, but decreased in expression by 36% (±4%) at P10 [Fig. 3(B)]. Temporal changes in the expression levels of these adhesion proteins in the null mice from lower than normal to higher were also correlated with the collapse of the meandering OC innervation along the OHC soma to the basal pole (Fig. 1) and consequent hypertrophy of the synaptic terminal observed approximately from P18 through adulthood. One-way ANOVA analysis of the full time series for each protein revealed significant expression level changes. A Tukey post-hoc multiple comparisons test also revealed significant changes among the protein expression levels across age relative to the wild type levels, indicating abnormal fluctuations of these proteins during the time of active synapse formation.
Altered Expression of Presynaptic Active Zone and Vesicle Associated Proteins in α9 Null Mice
It has been demonstrated that the OC terminals are neurochemically complex, and co-express numerous neurotransmitters such as ACh, GABA, and peptides within the same terminal (Maison et al., 2003). Thus, it becomes important for understanding whether any non-cholinergic-based OC function could be spared following the α9 null mutation and to determine whether proteins involved in the release of these transmitters may be altered in expression by loss of the postsynaptic nAChR. We therefore examined expression of various presynaptic active zone proteins and synaptic vesicle binding proteins by immunohistochemistry and quantitative western blot analysis of the α9 null mice (Figs. 4 and 5).
Figure 4.
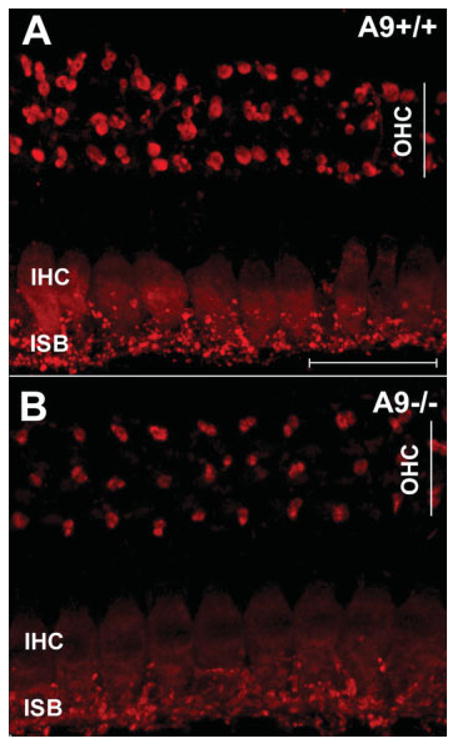
Decrease in CSP immunoreactivity in IHCs and efferent terminals of α9 null mice. (A) CSP immunostaining of adult wild-type cochleae (whole mount configuration) confirms previous reports of the expression in OC terminals at the base of the OHCs, and immunoreactivity of the IHC soma and the inner spiral bundle (ISB), a region basal to the IHC that contains a mixture of afferent and efferent fibers. (B) CSP immunostaining in the α9 null mice reveal less intense immunopositive OC terminals under OHCs, and a sharp reduction of expression in the IHC soma and the inner spiral bundle. Scale bar 25 μm applies to both panels. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Figure 5.

Loss of α9 nAChR results in changes in presynaptic active zone proteins and synaptic vesicle binding proteins. (A) Total cochlear lysates from α9 null and wild-type control mice at P7, P10, and adult (2 months) ages were probed by western blotting with antibodies against trans-synaptic and signaling proteins as indicated. Western blots reveal that loss of α9 nAChR activity results in marked changes in expression of presynaptic SNARE proteins, scaffolding proteins and synaptic vesicle binding proteins in the α9 nulls at early postnatal and adult ages. Additionally, lysates from P7 and adult cochleae were probed to assess expression levels of the high affinity choline transporter ChT1. (B–D) Time series bar graphs show percent change in protein expression levels in α9 nulls relative to the wild type as discussed for Figure 4. These data demonstrate that α9 nAChR is required for the normal expression of presynaptic components that are essential for normal synapse assembly and function. Although synaptophysin levels remain essentially unchanged in the α9 null mice (D), the other proteins probed fall into distinct families of expression profiles. Thus, synapsin and CASK both begin expression at levels lower than that of wild type values and steadily increase in expression levels to adult ages (B,D). Mint-1, CSP, and ChT1 are normally expressed at early ages and then decrease to approximately 50% or less relative to wild type values by adult ages (B,C). SNAP-25 is the only protein probed that starts at higher than normal levels of expression (C). One-way ANOVA and Tukey multiple comparisons analyses revealed significant changes in expression over time in α9 null mice for all proteins examined except for synaptophysin, which was just outside of the significance margin (*p < 0.05; **p < 0.005; ***p < 0.0001).
Immunohistochemical staining for synaptophysin, a protein involved in vesicle trafficking and exocytosis, has been used to stain the OC terminals contacting cochlear hair cells previously (Vetter et al., 1999, 2007; Murthy et al., 2009) without any apparent change in staining intensity between null and wild-type mice. CSP, a vesicle associated protein that functions as a chaperone coordinating protein interactions during vesicle exocytosis (Seagar et al., 1999; Dawson-Scully et al., 2000) has also been shown to be expressed in OC nerve terminals and IHCs of the cochlea (Eybalin et al., 2002). Immunostaining of adult wild-type cochleae for CSP (Fig. 4) confirmed previous reports of its expression pattern. Immunostaining of α9 null cochleae revealed a reduced intensity of OC terminal immunostaining under OHCs for CSP [Fig. 4(A,B)]. In addition, presumptive OC nerve fibers innervating the spiral ganglion cell processes below the IHCs in the inner spiral bundle (ISB) and the IHC soma were less intensely stained for CSP in the α9 null mice compared to wild types.
To better quantify protein expression changes with regard to presynaptic function that take place following loss of α9 gene expression, a quantitative immunoblot analysis was performed. Temporal expression dynamics of: (1) CSP; (2) SNAP25, involved in synaptic vesicle docking and fusion, and previously shown to be expressed by OC nerve terminals (Eybalin et al., 2002); (3) synaptic vesicle binding proteins synapsin and synaptophysin, (Safieddine and Wenthold, 1999; Eybalin et al., 2002); and (4) the presynaptic active zone scaffolding proteins MINT, and CASK [Fig. 5(A–D)], was assessed. As a group, these proteins organize synaptic vesicle pools, regulate recruitment of vesicles to the readily releasable pool, and are essential for vesicle fusion and exocytosis (Olsen et al., 2006; Schoch and Gundelfinger, 2006; Atasoy et al., 2007).
The expression of synaptic vesicle binding proteins synapsin and synaptophysin showed a differential temporal expression pattern in the α9 null mice [Fig. 5(A–D)]. At P7 and P10, the expression of the scaffolding protein MINT1 in α9 null mice was comparable to that of wild type [Fig. 5(A,B)], but in the adult α9 null mice, expression of MINT was reduced by 73% (±6%) relative to wild-type control. One-way ANOVA analysis revealed a highly significant difference across ages for the α9 null mice (p = 0.0071). Expression of CASK was decreased in the α9 null mice by approximately 49% (±5%) at P7, but was normal to wild type by P10, and increased by 95% (±18%) in adult α9 null mice compared to wild-type mice [Fig. 5(B)]. One-way ANOVA analysis revealed that the alteration in CASK expression across ages in the α9 nulls compared to that of wild type mice was statistically significant (p = 0.0113).
Expression of CSP in the α9 nulls compared to wild type mice at P7 and P10 [Fig. 5(C)] were downregulated by 19% at P7, but increased to the wild type level at P10. However, at adult ages, the α9 nulls demonstrated a 70% (±14%) downregulation in CSP expression [Fig. 5(C)]. One-way ANOVA analysis revealed a significant change (p = 0.0350) across ages. This was reflected in the decreased CSP immunostaining in the IHCs and lighter CSP immunoreactivity of OC terminals under OHCs of α9 nulls [Fig. 4(A,B)]. Expression of SNAP25 was upregulated at P7 by 75% (±15%) that then was downregulated by 56% (±3%) in P10, and 61% (±9%) in adult α9 nulls relative to wild type levels [Fig. 5(C)]. One-way ANOVA analysis demonstrated a highly significant change in expression across the ages examined (p = 0.0001).
Synapsin 1 was downregulated by 77% (±5%) at P7 in α9 null mice relative to that of the wild type level [Fig. 5(A,D)]. Synapsin 1 expression in the null mice was comparable to wild type at P10, and significantly (albeit variably) upregulated twofold, by 108% (±51%) in adult α9 null mice [Fig. 5(D)]. A one-way ANOVA revealed a significant change in synapsin 1 expression across ages (p = 0.0160). Synaptophysin levels were comparable between null and wild-type mice in adult stages, but upregulated (albeit variably) by 23% (±35%) at P10. Expression at P7 in null mice was comparable to wild type levels [Fig. 5(D)]. ANOVA analyses revealed that the changes in Synaptophysin expression in the nulls were not significant over time.
ACh release during rapid cholinergic transmission between neurons at central and peripheral synapses requires constant synthesis of ACh from choline and acetyl-CoA by choline acetyltransferase and concentration of ACh into synaptic vesicles by the vesicular ACh transporter (VAT). The rate-limiting step for neurotransmitter synthesis is choline uptake from the extracellular fluid which is mediated by high affinity choline transporter (ChT1). We have previously demonstrated that VAT immunostaining intensity of OC terminals at OHCs in α9 null mice is similar to wild-type mice (Vetter et al., 1999; Fig. 5). However, to gain further insight into the cholinergic differentiation of presynaptic OC terminals in α9 null mice, we examined the expression of another cholinergic marker ChT1, previously shown to be localized to presynaptic OC terminals in mice (Bergeron et al., 2005). Quantitative immunoblot analyses revealed a modest increase (15% ± 10%) in ChT1 expression in P7 α9 null mice and a significant decrease (77% ± 10%) in ChT1 expression in adult null mice [Fig. 5(B)].
The N-Cadherin-CREB Binding Protein Transcriptional Activation Pathway Is Altered in α9 Null Mice
N-Cadherin, the expression of which was found to be abnormal in the α9 null mice [Figs. 2 and 4(A–C)], undergoes proteolytic processing that releases its C-terminal tail into the cytoplasm, where it interacts with CREB binding protein (CBP) to target it for proteosomal degradation, thereby regulating CREB induced gene expression (Marambaud et al., 2003). Perturbation of this pathway in α9 null mice may represent a mechanism by which some of the above mentioned proteins undergo expression changes in the α9 null mice.
As detailed in Figures 3(A,B) and 6(B), quantitative immunoblot analysis revealed that while N-cadherin expression levels in the α9 null mice decreased by 48% (±4%) at P7 compared to wild types, there was a concomitant increase in CBP expression by 21% (±9%) in the α9 nulls compared to wild-type mice. Conversely, adult α9 null mice exhibited a 47% (±20%) upregulation of N-Cadherin while at the same time exhibiting a 77% (±13%) decrease in total CBP levels compared to wild-type expression [Fig. 6(B)]. Given the apparent coordinated change in CBP and N-Cadherin expression and the previously demonstrated functional link between these proteins, we then performed a two-way ANOVA to determine whether a statistically significant interaction between N-Cadherin and CBP takes place with age in the α9 null mice. This analysis revealed a highly significant interaction between levels of protein expression and age (p = 0.0005). Thus, given the known role of N-Cadherin proteolytic processing in controlling CBP expression levels, it is highly probable that the altered CBP expression found in the α9 null mice are caused by the abnormal and dynamic shifts in N-Cadherin expression following loss of α9 gene expression. Interestingly, the expression level of Presenilin, part of the γ-secretase associated with N-Cadherin processing, was unchanged in the α9 null mice compared to wild type mice [Fig. 6(A,B)]. The expression level of total CREB in P7 α9 null mice was decreased 32% (±3%), but was slightly (albeit variably) increased 26% (±20%) compared to wild-type mice in adults. While the Presenilin changes were not significant, the changes in expression level of total CREB in the α9 null mice did reach significance (p = 0.0459) by Student’s t-test analysis. Variability of total CREB expression combined with the large number of cells expressing CREB in the cochlea make it difficult to interpret the results, but may reflect compensatory changes.
Figure 6.

Loss of α9 nAChR activity perturbs a synapse specific CREB pathway. (A) Western blots of total cochlear lysate from α9 null and wild-type control mice at P7 and adult (2 months) ages, probed with antibodies against CREB pathway proteins as indicated. (B) Quantitative western blot analyses reveal a temporal interplay between N-Cadherin (data replotted from Fig. 4 for ease of comparison) and CREB Binding Protein (CBP) at P7 and adult ages in α9 null cochleae normalized to wild type expression levels. Note the downregulation of N-cadherin and upregulation of CBP at P7, followed by an increase of N-Cadherin expression and a reduction of CBP at the adult stage. Expression of presenilin, the γ-secretase responsible for final cleavage of the C-terminal tail of N-Cadherin and its release into the cytosol, does not significantly change across the ages examined. Total CREB expression increases in adult α9 null mice. Student’s t-test indicated significant changes with age for all proteins except presenilin. A two-way ANOVA analysis was run for the N-Cadherin and CBP data, given published functional links between these two proteins. This analysis demonstrated a highly significant interaction between expression of N-Cadherin and CBP across ages (p = 0.0005).
Efferent Innervation Is Increased in α9 KI Mutant Cochleae
The α9 null mice are incapable of ACh-induced activity [see Fig. 3(B) in (Vetter et al., 2007)], and thus our hypothesis for the myriad changes observed in the α9 nulls has been that altered ACh activity drives the structural changes observed. Our data indicate that this can be the product of indirect consequences following loss of ACh-induced activity, as shown by alteration of the N-Cadherin/CBP signaling pathway. If loss of cholinergic activity is responsible for the observed changes, and not simply loss of the protein upon which protein–protein interactions take place regardless of activity, then inducing a functional change in the physiological activity pattern of nAChRs containing the α9 subunit might also lead to alterations in innervation. To begin to assess this important point, we have used the recently described α9KI model (Taranda et al., 2009), which has been shown to have normal α9, α10, and SK2 gene expression, but abnormal nAChR physiological activity.
Cochleae isolated from α9KI mice were immunostained with synaptophysin, and the counts and areas of immunopositive terminals were measured at the mid-turn at 6 weeks of age (Fig. 7). As we have demonstrated previously in wild-type mice, OC terminals typically occurred as clusters under OHCs [Fig. 7(A)]. Knock-in mutants showed an increase (p < 0.02) in the mean number of efferent terminals per OHC from 2 (±0.08, n = 538 cells) in wild-type mice, to 2.9 (±0.03, n = 614) and as many as six terminals on some OHCs [arrows in Fig. 7(B,C)] in α9KI mice. The mean volume of terminals per OHC nearly doubled (p < 0.02) from 36.9 μm3 (±1.35, n = 95 cells) in the wild-type to 63.9 μm3 (±1.82, n = 119 cells) in the homozygous knock-in mice [Fig. 7(D)]. Thus, combining data detailing the structural consequences that follow both ablation of the α9 gene, and genetic manipulation of the gene to induce increased activity, the hypothesis that ACh-induced activity plays a role in OC synapse formation at OHCs is strengthened.
Figure 7.

α9KI L9′T point mutation results in profound innervation changes to OHCs. (A) Wild-type mice exhibit the expected innervation pattern of medial olivocochlear fibers to the OHCs. Each OHC receives, on average two synaptic terminals (arrow), but instances of greater innervation complexity can be found. (B) In the α9KI mice, each OHC is contacted by a larger than normal contingent of olivocochlear terminals (arrows). On average, three terminals contact each OHC. (C) Counting terminals under OHCs of wild type and KI mice reveals the percent occurrence of different numbers of efferent contacts. Fifty percent of the wild-type OHCs are contacted by two terminals, while 50% of the KI mouse OHC’s are contacted by three terminals. Three times as many α9KI OHCs are contacted by four terminals compared to wild-type mice. (D) An estimation of the total volume of the synaptic terminals under OHCs of wild type and KI mice reveals that synapses of the KI occupy greater than twice the volume of those of wild-type mice. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
DISCUSSION
Altered Activity via the Hair Cell nAChR, But Not the Loss of nAChR Structure, Drives Observed Changes in Protein Expression and Presynaptic Structural Defects
Structural consequences following α9 gene ablation may be the product of physical loss of the α9 nAChR subunit, and thus the loss of some unknown physical interactions involving the α9 subunit that are normally required for proper synapse assembly, or the result of loss of nAChR-mediated activity, because expression of α9 is an absolute requirement for nAChR-mediated activity in hair cells (Elgoyhen et al., 2001; Vetter et al., 1999, 2007). To test whether nAChR activity drives change in the synaptic structure/innervation observed in α9 null mice, we examined the α9KI mouse, previously shown to express hyperactive nAChRs (Taranda et al., 2009). Despite normal nAChR gene transcript levels, the α9KI mouse line exhibits OC hyper-innervation of hair cells. One may posit that the α9KI mouse expresses greater levels of α9 protein on the OHC membrane than one is led to believe from the gene transcript data that may explain the hyperinnervation in these mice, thus suggesting that mere presence of the protein, and not the abnormal nAChR activity plays a role in synaptic dynamics. While a pore-dead α9 protein may be required to directly test this possibility, one may also assess this possibility indirectly by examining the electrophysiological behavior of the α9KI OHCs. In the α9KI mouse, OHCs express significantly lower peak amplitude (207 ± 25 pA in wild types and 129 ± 11 pA in α9KI mice) at −90 mV holding potential, while possessing twice the charge transfer of wild type channels due to the prolonged τdecay (four times greater than wild type). These data suggest that the α9KI mutation, which is resident within one of the rings of amino acids responsible for the gating mechanism within the pore of the channel, does not alter α9 surface expression compared to wild-type mice. To begin elucidating mechanisms underlying the structural changes induced by altered ACh activity, we carried out a series of experiments designed to probe protein expression levels of key components of the OC synapse in the α9 null mice.
α9 nAChRs Regulate Expression of Adhesion Molecules
From the earliest stage examined, the medial olivocochlear axons behave abnormally in α9 nulls, indicating that α9 nAChR subunit expression is required for normal spatiotemporal development and stabilization of olivocochlear synaptic innervation to hair cells during periods of synaptic refinement. This stands in sharp contrast to the well-known model of synapse formation at the neuromuscular junction (NMJ) demonstrating that global block of cholinergic activity does not alter synaptic structure (Balice-Gordon and Lichtman, 1993, 1994; Sanes and Lichtman, 2001). Such contrasts may highlight fundamental differences in synapse formation/maintenance between structures that do or do not express a basal lamina. However, a comparison between the OHC OC synapse and the NMJ is still of interest, given the expression of a number of molecules in the OHCs known to be important for synapse formation at the NMJ, such as MUSK and Rapsyn (Osman et al., 2008). Additionally, it has been shown that loss of another adhesion molecule, NCAM, from the NMJ results in altered transmission vesicle dynamics, and endocytotic machinery (Polo-Parada et al., 2001). Finally, parallels can be drawn between our study and a recent study by Krishnaswamy and Cooper (2009), which demonstrated that neuronal nicotinic synapses are maintained in the absence of activity, albeit with structural changes. Similar to the hypertrophy of synaptic boutons under OHCs in α9 nulls, genetic ablation of α3 nAChR in superior cervical ganglia (SCG) of mice results in larger presynaptic terminals and presynaptic clusters. Additionally, ChT1 expression is lost following α3 deletion in the SCG, just as we demonstrate for the adult OHC-OC terminal synapse.
α9 nAChR Activity Influences Olivocochlear Presynaptic Terminal Differentiation
Trans-synaptic adhesion molecules hold the postsynaptic density and the presynaptic cytomatrix around the active zone in register, and hence bidirectionally coordinate synapse differentiation (reviewed in Ziv and Garner, 2004), target specification and stabilization of early synaptic contacts. Previous studies have shown that deficiencies in CAM expression result in altered synaptic innervation and stabilization (Inoue and Sanes, 1997; Lee et al., 2001; Waites et al., 2005). Neuroligins provide inductive signals for active zone formation and trigger initial stages of glutamatergic as well as nicotinic synapse formation in vitro (Dean et al., 2003; Conroy et al., 2007). Trans-synaptic adhesion proteins and their associated cytoplasmic signaling proteins also play a role in pre-synaptic development, and are involved in synaptic vesicle dynamics (Uchida et al., 1996; Moscoso et al., 1998; Rafuse et al., 2000; Nishimura et al., 2002; Sytnyk et al., 2002; Bruses, 2000, 2006), and induce pre-synaptic differentiation in-vitro (Scheiffele et al., 2000; Dean et al., 2003) by stimulating clustering of synaptic vesicles, and active zone formation by binding to scaffolding proteins CASK, MINT1 and synaptotagmin (Petrenko et al., 1991; Hata et al., 1993). Because altered cell adhesion may result in abnormal presynaptic structure, we quantified the expression levels of these molecules in the α9 null mice.
Our data demonstrate changes in pre-synaptic active zone proteins in the α9 null mice. In addition to the SNARE protein SNAP25, and CSP, which interacts with SNARE proteins, structural molecules of the active zone such as CASK (a CaMKII domain containing MAGUK) and MINT1 (found in complex with CASK and Veli, (Zhen and Jin, 2004; Ziv and Garner, 2004; Olsen et al., 2006), are perturbed in the α9 null mice despite the fact that the genetic manipulation involves only the postsynaptic hair cells, and not the central neurons that give rise to the efferent boutons in question. Interestingly, Synapsin, a synaptic vesicle binding protein that anchors the synaptic vesicles to actin and is involved in regulation of reserve pool of synaptic vesicles (Chi et al., 2001; Bloom et al., 2003) is upregulated in adult α9 null mice compared to wild-type mice. These results indicate that the misregulation of active zone proteins and synaptic vesicle binding proteins in the α9 null mice might cause aberrant organization and recruitment of synaptic vesicles in the active zone, thus impairing general neurotransmission.
Finally, the high affinity choline transporter ChT1 is crucial for the continual resynthesis and release of ACh during sustained neurotransmission, and it has been implicated as a marker for final differentiation of OC fibers in the cochlea (Bergeron et al., 2005). In α9 null mice, ChT1 expression is upregulated at P7, but significantly downregulated in adults compared to age-matched wild-type mice. Lack of ChT1 in adult α9 null cochleae suggests an aberrant maintenance of presynaptic cholinergic identity in the absence of α9 nAChR activity. Similarly, lack of cholinergic post-synaptic receptor activity in the SCG has also been shown to decrease ChT1 expression (Krishnaswamy and Cooper, 2009). Because vesicular ACh transporter was previously shown to be qualitatively normal in expression in the α9 null mice (Vetter et al., 1999), downregulation of ChT1 may indicate a lack of synaptic release of ACh, and with it, any cotransmitters normally also released. Together with the data demonstrating altered presynaptic proteins involved in vesicle dynamics, this may indicate a general loss of presynaptic function in the α9 null mice.
α9-Mediated nAChR Activity Influences Innervation Patterns to OHCs
nAChR activity may normally be required for generation of permissive zones within which OC terminals can be maintained. Normal ACh-induced activity may generate an as yet undefined permissive signal in the OHCs that modulates adhesion molecule expression and thereby establishes zones within which the normal poly-innervation of OHCs can form by limiting the size of the presynaptic terminal. In the α9 null, the ACh-induced activity is missing, and hence also the instruction needed to delineate normal synaptic boundaries, resulting in hypertrophy of a single bouton. Under different nAChR activity conditions such as generated by the α9KI mutation, these permissive signals may be greater, either in distribution area or expression level, leading to a hyper-innervation of OHCs. While the identity of the permissive signal(s) is currently unclear, what is apparent is that formation and maintenance of OC terminals on OHCs is a competitive process between OC terminals that intimately involves nAChR-based activity. Because each OC bouton under an OHC derives from a separate axon (Wilson et al., 1991; Warr et al., 1997), cooperativity for establishing synapses seems to rule under certain circumstances (heightened nAChR activity), while competition rules under other conditions (complete silencing of nAChR-based activity). These observations stand in contrast to the classic work done at the neuromuscular junction (Balice-Gordon and Lichtman, 1994), but largely reflect recent results from non-NMJ cholinergic synapses such as that in the SCG (Krishnaswamy and Cooper, 2009).
CREB Pathway and OC Nicotinic Synapse Formation
Evidence from studies on ciliary ganglion and hippocampal neurons indicates that nicotinic signaling regulates gene expression by altering the intracellular calcium concentration, which activates downstream CREB signaling pathways (Chang and Berg, 2001; Hu et al., 2002; Dajas-Bailador and Wonnacott, 2004). In light of these reports, and given that the hair cell nAChRs are highly permeable to Ca2+ (Jagger et al., 2000; Katz et al., 2000; Weisstaub et al., 2002), and that most of the synaptic proteins misexpressed in the α9 null cochleae, with the exception of MINT1 and SNAP25, are regulated by CREB transcriptional pathway (as revealed by a CREB target gene database search, http://natural.salk.edu/CREB/), we investigated the possibility that the CREB signaling pathway becomes perturbed in the α9 null mice.
N-Cadherin expression is dynamically altered with age and is correlated with the behavior of the OC fibers over the course of early postnatal innervation in the α9 null mice. As schematically summarized in Figure 8, proteolytic processing of N-Cadherin occurs (Marambaud et al., 2003) and is carried out by the matrix metalloproteinase ADAM10, and the γ-secretase complex (which includes Presenilin 1), releasing a C-terminal tail fragment (CTF2) of N-Cadherin into the cytoplasm. CTF2 binds the essential transcriptional co-activator CBP and promotes its ubiquitination and proteosomal degradation, thereby decreasing cellular CBP and therefore CBP-regulated CREB-mediated transcription. Because nAChRs containing the α9 subunit are highly calcium permeable, and altered CBP levels were detected in the α9 null mice, it is possible that calcium signals play a role in regulating these processes. It has been demonstrated that calpain, a calcium-sensitive proteinase, is expressed in hair cells (Wang et al., 2007), and can digest the CTF2, thus regulating the amount of CTF2 available for CBP binding. Thus, a fine balance between OC nAChR activity, generation and degradation of the N-Cadherin CTF2, and CBP proteosomal processing may exist in cochlear hair cells. Since CREB induces the transcription of many components of the synaptic machinery (Lonze and Ginty, 2002), (including all proteins that we have examined except MINT1 and SNAP25) proteolytic processing of N-Cadherin in the α9 null mice may serve as an aberrant signal resulting in abnormal synapse maturation via suppression of CREB-mediated transcriptional activity (Rubio et al., 2005). While not addressed in this study, one may hypothesize that the α9KI mouse with its hyperactive nAChRs may induce higher calpain activity, leading to degradation of the CTF2 before it is able to bind and ultimately degrade CBP. In this scenario, the α9KI may induce prolonged periods of excessive CBP/CREB transcriptional activity in a manner similar to the early stages of the α9 null mice, leading to the hyper-innervation demonstrated in Figure 7. Further studies are required to address this hypothesis.
Figure 8.

Summary of potential mechanism by which altered OC nAChR activity may drive structural and functional changes observed in cochleae of α9 null mice. N-Cadherin is cleaved first by ADAM10, a metaloprotease enzyme, to generate an N-term fragment of N-Cadherin. Following MMP cleavage, the γ-secretase/PS1 complex cleaves the C-terminal fragment (CTF) to release the CTF2 fragment into the cytoplasm. CTF2 has been shown to be proteolytically cleaved by calcium-activated calpain, but if intact can also bind CREB-binding protein, where the entire complex is then ubiquitinated and sent for proteosomal degradation. Loss of CBP via this pathway would alter CREB-associated transcriptional activity. In α9 nAChR subunit null mice, less calcium enters the hair cells as a result of loss of ACh-inducible activity (dashed lines below nAChR complex). This would result in a loss of calcium-induced calcium release (CICR) from the subsynaptic cistern, and a decrease in calpain activity. The result would be an accumulation of CTF2 upon the normal γ-secretase activity acting on the over expressed levels of N-Cadherin (thick solid arrow). Accumulated CTF2 in the null mice binds a greater proportion of CBP, sending it off for degradation and at the same time inhibiting translocation of normal levels of CBP into the nucleus. Hyperactivity of the α9KI channel may induce greater calpain activity as a result of excessive calcium signaling, resulting in a greater proportion of CTF2 degradation in adults before it binds to CBP. Thus, CBP levels may be higher in adults, reflecting the early stage innervation of the α9 nulls in which exuberant contacts were observed between the OC fibers and the OHCs, and potentially explaining the hyper-innervation demonstrated in Figure 7. Dashed lines with arrows represents downregulated pathways; thin lines with arrows represents normally active pathways; thick lines with arrows represents upregulated overactive pathways; black elements represents downregulated or missing proteins (e.g. α9 nAChR subunit, CBP); gray elements represents upregulated proteins.
Acknowledgments
ABE is supported by a Howard Hughes International Scholars Grant. This article is submitted as partial fulfillment of requirements for a Ph.D. to VM.
Contract grant sponsor: NIH; contract grant number: R01DC006258.
Contract grant sponsor: The Tufts Center for Neuroscience Research; contract grant number: P30 NS047243.
Contract grant sponsor: Mass Eye and Ear Infirmary Eaton-Peabody Lab; contract grant number: P30 DC05209.
References
- Arikkath J, Reichardt LF. Cadherins and catenins at synapses: Roles in synaptogenesis and synaptic plasticity. Trends Neurosci. 2008;31:487–494. doi: 10.1016/j.tins.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atasoy D, Schoch S, Ho A, Nadasy KA, Liu X, Zhang W, Mukherjee K, Nosyreva ED, Fernandez-Chacon R, Missler M, Kavalali ET, Südhof TC. Deletion of CASK in mice is lethal and impairs synaptic function. Proc Natl Acad Sci USA. 2007;104:2525–2530. doi: 10.1073/pnas.0611003104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balice-Gordon RJ, Lichtman JW. In vivo observations of pre- and postsynaptic changes during the transition from multiple to single innervation at developing neuromuscular junctions. J Neurosci. 1993;13:834–855. doi: 10.1523/JNEUROSCI.13-02-00834.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balice-Gordon RJ, Lichtman JW. Long-term synapse loss induced by focal blockade of postsynaptic receptors. Nature. 1994;372:519–524. doi: 10.1038/372519a0. [DOI] [PubMed] [Google Scholar]
- Bamji SX, Shimazu K, Kimes N, Huelsken J, Birchmeier W, Lu B, Reichardt LF. Role of beta-catenin in synaptic vesicle localization and presynaptic assembly. Neuron. 2003;40:719–731. doi: 10.1016/s0896-6273(03)00718-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron AL, Schrader A, Yang D, Osman AA, Simmons DD. The final stage of cholinergic differentiation occurs below inner hair cells during development of the rodent cochlea. JARO. 2005;6:401–415. doi: 10.1007/s10162-005-0018-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom O, Evergren E, Tomilin N, Kjaerulff O, Low P, Brodin L, Pieribone VA, Greengard P, Shupliakov O. Colocalization of synapsin and actin during synaptic vesicle recycling. J Cell Biol. 2003;161:737–747. doi: 10.1083/jcb.200212140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce LL, Christensen MA, Warr WB. Postnatal development of efferent synapses in the rat cochlea. J Comp Neurol. 2000;423:532–548. doi: 10.1002/1096-9861(20000731)423:3<532::aid-cne14>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Bruses JL. Cadherin-mediated adhesion at the inter-neuronal synapse. Curr Opin Cell Biol. 2000;12:593–597. doi: 10.1016/s0955-0674(00)00137-x. [DOI] [PubMed] [Google Scholar]
- Bruses JL. N-cadherin signaling in synapse formation and neuronal physiology. Mol Neurobiol. 2006;33:237–252. doi: 10.1385/MN:33:3:237. [DOI] [PubMed] [Google Scholar]
- Chang KT, Berg DK. Voltage-gated channels block nicotinic regulation of CREB phosphorylation and gene expression in neurons. Neuron. 2001a;32:855–865. doi: 10.1016/s0896-6273(01)00516-5. [DOI] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan TA. Synapsin dispersion and reclustering during synaptic activity. Nat Neurosci. 2001;4:1187–1193. doi: 10.1038/nn756. [DOI] [PubMed] [Google Scholar]
- Conroy WG, Nai Q, Ross B, Naughton G, Berg DK. Postsynaptic neuroligin enhances presynaptic inputs at neuronal nicotinic synapses. Dev Biol. 2007;307:79–91. doi: 10.1016/j.ydbio.2007.04.017. [DOI] [PubMed] [Google Scholar]
- Dajas-Bailador F, Wonnacott S. Nicotinic acetylcholine receptors and the regulation of neuronal signalling. Trends Pharmacol Sci. 2004;25:317–324. doi: 10.1016/j.tips.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Dawson-Scully K, Bronk P, Atwood HL, Zinsmaier KE. Cysteine-string protein increases the calcium sensitivity of neurotransmitter exocytosis in Drosophila. J Neurosci. 2000;20:6039–6047. doi: 10.1523/JNEUROSCI.20-16-06039.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean C, Scholl FG, Choih J, DeMaria S, Berger J, Isacoff E, Scheiffele P. Neurexin mediates the assembly of presynaptic terminals. Nat Neurosci. 2003;6:708–716. doi: 10.1038/nn1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgoyhen AB, Johnson DS, Boulter J, Vetter DE, Heinemann S. Alpha 9: An acetylcholine receptor with novel pharmacological properties expressed in rat cochlear hair cells. Cell. 1994;79:705–715. doi: 10.1016/0092-8674(94)90555-x. [DOI] [PubMed] [Google Scholar]
- Elgoyhen AB, Vetter DE, Katz E, Rothlin CV, Heinemann SF, Boulter J. alpha10: A determinant of nicotinic cholinergic receptor function in mammalian vestibular and cochlear mechanosensory hair cells. Proc Natl Acad Sci USA. 2001;98:3501–3506. doi: 10.1073/pnas.051622798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eybalin M, Renard N, Aure F, Safieddine S. Cysteine-string protein in inner hair cells of the organ of Corti: Synaptic expression and upregulation at the onset of hearing. Eur J Neurosci. 2002;15:1409–1420. doi: 10.1046/j.1460-9568.2002.01978.x. [DOI] [PubMed] [Google Scholar]
- Hata Y, Davletov B, Petrenko AG, Jahn R, Sudhof TC. Interaction of synaptotagmin with the cytoplasmic domains of neurexins. Neuron. 1993;10:307–315. doi: 10.1016/0896-6273(93)90320-q. [DOI] [PubMed] [Google Scholar]
- Inoue A, Sanes JR. Lamina-specific connectivity in the brain: Regulation by N-cadherin, neurotrophins, and glycoconjugates. Science. 1997;276:1428–1431. doi: 10.1126/science.276.5317.1428. [DOI] [PubMed] [Google Scholar]
- Jagger DJ, Griesinger CB, Rivolta MN, Holley MC, Ashmore JF. Calcium signalling mediated by the 9 acetylcholine receptor in a cochlear cell line from the Immortomouse. J Physiol. 2000;527:49–54. doi: 10.1111/j.1469-7793.2000.t01-1-00049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang Y-N, Jung Y-S, Lee SH, Moon C-H, Kim C-H, Baik EJ. Calpain-mediated N-Cadherin proteolytic processing in brain injury. J Neurosci. 2009;29:5074–5084. doi: 10.1523/JNEUROSCI.6178-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz E, Elgoyhen AB, Gomez-Casati ME, Knipper M, Vetter DE, Fuchs PA, Glowatzki E. Developmental regulation of nicotinic synapses on cochlear inner hair cells. J Neurosci. 2004;24:7814–7820. doi: 10.1523/JNEUROSCI.2102-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz E, Verbitsky M, Rothlin CV, Vetter DE, Heinemann SF, Elgoyhen AB. High calcium permeability and calcium block of the alpha9 nicotinic acetylcholine receptor. Hear Res. 2000;141:117–128. doi: 10.1016/s0378-5955(99)00214-2. [DOI] [PubMed] [Google Scholar]
- Krishnaswamy A, Cooper E. An activity-dependent retrograde signal induces the expression of the high-affinity choline transporter in cholinergic neurons. Neuron. 2009;61:272–286. doi: 10.1016/j.neuron.2008.11.025. [DOI] [PubMed] [Google Scholar]
- Lee CH, Herman T, Clandinin TR, Lee R, Zipursky SL. N-cadherin regulates target specificity in the Drosophila visual system. Neuron. 2001;30:437–450. doi: 10.1016/s0896-6273(01)00291-4. [DOI] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–623. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- Maison SF, Adams JC, Liberman MC. Olivocochlear innervation in the mouse: Immunocytochemical maps, crossed versus uncrossed contributions, and transmitter colocalization. J Comp Neurol. 2003;455:406–416. doi: 10.1002/cne.10490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marambaud P, Wen PH, Dutt A, Shioi J, Takashima A, Siman R, Robakis NK. A CBP binding transcriptional repressor produced by the PS1/epsilon-cleavage of N-cadherin is inhibited by PS1 FAD mutations. Cell. 2003;114:635–645. doi: 10.1016/j.cell.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Moscoso LM, Cremer H, Sanes JR. Organization and reorganization of neuromuscular junctions in mice lacking neural cell adhesion molecule, tenascin-C, or fibroblast growth factor-5. J Neurosci. 1998;18:1465–1477. doi: 10.1523/JNEUROSCI.18-04-01465.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller D, Mendez P, De Roo M, Klauser P, Steen S, Poglia L. Role of NCAM in spine dynamics and synaptogenesis. Neurochem Res. 2008 doi: 10.1007/s11064-008-9653-5. [DOI] [PubMed] [Google Scholar]
- Murthy V, Maison SF, Taranda J, Haque N, Bond CT, Elgoyhen AB, Adelman JP, Liberman MC, Vetter DE. SK2 channels are required for function and long-term survival of efferent synapses on mammalian outer hair cells. Mol Cell Neurosci. 2009;40:39–49. doi: 10.1016/j.mcn.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura W, Yao I, Iida J, Tanaka N, Hata Y. Interaction of synaptic scaffolding molecule and beta-catenin. J Neurosci. 2002;22:757–765. doi: 10.1523/JNEUROSCI.22-03-00757.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen O, Moore KA, Nicoll RA, Bredt DS. Synaptic transmission regulated by a presynaptic MALS/Liprin-alpha protein complex. Curr Opin Cell Biol. 2006;18:223–227. doi: 10.1016/j.ceb.2006.02.010. [DOI] [PubMed] [Google Scholar]
- Osman AA, Schrader AD, Hawkes AJ, Akil O, Bergeron A, Lustig LR, Simmons DD. Muscle-like nicotinic receptor accessory molecules in sensory hair cells of the inner ear. Mol Cell Neurosci. 2008;38:153–169. doi: 10.1016/j.mcn.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrenko AG, Perin MS, Davletov BA, Ushkaryov YA, Geppert M, Sudhof TC. Binding of synaptotagmin to the alpha-latrotoxin receptor implicates both in synaptic vesicle exocytosis. Nature. 1991;353:65–68. doi: 10.1038/353065a0. [DOI] [PubMed] [Google Scholar]
- Polo-Parada L, Bose CM, Landmesser LT. Alterations in transmission, vesicle dynamics, and transmitter release machinery at NCAM-deficient neuromuscular junctions. Neuron. 2001;32:815–828. doi: 10.1016/s0896-6273(01)00521-9. [DOI] [PubMed] [Google Scholar]
- Rafuse VF, Polo-Parada L, Landmesser LT. Structural and functional alterations of neuromuscular junctions in NCAM-deficient mice. J Neurosci. 2000;20:6529–6539. doi: 10.1523/JNEUROSCI.20-17-06529.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio ME, Curcio C, Chauvet N, Bruses JL. Assembly of the N-cadherin complex during synapse formation involves uncoupling of p120-catenin and association with presenilin 1. Mol Cell Neurosci. 2005;30:611–623. [PubMed] [Google Scholar]
- Safieddine S, Wenthold RJ. SNARE complex at the ribbon synapses of cochlear hair cells: Analysis of synaptic vesicle- and synaptic membrane-associated proteins. Eur J Neurosci. 1999;11:803–812. doi: 10.1046/j.1460-9568.1999.00487.x. [DOI] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW. Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat Rev Neurosci. 2001;2:791–805. doi: 10.1038/35097557. [DOI] [PubMed] [Google Scholar]
- Scheiffele P, Fan J, Choih J, Fetter R, Serafini T. Neuroligin expressed in nonneuronal cells triggers pre-synaptic development in contacting axons. Cell. 2000;101:657–669. doi: 10.1016/s0092-8674(00)80877-6. [DOI] [PubMed] [Google Scholar]
- Schoch S, Gundelfinger ED. Molecular organization of the presynaptic active zone. Cell Tissue Res. 2006;326:379–391. doi: 10.1007/s00441-006-0244-y. [DOI] [PubMed] [Google Scholar]
- Seagar M, Leveque C, Charvin N, Marqueze B, Martin-Moutot N, Boudier JA, Boudier JL, Shoji-Kasai Y, Sato K, Takahashi M. Interactions between proteins implicated in exocytosis and voltage-gated calcium channels. Philos Trans R Soc Lond B Biol Sci. 1999;354:289–297. doi: 10.1098/rstb.1999.0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons DD. Development of the inner ear efferent system across vertebrate species. J Neurobiol. 2002;53:228–250. doi: 10.1002/neu.10130. [DOI] [PubMed] [Google Scholar]
- Simmons DD, Mansdorf NB, Kim JH. Olivocochlear innervation of inner and outer hair cells during postnatal maturation: Evidence for a waiting period. J Comp Neurol. 1996;370:551–562. doi: 10.1002/(SICI)1096-9861(19960708)370:4<551::AID-CNE10>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Simmons DD, Moulding HD, Zee D. Olivocochlear innervation of inner and outer hair cells during postnatal maturation: An immunocytochemical study. Brain Res Dev Brain Res. 1996b;95:213–226. doi: 10.1016/0165-3806(96)00084-3. [DOI] [PubMed] [Google Scholar]
- Sytnyk V, Leshchyns’ka I, Delling M, Dityateva G, Dityatev A, Schachner M. Neural cell adhesion molecule promotes accumulation of TGN organelles at sites of neuron-to-neuron contacts. J Cell Biol. 2002;159:649–661. doi: 10.1083/jcb.200205098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taranda J, Maison S, Ballestero J, Katz E, Savino J, Vetter DE, Boulter J, Liberman MC, Fuchs P, Elgoyhen E. A point mutation in the cochlear hair cell nicotinic acetylcholine receptor prolongs efferent inhibition to enhance noise protection. PLoS Biol. 2009;7:e1000018. doi: 10.1371/journal.pbio.1000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida N, Honjo Y, Johnson KR, Wheelock MJ, Takeichi M. The catenin/cadherin adhesion system is localized in synaptic junctions bordering transmitter release zones. J Cell Biol. 1996;135:767–779. doi: 10.1083/jcb.135.3.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetter DE, Katz E, Maison SF, Taranda J, Turcan S, Ballestero J, Liberman MC, Elgoyhen AB, Boulter J. The alpha10 nicotinic acetylcholine receptor subunit is required for normal synaptic function and integrity of the olivocochlear system. Proc Natl Acad Sci USA. 2007;104:20594–20599. doi: 10.1073/pnas.0708545105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetter DE, Liberman MC, Mann J, Barhanin J, Boulter J, Brown MC, Saffiote-Kolman J, Heinemann SF, Elgoyhen AB. Role of alpha9 nicotinic ACh receptor subunits in the development and function of cochlear efferent innervation. Neuron. 1999;23:93–103. doi: 10.1016/s0896-6273(00)80756-4. [DOI] [PubMed] [Google Scholar]
- Walsh EJ, McGee J, McFadden SL, Liberman MC. Long-term effects of sectioning the olivocochlear bundle in neonatal cats. J Neurosci. 1998;18:3859–3869. doi: 10.1523/JNEUROSCI.18-10-03859.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Pignol B, Chabrier PE, Saido T, Lloyd R, Tang Y, Lenoir M, Puel JL. A novel dual inhibitor of calpains and lipid peroxidation (BN82270) rescues the cochlea from sound trauma. Neuropharmacology. 2007;52:1426–1433. doi: 10.1016/j.neuropharm.2007.02.007. [DOI] [PubMed] [Google Scholar]
- Warr WB, Boche JB, Neely ST. Efferent innervation of the inner hair cell region: Origins and terminations of two lateral olivocochlear systems. Hear Res. 1997;108:89–111. doi: 10.1016/s0378-5955(97)00044-0. [DOI] [PubMed] [Google Scholar]
- Weisstaub N, Vetter DE, Elgoyhen AB, Katz E. The alpha9alpha10 nicotinic acetylcholine receptor is permeable to and is modulated by divalent cations. Hear Res. 2002;167:122–135. doi: 10.1016/s0378-5955(02)00380-5. [DOI] [PubMed] [Google Scholar]
- Whitlon D, Zhang X, Pecelunas K, Greiner M. A temporospatial map of adhesive molecules in the organ of Corti of the mouse cochlea. J Neurocytol. 1999;28:955–968. doi: 10.1023/a:1007038609456. [DOI] [PubMed] [Google Scholar]
- Wilson JL, Henson MM, Henson OW., Jr Course and distribution of efferent fibers in the cochlea of the mouse. Hear Res. 1991;55:98–108. doi: 10.1016/0378-5955(91)90096-r. [DOI] [PubMed] [Google Scholar]
- Zhen M, Jin Y. Presynaptic terminal differentiation: Transport and assembly. Curr Opin Neurobiol. 2004;14:280–287. doi: 10.1016/j.conb.2004.05.013. [DOI] [PubMed] [Google Scholar]
- Ziv NE, Garner CC. Cellular and molecular mechanisms of presynaptic assembly. Nat Rev Neurosci. 2004;5:385–399. doi: 10.1038/nrn1370. [DOI] [PubMed] [Google Scholar]