

Abstract
Long-lived plasma cells (PCs) and memory B cells (Bmem) constitute the cellular components of enduring humoral immunity, whereas short-lived PCs that rapidly produce Ig correspond to the host's need for immediate protection against pathogens. In this study we show that the innate affinity of the BCR for Ag imprints upon naive B cells their differentiation fate to become short-or long-lived PCs and Bmem. Using BCR transgenic mice with varying affinities for Ag, naive B cells with high affinity lose their capacity to form germinal centers (GCs), develop neither Bmem nor long-lived PCs, and are destined to a short-lived PC fate. Moderate affinity interactions result in hastened GC responses, and differentiation to long-lived PCs, but Bmem remain extinct. In contrast, lower affinity interactions show tempered GCs, producing Bmem and affinity-matured, long-lived PCs. Thus, a continuum of elementary to comprehensive humoral immune responses exists that is controlled by inherent BCR affinity.
Short- and long-lived humoral immunity are two mechanistically distinct arms of the Ab-dependent immune response that rely on the emergence of different types of B cells, the expression of different B cell transcription factors, and the production of Ig with varying affinities, specificities, and isotypes. Furthermore, these two pathways serve unique functions, with the former providing rapid protective Ab to limit infection, whereas the latter facilitates the persistent generation of affinity-matured, isotype-switched Abs.
Although numerous reports have characterized the molecular and cellular events that influence the affinity of Ag-experienced B cells within germinal centers (GCs)4 (1–3), few have addressed whether the affinity of the BCR on naive B cells influences their differentiation fate. It is known that engagement of the BCR to a T cell-dependent (TD) Ag results in several oligoclonal B cells giving rise to GCs as early as five days after encountering the Ag 1, 2). Within GCs, activated B cells proliferate rapidly and undergo somatic hypermutation (SHM) coupled with isotype switching that engenders BCRs with varying specificities to the activating Ag, an event that is critical for the generation of long-lived plasma cells (PCs) and memory B cells (Bmem). B cells that exit the GC show varying affinity for Ag and are subject to selection (4–7). Those that demonstrate enhanced affinity, relative to their lower affinity counterparts will terminally differentiate to PCs and Bmem (1, 3). This suggests that whereas a heterogeneous population of B cells specific for a single Ag can become activated, the enhanced specificity of the humoral immune response observed late after immunization is due to the competitive selection of higher affinity clones that ultimately become long-lived. Indirect evidence in support of this hypothesis comes from studies demonstrating that the affinity of serum Abs steadily increases over time after immunization (8–10).
A number of studies have evaluated the role of BCR affinity in the recruitment of Ag-specific B cells into GCs using Ig transgenic (Tg) systems that vary in affinity (11–13). In sum, these studies show that low-affinity B cells can form GCs, and there does not appear to be an affinity threshold for entry into the GC. Furthermore, once entering the GC, these B cells have the capacity to become Bmem and PCs. Hence, the appraisal of BCR affinity in B cell fate has been drawn from the analyses of GC B cells via histology and SHM techniques. These pioneering studies have provided a strong basis for affinity-independent cell entry into GCs, with clonal competition among higher affinity clones driving the selection of post-GC B cells. However, it has yet to be established whether the heterogeneity of BCR affinity at the time of Ag exposure ultimately governs humoral immune longevity through controlling the emergence of short- and/or long-lived PCs and Bmem.
To evaluate whether the intrinsic BCR signal strength controls the humoral fate of naive B cells, we analyzed the in vivo differentiation capacity of heterogeneous B cell populations from Ig Tg mice that recognize the same Ag yet vary in their average affinity. These Tg mice share a rearranged VH chain that confers recognition to the Ag, (4-hydroxy-3-nitrophenyl)acetyl (NP), and faithfully recapitulate the early humoral immune response to T cell-independent (TI) and TD Ag in terms of Tg B cell proliferation, Ab production, and the capacity to undergo isotype-switching (14, 15). Following passive transfer, low-affinity binding of Ag to the BCR results in protracted GC formation, SHM, and the generation of Bmem and long-lived bone marrow (BM) PCs. Moderate BCR affinity hastens the GC response, reduces Bmem production, and steers the immune response toward the production of long-lived PCs. Finally, high-affinity BCR engagement induces B cell proliferation and Ab secretion yet terminates GC formation, the generation of Bmem, and long-lived BM PCs. Furthermore, the loss of long-lived PCs results from an inability of Ag-experienced B cells, which are direct precursors to PCs (PCpre), to terminally differentiate into end-stage Ab-secreting cells. These data indicate that, within an Ag-specific polyclonal population, the overall intrinsic BCR affinity controls B cell fate and survival by determining the differentiation program that provides the host with a range of acute to persistent Ab protection.
Materials and Methods
Mice
Eight- to 10-wk-old quasi-monoclonal (QM) and C57B6-Ly5.2 congenic mice were maintained in the specific pathogen-free animal facility at Dartmouth Medical School (Lebanon, NH). Generation of the QM mice has been previously described (16); they have been backbred to the C57B6 JH −/−Jκ −/− strain for nine generations to generate Tg Ig that bear the λ L chain. To generate Tg B cells that bear κ L chains, QM mice were backbred to wild-type C57B6. All of the experiments involving the use of mice were performed in accordance with protocols approved by the Animal Care and Use Committee of the University of Virginia (Charlottesville, VA).
Adoptive transfer and immunization
For each primary (1°) transfer, the donor cells were prepared from the spleens of QM κ or λ mice, RBC were lysed with ammonium-chloride Tris buffer, and T cells were depleted using complement-mediated lysis with mAbs to Thy1.2, CD4, and CD8. Percentages of Tg+ B cells were determined by flow cytometry with fluorescently labeled mAbs specific for the VH17.2.25 Id and B220, and then 3 × 106 splenic Tg B cells (B220+Id+) were injected i.v. into Ly5.2 recipients. One day following transfer, recipients were immunized i.p. with 100 μg of either NP36-keyhole limpet hemocyanin (KLH) or (4-hydroxy-5-iodo-3-nitrophenyl)acetyl (NIP)23-KLH (Biosearch Technologies) emulsified in CFA. Recipient spleen and BM were harvested 5–30 days following challenge. Tg PCs and PCpre were purified to >95% by magnetic bead selection based on CD138 and Ly5.1 expression as previously described (17). PCpre (1 × 105) were transferred i.v. into secondary (2°) naive Ly5.2 recipients, and after 2 wk anti-NP IgGa ELISPOT assays were performed on total BM cells. For detecting Bmem, immune splenic B cells (30 × 106) from 1° recipients that received Tg B cells were transferred into 2° recipients that had been KLH primed. After 24 h, mice were challenged i.p. with either alum or 10 μg of NP-KLH precipitated in alum, which had been previously determined not to elicit an endogenous response (14). Sera were collected and assayed for anti-NP IgG by ELISA 7 days later. All experiments consisted of 3–4 mice per group.
Abs and reagents
Monoclonal Abs to B220, IgG1a, IgG1b, IgG2aa, IgMa, CD45.2 (Ly5.1), Ly6C, CD138, GL7, and CD38 were purchased from BD Biosciences. The VH17.2.25 Id-specific cell line R2348.8, was generously provided by Dr. T. Imanishi-Kari (Tufts University School of Medicine, Boston, MA). Peanut agglutinin (PNA) was purchased from Sigma-Aldrich. Purified rat IgG was used as an isotype control. Samples were collected on a BD FACSCalibur flow cytometer.
ELISA and ELISPOT
To measure Ab affinities, Tg Ig was purified from pooled serum of 6–8 naive Tg λ and κ mice using an anti-Id-coupled AminoLink Plus agarose column (Pierce) that binds all Tg Ig. Bound Ab was eluted with 0.2 M glycine (pH 2.3) and immediately neutralized with 0.5 M Tris (pH 7.6), and the concentration was determined by absorbance at 280 nm. Hapten inhibition assays were performed as previously described (18) using microtiter plates coated with serial dilutions (.005–10 μg/ml) of NP25-or NIP23-BSA in 1% BSA PBS. Purified Tg λ or κ Ig was added at 2 μg per well with or without soluble monomeric NP hapten (3-nitro-4-hydroxy-phenylacetyl aminocaproic acid; Biosearch Technologies) or its NIP analog at 10-fold dilutions from 10−2 to 10−8 M. Total bound Tg Ig was detected by anti-mouse Ig alkaline phosphatase. Affinities were estimated by measuring the free hapten concentration that is required for 50% inhibition of Ig binding to the solid phase. Serum Ab titers and the number of splenic or BM PCs were quantified by incubating serum or cells in NP25-BSA- or NP4-BSA-coated Millipore plates to measure low- and high-affinity anti-NP, respectively, as previously described (15). Allotype-specific IgG1a, IgG2aa, IgMa, and IgG1b mAbs were used to detect Abs derived from Tg or endogenous B cells, respectively. Data shown are the mean ± SD of three or four mice per group and are representative of at least three independent experiments.
Confocal image analysis
Five-micrometer frozen spleen sections were acetone fixed and stained in 1% BSA PBS with fluorochrome-coupled rat anti-mouse mAbs. Samples were analyzed on a Bio-Rad 1024 confocal microscope. Images are representative of three experiments with >30 scanned images of three or four mice per group (objective magnification, ×10).
Somatic hypermutation analysis
Thirty days following the immunization of transfer recipients, total BM PCs were labeled with CD138 mAb and purified by magnetic bead selection (Miltenyi Biotec). cDNA from purified RNA was amplified using VH17.2.25-specific primers and sequenced as previously described (16). Mutations from germline sequence were verified by comparing sense and antisense strand sequences. The error rate of avian myeloblastosis virus high fidelity DNA polymerase is 1–2 × 10−6 misincorporations per base pair. Background levels of mutation rate were determined by amplification of VH17.2.25 from naive Tg B cells, with 16 sequences showing no mutations.
Real-time RT-PCR
cDNA was generated from RNA isolated from Id-purified splenic Tg B cells of day 7 immune recipients (Qiagen). Amplification of cDNA using SYBR Green (Applied Biosystems) was performed on a Bio-Rad iCycler. Sequences of PCR primers and conditions are available from the authors on request. The relative level of expression for each primer target was calculated by the formula 2−(ΔΔCT) × 1000, where ΔΔCT = (CT gene of interest − CT β-actin in experimental sample) − (CT gene of interest − CT β-actin in a no-template control sample), where CT is cycle threshold.
Results
B cells specific for a single Ag vary in BCR affinity
Tg B cells from QM mice bear Ag specificity to NP and, when transferred into Ly5.2 recipients, can be serologically identified by the expression of the VH17.2.25 Id, IgGa allotype and the Ly5.1 congenic marker (14–16). The original QM mice were bred onto C57B6 Jκ −/− mice, thereby allowing only the production of high-affinity NP-specific λ-bearing Ab (Tg λ). Upon adoptive transfer and challenge, Tg λ B cells proliferate, differentiate, isotype switch, and produce Ig in response to NP-conjugated TI and TD Ags (14, 15). Because these prior studies demonstrated that Tg λ B cells rapidly respond to Ag in vivo with the peak of expansion at day 5 after immunization, we hypothesized that their hastened pace of differentiation was controlled by their high BCR affinity. To address issues of how a high intrinsic BCR affinity influenced the in vivo behavior of Tg λ B cells, we produced mice that expressed a lower intrinsic affinity. To this end, QM mice were intercrossed nine generations with wild-type C57B6 mice to generate a strain that had κ L chain-bearing (Tg κ), VH17.2.25-expressing B cells (Fig. 1A). Because it has previously been shown that homozygous (VHT/VHT) QM λ mice have an enlarged marginal zone B cell compartment (19), flow cytometric analysis was performed to compare splenic B cells of QM λ and κ mice that serve as donor cells. Findings revealed that splenic B cells of both Tg strains had equivalent proportions of follicular (B220+CD23highCD21intIgMint) (where int is intermediate) and marginal zone (B220+CD23lowCD21highIgMhigh) subsets, albeit higher than non-Tg, as previously defined (20). Frequencies of newly formed immigrating B cells (B220+CD23−CD21−IgMint) were found to be normal compared with controls.
FIGURE 1.
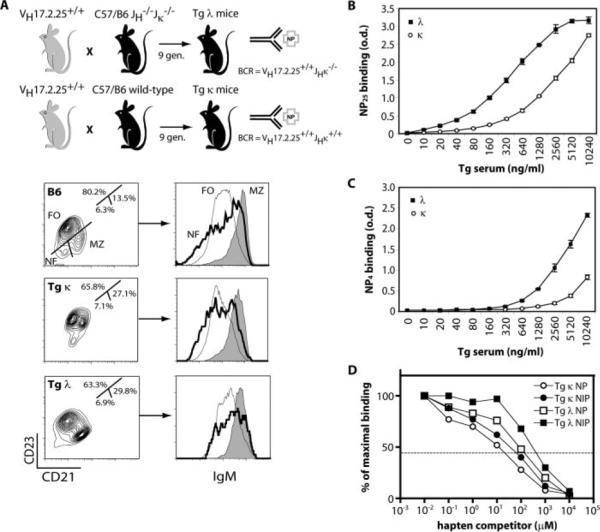
Tg λ and κ mice show disparate BCR affinities. A, Generation strategy and B cell phenotype of Tg λ and κ mice. Flow cytometric analysis of donor splenic B cells revealed that Tg mice have an equivalent yet higher frequency of marginal zone (MZ; B220+CD23lowCD21highIgMhigh) compared with follicular (FO; B220+CD23highCD21intIgMint) subsets than non-Tg mice. Newly formed (NF; B220+CD23−CD21−IgMint) immigrants were unchanged. Dot plots shown are gated on the B220+ population. B and C, Chromatographically purified VH17.2.25 Id+ Igs from the sera of naive Tg λ and κ mice were standardized and serial dilutions were tested for their ability to bind a high or low hapten ratio (NP25 vs NP4) by ELISA. D, A competitive ELISA was performed using the purified serum Tg Ig to measure average affinities of λ and κ Abs for NP and NIP as described in Materials and Methods. Hapten inhibition curves are shown that represent the percentage of maximal Ab binding to a constant amount of Ag when serial dilutions of competing soluble NP or NIP monomeric hapten (μM) is present. Measurements of the IC50 values are depicted by the dotted horizontal line (NP, Tg κ = 1.86 × 10−3 M, Tg λ = 2.21 × 10−4 M; NIP, Tg κ = 4.69 × 10−4 M, Tg λ = 2.07 × 10−5 M). Results are from three independent experiments.
Chromatographically purified Tg Ig was generated from the serum of naive animals using an anti-Id-coupled agarose column that binds all Tg Ig isotypes and yet does not inhibit the recognition of NP. Total Tg λ Ig showed a greater capacity to bind NP at lower concentrations as compared with Tg κ Ig (Fig. 1B). The inherent higher average affinity of Tg λ Ig was further demonstrated by its binding capacity to a low number of NP haptens (Fig. 1C). Direct measurement of the average affinity of Tg Ig to NP and NIP was performed by hapten inhibition ELISA as previously described (18). This method calculates an affinity distribution by quantifying the proportion of Ab bound to a constant concentration of immobilized NP-BSA or NIP-BSA in the presence of log dilutions of competing soluble monomeric hapten. Thus, affinity here is the reciprocal concentration of a soluble NP or NIP haptenic competitor required for 50% inhibition of Tg Ig binding to immobilized Ag. Using this assay, inhibition curves were calculated to initially determine the IC50 values of Tg Ig for NP and were found to be 1.86 × 10−3 M (Tg κ; Fig. 1D, ◯) and 2.21 × 10−4 M (Tg λ; Fig. 1D, ◻). Previous studies have estimated the IC50 of Tg λ serum Abs to be 3 × 10−4 M for NP hapten (21). Thus, our measurement of the innate Tg λ BCR affinity for NP approximates those values observed by others. To further distinguish the range of affinity for Tg λ and κ Abs, the average affinity of Tg Ig to the NP analog NIP was measured. NIP has been previously shown to have a 15-fold higher affinity than NP for Tg λ B cells (21). The IC50 levels for NIP were estimated to be 4.69 × 10−4 M (Tg κ; Fig. 1D, ●) and 2.07 × 10−5 M (Tg λ; Fig. 1D, ∎). Therefore, the increase in relative affinities for NIP compared with NP is 11-fold for Tg λ Ig and 4-fold for Tg κ Ig. These results are not a product of affinity maturation in the VHDJH region of Tg λ Ig, as sequencing VH17.2.25 in the absence of Ag does not show SHM (data not shown). This system allows us to modulate BCR signal strength in graded affinities from 1.86 × 10−3 to 2.07 × 10−5 M depending on the source of Tg B cells and Ag.
Higher affinity B cells show accelerated proliferation and a loss in GC formation
To investigate whether varying BCR affinities impact humoral responses in vivo, primary transfers were performed using equal numbers of either QM κ or QM λ donor Tg B cells. Recipients were immunized i.p. with NP, NIP or CFA alone (naive), and the numbers of splenic Tg B cells were determined by flow cytometry at various days posttransfer. Fig. 2A demonstrates that regardless of the form of Ag, Tg λ B cells expand earlier than Tg κ B cells even though total cellular accumulation was equivalent between the two populations 7 days after immunization. Interestingly, profound differences between the phenotype of the transferred Tg λ and Tg κ B cells were observed in response to NIP (Fig. 2B). By day 7 after NP challenge of recipients, both Tg λ and κ B cells exhibited a GC phenotype as measured by positive staining with the GC-specific marker, PNA (22). GC B cells were also observed 10 days after immunization but at fewer numbers that correspond with decreased total Tg B cells. In contrast to NP challenge, transferred Tg λ B cells failed to acquire a GC phenotype after NIP immunization even though expansion was similar to that of Tg κ B cells. This was not due to accelerated or delayed GC reaction kinetics, as Tg λ GC B cells were not observed on either day 5 or day 10 following challenge. These data indicate that a range in BCR signal strength exists for GC differentiation that exceeds this signaling threshold as observed in the case of Tg λ B cells engaging NIP, which results in expansion without ensuing GC responses. Thus, based on our measurements of the average affinity of naive Tg λ and κ B cells and those previously reported for NP, Tg BCR affinities ranging from 1.86 × 10−3 to 3 × 10−4 M allow GC responses. Increasing the BCR affinity with NIP, which has an IC50 of 2.07 × 10−5 (Fig. 1D), surpasses this permissible signal strength for GC differentiation.
FIGURE 2.

Adoptively transferred Tg λ B cells show accelerated proliferation and a failure to acquire GC phenotype and exhibit altered levels of B cell maturation-related gene expression in vivo. A, Donor Tg B cells were quantified by flow cytometry from recipient spleens on days 5, 7, and 10 after immunization with NP-KLH (NP), NIP-KLH (NIP), or CFA alone (naive). B, Ex vivo flow cytometric analysis on spleen cells from NP or NIP immune recipients. Fluorescence intensity of the GC marker PNA on Tg cells (B220+Id+) is shown by open histograms. Naive controls are shown in shaded histograms. C, Relative levels of mRNA expression of the indicated genes was measured by real time RT-PCR on purified donor Tg B cells from day 7 immune recipient spleens. The data are given as mean ± SD (n = 3) and are representative of three experiments.
BCR affinity influences the expression of transcription factors that control B cell differentiation
We next investigated whether BCR affinity influences the expression of a panel of B cell maturation-related genes known to be critically involved in controlling PC and Bmem differentiation. Real time RT-PCR was performed on splenic Tg λ or κ B cells that were anti-Id purified from day 7 immune recipients. Results showed that Pax5 expression was reduced in both transferred Tg λ and κ B cell populations regardless of the form of Ag as compared with naive controls (Fig. 2C). Because Pax5 is expressed throughout B cell ontogeny until the PC stage (23, 24), the reduced expression of Pax5 suggested that other genes involved in B cell activation and differentiation may be altered. Thus, we compared the expression of the transcription factors BCL-6 and Bach2, which are critical for GC responses (25–28). The expression profiles of Bach2 and BCL-6 correlated with the presence of a GC phenotype. Transferred Tg κ B cells that exhibit GC responses regardless of Ag stimulation showed increased levels of both genes, whereas Tg λ B cells showed elevated levels of expression in response to NP but not to NIP.
We further investigated the expression levels of transcription factors that drive the terminal differentiation of B cells to Ab-secreting cells. Heightened levels of Blimp-1 (29, 30) and the spliced isoform of XBP-1 (31, 32) were observed from immune-transferred Tg B cells regardless of Ag. Activation-induced cytidine deaminase (AID), which is critical for class-switching and SHM (33), was significantly heightened in Tg κ B cells responding to NP and NIP. These results corresponded with the production of serum Ag-specific IgG1a and IgG2aa by Tg κ B cells (Fig. 3). In contrast, Tg λ B cells do not significantly up-regulate AID expression regardless of Ag and is consistent with the poor titers of serum IgG1a and the frequency of PCs. Production of IgG2aa from Tg λ B cells was also significantly reduced compared with Tg κ B cells, with a pronounced decrease in response to NIP challenge. Diminished levels of IgG from Tg λ B cells were not due to an inability to respond to Ag, because they produced high titers of anti-NP IgMa upon NP and NIP immunization. Together, these results indicate that innate BCR affinity determines early differentiation events of Ag-reactive B cells by initiating distinct transcriptional programs that can direct follicular GC vs non-GC responses and isotype switching.
FIGURE 3.
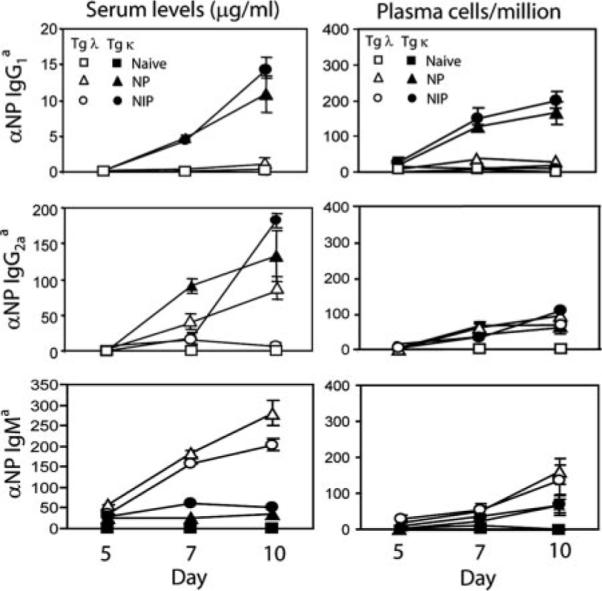
Serum IgG1 and IgG2a titers as well as the frequency of splenic PCs produced by transferred Tg λ B cells are reduced early on in the humoral immune response. Tg κ or λ cells were adoptively transferred and recipients immunized with NP, NIP, or CFA alone (naive). Serum and spleens from recipients were collected at days 5, 7, and 10 following challenge. Left panels show total levels of Tg NP-specific serum IgG1, IgG2a, and IgM as measured by ELISA using allotype-specific detection mAbs. Right panels show splenic Tg anti-NP IgG1, IgG2a, and IgM PCs quantified by ELISPOT from the same recipients. The data shown are mean ± SD (n = 3) and are representative of three experiments.
Higher affinity B cells fail to undergo a memory B cell differentiation program
To verify that the differential display of genes in immune Tg B cells functionally influenced cellular fate, adoptive transfer experiments into 2° recipients were conducted to measure Bmem formation. Levels of total and high-affinity anti-NP IgG1 from the various groups of mice are shown in Table I. Mice that received immune splenic B cells from 1° recipients, in which the Tg λ B cells were adoptively transferred, produced significantly low titers of high-affinity NP-specific IgG1a upon challenge. In contrast, 2° hosts, which were given B cells from 1° recipients that received Tg κ B cells, generated considerable levels of Ab when rechallenged. As an internal positive control, recall responses from the endogenous Bmem compartment (IgG1b allotype) generated during primary immunization was robustly detected after recall challenge. Because low-affinity IgG1 Bmem can be generated even in the absence of GC formation (34), we tested whether Tg λ B cells could make secondary IgG1 responses with low affinity for NP. The results demonstrated that Tg λ B cells produced 2-fold more NP30- to NP3-binding IgG1, yet levels were dramatically reduced compared with Tg κ B cells. Secondary high and low anti-NP IgG2aa responses showed comparable results (data not shown). Thus, while both Tg λ and κ B cell populations acquire a GC phenotype in response to NP Ag, differences in their overall BCR affinity significantly alter the differentiation program of post-GC B cells. Here, only those Tg B cells that exhibit an affinity for Ag below 2.21 × 10−4 M, as in the case of Tg κ B cells, mature into the long-lived Bmem pool.
Table I.
BCR affinity determines memory B cell responsesa
| NP3 Binding |
NP30 Binding |
|||||
|---|---|---|---|---|---|---|
| Tg B Cells | 1° Recipients | 2° Recipients | IgG1a | IgG1b | IgG1a | IgG1b |
| λ | Naive | Naive | NDb | ND | ND | ND |
| λ | Naive | Immune | ND | 0.56 ± 0.35 | ND | 2.4 ± 2.3 |
| λ | Immune | Immune | 0.82 ± 0.18 | 9.1 ± 1.7 | 1.8 ± 0.53 | 24.3 ± 4.0 |
| κ | Naive | Naive | ND | ND | ND | ND |
| κ | Naive | Immune | ND | ND | 0.38 ± 0.19 | 2.8 ± 1.8 |
| κ | Immune | Immune | 6.4 ± 0.9 | 9.5 ± 1.9 | 8.1 ± 1.6 | 27.1 ± 2.4 |
Splenic B cells from naive and NP-immune 1° recipients were adoptively transferred into carrier-primed 2° recipients and challenged with alum (naive) or NP-KLH alum (immune). After 7 days, titers of high and low affinity serum IgG1a or IgG1b allotype levels were measured by ELISA to detect Tg-derived or endogenous Ig, respectively. The results are expressed as mean μg/ml ± SD from three independent experiments.
ND, Not detected (<0.2 μg/ml).
Generation of PCs and their precursors is controlled by BCR affinity
We and various other groups have identified post-GC B cells that are direct precursors to PCs (17, 35–37) and that reside in the BM long term, are self-renewing, and do not secrete Ig but can mature into end-stage PCs (17). In contrast to mature BM PCs, PCpre retain expression of surface IgG, Ly6C, and CD45 (17). To determine whether differences in the average affinity of Tg λ and κ B cells influences the formation of PCpre and PCs, equal numbers of Tg B cells were transferred into primary recipients and immunized with Ag. After 3 wk, the generation of PCpre in recipient BM was measured. Results showed that PCpre (Ly5.1+IgG+Ly6C+) were derived from both transferred Tg λ and κ B cells regardless of immunization protocol (Fig. 4, A and B). Strikingly, however, PCpre express altered levels of CD138 expression depending on antigenic challenge. Tg λ PCpre from NP immune recipients uniformly express CD138. In contrast, Tg λ PCpre generated in response to NIP showed a complete loss of CD138 expression.
FIGURE 4.
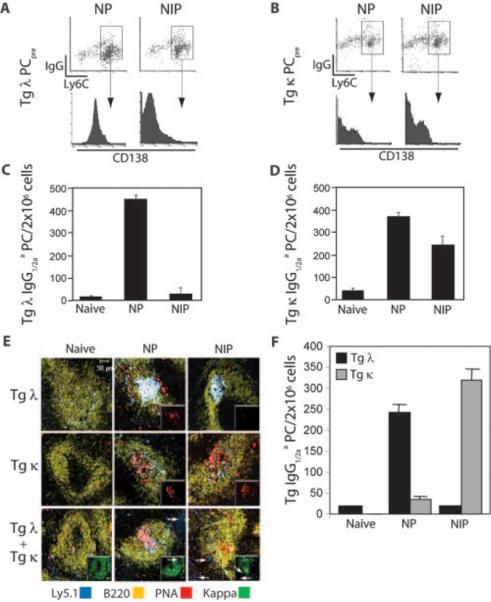
BCR affinity determines the generation of long-lived BM PCs in noncompeting and competing environments. A and B, Recipient mice received either Tg λ (A) or Tg κ (B) donor B cells and were immunized with NP or NIP. Three weeks later, BM cells were analyzed by flow cytometry for fluorescence intensity of CD138 on PCpre (Ly5.1+sIgG+Ly6C+). C and D, PCpre, shown in A and B, were sort purified and transferred into naive 2° recipients. Two weeks later, the capacity of transferred Tg PCpre to produce anti-NP IgG1/2aa BM PCs was quantified by ELISPOT. E, Equal numbers of Tg λ and κ B cells were transferred into 1° recipients singly or combined. Recipients were immunized with NP, NIP, or CFA alone (naive). Confocal image analysis of recipient spleens was performed 7 days later using fluorescently labeled mAbs to Ly5.1, B220, κ, and PNA. Insets in upper two rows show PNA staining only, whereas insets in lower row show κ staining only. F, Tg λ and κ B cells were cotransferred into 1° recipients and immunized with NP, NIP, or CFA (naive). After 30 days, the frequency of BM PCs produced from transferred Tg B cells was quantified by ELISPOT to dually detect anti-NP IgG1/2aa+κ+- or IgG1/2aa+λ+-secreting cells. Data shown are representative of four experiments.
To test whether the difference in CD138 expression on PCpre directly correlated with their capacity to produce terminally differentiated PCs, equal numbers of Ly5.1 purified PCpre from immune BM were transferred into naive 2° recipients, and the number of BM Tg PCs (anti-NP IgG1/2aa) was quantified after 2 wk. Tg λ PCpre generated in response to NIP showed a dramatic loss when further differentiating into end-stage PCs (Fig. 4C). In contrast, Tg κ B cells generated PCpre that expressed CD138 and were functionally capable of producing BM PCs regardless of immunization (Fig. 4, B and D). These data indicate that the generation of long-lived BM PCs is regulated by the quality of PCpre that is formed, and the quality of PCpre is determined by BCR affinity.
Affinity-driven competition determines B cell fate
Adoptive transfer experiments with Tg B cell populations combined were performed to examine whether diverse BCR affinities influence B cell fate decisions at the initiation of an immune response. To this end, Ly5.2 mice received Tg λ and κ B cells either singly or combined and were challenged with NP or NIP. Because both Tg populations express Ly5.1, the presence or absence of κ staining was used to discriminate Tg κ B cells (Ly5.1+) from Tg λ B cells (Ly5.1+κ−). A vast proportion of endogenous murine B cells will also be κ+ but will be Ly5.1−. The transfer of single Tg B cell populations resulted in the expansion and the development of histologically apparent splenic GCs (PNA+) in response to NP (Fig. 4E, two upper middle panels, insets). In contrast to Tg κ B cells, Tg λ B cells expanded yet failed to generate GCs in response to NIP as measured by the lack of PNA binding (Fig. 4E, top right panel, inset). Of interest, NP immunization of recipients that received both Tg λ and κ B cells resulted in GC formation that was comprised of Tg λ B cells (Ly5.1+PNA+κ−), whereas Tg κ B cells localized to extrafollicular areas of the spleen (Fig. 4E, lower middle panel, white arrows). However, the inverse was observed when recipients were challenged with NIP, where GCs consisted of Tg κ B cells and Tg λ B cells were excluded (Fig. 4E, lower right panel, white arrows).
To determine whether GC exclusion of Tg B cells ultimately controls their fate, the development of long-lived BM PCs was examined when Tg B cell populations were cotransferred. Results showed that in NP immune recipients the frequency of BM PCs derived from Tg λ B cells is significantly greater than those originating from Tg κ B cells (Fig. 4F). In contrast, when BCR signal strength is increased with NIP, Tg λ B cells do not proceed to become long-lived BM PCs, a fate that was similarly observed when Tg λ B cells were singularly transferred (Fig. 4C). With NIP, however, Tg κ B cells continue to produce BM PCs. Thus, although competition does occur as demonstrated upon NP challenge, there is a limit to affinity-driven competition. Ag-experienced B cells exceeding a certain affinity threshold (≥2 × 10−5 M), as in the case of Tg λ B cells exposed to NIP, preclude the responding cells from entering a GC and are diverted to short-term responses.
BCR affinity determines the extent of affinity maturation
We next evaluated whether the initial BCR affinity influences affinity maturation of Ag-experienced Tg B cells. Because it has been shown that GC B cells begin to accumulate SHM to NP as early as day 7 postimmunization (8, 38, 39), splenic Tg B cells (B220+Id+) from day 7 immune recipients were tested for their ability to bind graded doses of PE-conjugated NP and NIP. As shown in Fig. 5A, not all naive Tg κ B cells bind NP at the highest concentration of 1/50 (1 μg) and significantly lose their binding capacity at 1/800 (62 ng) titration. Although the overall intrinsic affinity of Tg κ Ig is low, the bimodal distribution of NP binding likely reflects a spectrum of BCR affinity due to greater diversity within this Ig Tg heterogeneous population, with ~50 κ light chains available. In contrast, naive Tg λ B cells exhibit a unimodal capacity to bind NP owing to three functional λ light chains available. Interestingly, immune Tg λ B cells show a similar pattern of NP binding capacity and suggest that upon Ag exposure the average BCR affinity of these cells is not improved. NIP binding showed higher fluorescent intensities and yet a similar profile as expected. In contrast, immune Tg κ B cells bind NP with significantly greater intensity than either naive controls or immune Tg λ B cells, even at a 1/800 titration. This increased intensity in NP binding by immune Tg κ B cells is not due to differences in BCR surface expression, as both Tg B cell populations express equivalent levels of IgG. Titers of NIP binding to Tg κ B cells showed a similar pattern but exhibited more unimodal expression than with NP. This finding suggests that cells with varied affinities for NP are uniformly capable of binding Ag above a certain threshold. Together, these results imply that, within the Tg κ B cell population, some cells undergo affinity maturation upon an antigenic challenge that enhances the affinity of that Ab.
FIGURE 5.

Innate BCR affinity determines affinity maturation. A, Tg κ or Tg λ cells were adoptively transferred into 1° recipients and immunized with NP or CFA (naive). Spleens were removed at day 7 postimmunization, and transferred Tg B cells were stained with mAbs to B220, VH17.2.25 Id, IgG, and graded dilutions of fluorescently labeled NP or NIP. Histograms shown from flow cytometric analyses are gated on B220+Id+ Tg κ or λ cells. Surface IgG expression was equivalent between Tg λ and κ B cells (gray histogram, naive Tg B cells; black histogram, immune Tg B cells). B and C, SHM analysis of individual VH17.2.25 transcripts from Tg λ and κ BM PCs. Tg κ or λ B cells were adoptively transferred into 1° recipients and immunized with NP. After 4 wk, CD138+ BM PCs were purified by magnetic bead selection. cDNA from purified RNA was amplified using VH17.2.25-specific primers and transcripts were sequenced as described in Materials and Methods. Silent and replacement mutations are denoted by lowercase and uppercase nucleotides, respectively. Sequences shown are a composite of two independent experiments.
To identify SHM that could lead to improved affinity of immune Tg κ B cells, sequence analysis of individual VH17.2.25 transcripts was performed. Because Tg PCs lose membrane expression of allotype- and Id-marked Ig, long-lived BM PCs were isolated via CD138+ selection from 4-wk immune primary recipients that received either Tg λ or κ B cells (Fig. 5, B and C). This approach also serves to eliminate any selection bias based on BCR surface expression, because SHM could potentially alter anti-Id mAb binding. Of the 15 VH sequences derived from Tg κ PCs, there were 18 silent mutations and 59 replacement mutations. The percentage of replacement mutations within the framework and CDR of Tg κ PCs were 68 and 88%, respectively (Table II), and is consistent with a previous report (40). In contrast, 11 of 20 VH sequences derived from Tg λ PCs exhibited a germline sequence and nine sequences showing six silent and four replacement mutations. No SHM was observed within the CDRs of any sequences from Tg λ PCs. These results are consistent with the earlier finding that AID expression is virtually absent in Tg λ B cells (Fig. 2). The overall mutation frequency showed a significant difference between Tg λ and κ PCs, with a 5.8-fold increase in point mutations in the framework regions of the latter.
Table II.
Somatic hypermutation of long-lived bone marrow Tg plasma cells
| VH17.2.25 | Replacement | Silent | Percent Replacement (%) | Mutation Frequencya (%) |
|---|---|---|---|---|
| λ+ Plasma cells | ||||
| FRb | 4 | 6 | 40 | 0.14 (10/7200) |
| CDRc | 0 | 0 | 0 | 0 |
| κ+ Plasma cells | ||||
| FR | 30 | 14 | 68 | 0.81 (44/5400) |
| CDR | 29 | 4 | 88 | 0.61 (33/5400) |
| λ+ Naive B cells | 0 | 0 | 0 | 0 |
| κ+ Naive B cells | 0 | 0 | 0 | 0 |
Calculated by dividing the total number of mutations within the framework region or CDR by the total number of sequenced nucleotides from amplified PCR products (360 bp).
Number of mutations within framework regions (FR).
Number of mutations within CDRs.
Discussion
A central question of B cell biology has been to understand what controls GC development and commitment to a long-lived Bmem and/or PC fate. Although the impact of BCR affinity has been shown to influence B cell activation in vivo, the differences in the innate BCR affinity controlling B cell fate and lifespan remain unclear. The data presented here indicate that the intrinsic BCR affinity of naive B cells is one factor that regulates GC B cell differentiation and the subsequent development of short- or long-lived effector cells. The findings establish that there is a continuum of responses that is manifested by Ag-activated B cells and that continuum is at least partially controlled by BCR affinity. At one end of the spectrum, low-affinity engagement of the BCR results in protracted GC formation, SHM, and the generation of Bmem and long-lived PCs. This is characterized by the in vivo behavior of the Tg κ B cells. If the average BCR affinity is moderately increased, the tendency is to hasten the GC response, reduce the generation of Bmem, and direct the immune response toward the production of long-lived PCs. This scenario is represented by Tg λ B cells responding to NP challenge. Finally, even higher affinity interactions terminate GC formation and the generation of Bmem and long-lived PCs and are represented by NIP-activated Tg λ B cells.
It has previously been thought that low-affinity Ag-experienced B cells fail to respond because of an inability to reach an intrinsic affinity threshold or are lost during an immune response due to competition among B cells with higher affinity. This notion has arisen from earlier studies using Tg mice that vary in BCR affinity (7, 41, 42). Under conditions where competition is restricted, immune Tg mice with low BCR affinity (Kas 1.2 × 105 or 3 × 104 M−1) form GCs, produce Ig, and generate Bmem (13). However, these Tg B cells within GCs late in the immune response are replaced by Ag-experienced B cells expressing endogenous BCRs, presumably due to competition. This work indicates that a certain affinity threshold is not required for cell entry into early TD humoral immune responses. Our finding that low-affinity Tg κ B cells form GCs among a heterogeneous B cell population when singly transferred is in agreement with these results.
However, it remains unknown whether an oligoclonal B cell response – each responding to the same Ag yet varies in affinity – serves a specific biological purpose. We consider that noncompetitive and competitive pathways together coordinate a spectrum of Ab protection for the host. In this regard, the differentiation potential of an Ag-specific B cell is partially determined at the time it first encounters Ag before competition occurs. We show that low-affinity B cells not only are capable of responding to Ag but are critical for establishing complete participation in long-lived humoral immunity by generating Bmem and affinity-matured, long-lived BM PCs. Moderate- to high-affinity B cells serve a distinct function by providing long-term and immediate Ab protection from the establishment of long- and short-lived PCs, respectively. Furthermore, the cell fate of high-affinity B cells is independent of clonal competition. Although moderate affinity Tg λ B cells out-competed lower affinity Tg κ B cells in response to NP when cotransferred, the innate BCR affinity of the Tg λ B cells still determined their short-lived fate when exposed to NIP. Thus, within the BCR affinity continuum that permits entrance into B cell follicles and GC responses, a checkpoint exists that governs which long-lived effector B cells are created independently of Ag-driven selection. Although B cells with intrinsically high affinity may have a competitive advantage, their contribution to the longevity and specificity of the humoral immune response is dampened due to limited SHM and Bmem production, and function in a similar manner as the behavior of B cells that have engaged TI Ags. As the immune response matures, additional layers of Ab complexity would be provided.
How affinity influences GC responses and the longevity of the humoral immune response is unclear. One possibility is that signals mediated by the BCR are distinct between B cell subsets. For example, marginal zone B cells that constitutively exhibit properties of activation and are long-lived rapidly generate protective Abs to TI Ags as a first line of defense (20, 43). This is strikingly similar to the fate of Tg λ B cells, which we and others (19) have shown consist of a greater frequency of marginal zone B cells. Yet, differences in BCR affinity between marginal zone and follicular B cells cannot fully account for affinity-driven responses, because Tg κ B cells show a similar frequency of marginal zone and follicular B cell compartments to Tg λ B cells (Fig. 1). An alternative explanation for how affinity controls humoral immunity is that, during an immune response, circulating Abs with increasing affinity will compete for Ag and act as a feedback loop to temper B cell differentiation. Tarlinton and Smith (44) postulate that high-affinity GC B cells will preferentially differentiate to PCs until levels of circulating high-affinity Abs become saturating.
The impact of affinity on early TD responses has been previously shown using other Tg systems. Brink and coworkers (45) elegantly demonstrated that the initial BCR signal strength also controls extrafollicular PC development using the hen egg lysozyme (HEL) Ig system. Immunization with a panel of recombinant HEL-SRBC conjugates that varied in affinity showed >100-fold more splenic PCs in response to HEL Ag with moderate to high affinity compared with low-affinity HEL. However, a significant difference between these findings and our study is that the variation of Ag affinity did not affect early GC formation of HEL-binding B cells at the time point examined (day 5 postchallenge). One potential explanation for this disparity is that high affinity HEL could hasten or prematurely terminate the kinetics of GC responses. Whether the tempo of GC formation affects terminal differentiation to long-lived BM PCs and/or Bmem has not been examined in this system but would be of interest to test, because similar results were observed in an earlier study by Nussenzweig and colleagues (11) using the B1–8 Ig Tg system. This system, which uses the canonical or mutated B1–8 H chain that, when paired with λ light chains, confers low and high affinity for NP, respectively, produces GC responses to both TI (46) and TD (11) Ag regardless of affinity. Several reasons could account for the difference in GC B cell formation between the B1–8 and QM Tg systems. One possibility is that a certain number of Tg B cells are required to respond and compete for Ag to form GCs. B1–8 mice show that 3–5% of total B cells are λ+ and bind NP, whereas the remaining B cells are κ+ and do not bind to NP. In our system, QM Tg B cells represent only ~0.5% of the B cell population upon adoptive transfer in wild-type recipients whose endogenous B cell compartment can respond to NP and NIP. Thus, perhaps the loss of GC formation by QM Tg λ B cells in response to NIP results from not meeting the required number of cells. An alternative explanation would be that differences in the amount and/or persistence of Ag may influence GC initiation and maintenance. Interestingly, B1–8 high-affinity GC B cells showed little SHM compared with B1–8 low-affinity GC B cells (11). Although the long-lived fate of B1–8 Tg B cells was not assessed, these results are consistent with our observation that BM PCs derived from high-affinity Tg λ B cells accumulated fewer mutations. Together, our findings here corroborate the role of BCR affinity in early TD responses but further reveal that the initial interaction of BCR and Ag impacts the fate of responding B cells to long-lived Bmem and BM PCs.
At the molecular level, how BCR affinity controls B cell fate decisions is largely unknown. It is known that specific transcriptional regulators are involved in the differentiation of GC B cells to PCs. Two genes, Blimp-1 and XBP-1, play a critical role in PC differentiation by repressing genes required for GC function, including Bcl-6 and Pax5 (47). Our results show that the strength of BCR signaling influences the expression of genes that are involved in B cell maturation and differentiation (Fig. 2C). Under conditions where GCs are formed, both Tg λ and κ B cells expressed Bcl-6 and Bach-2, a newly identified molecule shown to be required for GCs (28). Expression of Pax 5 was also observed, but to a lesser degree in GC cells compared with naive B cells, and likely reflects its repression by the up-regulation of Blimp-1 and XBP-1 as cells commit to a PC fate; a subset of GC B cells that express some PC markers (CD138+Blimp-1+Bcl-6−Pax5−) has been identified (48). Of interest is the observation that AID, a gene whose expression is critical for class switching and SHM (33), may partially be controlled by BCR affinity. AID expression was dramatically elevated in GC Tg κ B cells and is consistent with their ability to produce high levels of IgG1 and IgG2a serum titers, early splenic PCs, and, ultimately, long-lived BM PCs that exhibited SHM. In contrast, with increased affinity as in the case of NP-responding Tg λ B cells, AID was nominally expressed within GCs. This pattern correlated with very low levels of IgG1 serum titers as well as early splenic PCs produced by Tg λ B cells. However, Tg λ B cells were capable of producing modest levels of IgG2a, albeit less than the amount generated from Tg κ B cells, which ultimately led to the generation of BM PCs with limited SHM. Thus, AID appears to be up-regulated in developing GC B cells when the initial affinity is low to modest with progressively less activity in GC B cells that have increasing affinity. Because AID is required for class switching (33), the capacity of GC Tg λ B cells to produce IgG2a serum titers and BM PCs may result from activated B cells that rapidly proceeded through the GC response. The expansion, contraction, and GC phenotype of Tg λ B cells occurs temporally faster than that of Tg κ B cells (Fig. 2). In addition to AID, accumulating evidence indicates that other factors such as c-Rel and the p50 subunit of NFκB can contribute to class switching (49–52). Perhaps the presence of isotype-specific switching factors varies depending on BCR affinity, which helps to shape the class-switching program. This leads to the question of whether the innate affinity of the BCR can also control SHM and the stringency of selection for a B cell. In the B1–8 Tg system, analyses of high-affinity GC B cells showed fewer VH mutations compared with low-affinity GC B cells, yet the frequency of mutation in the IgH intron was equivalent between both cell populations (11). The authors conclude that SHM is not controlled by the initial BCR affinity but is rather the result of a predetermined mutation program. Upon the completion of a fixed mutation program, GC B cells with increasing affinity would be selected. From our data we cannot exclude the possibility that selection of Tg λ B cells within the GC is less stringent due to their inherent high affinity as compared with Tg κ B cells. However, analyses of BM PCs from selected GC B cells showed fewer mutations in both the CDR and framework regions of Tg λ PCs as compared with Tg κ PCs (Table II), suggesting that affinity may partially control the rate of SHM, conceivably through the level of AID expression.
To date, the nature of the factors that control Bmem differentiation remain uncertain, if not disputed. What has been commonly agreed upon is the inextricable link between GC formation and the production of Bmem. To examine the role of GCs in Bmem or PC selection, several groups have made Tg mice overexpressing genes that inhibit apoptosis of GCs. Tg mice that express Bcl-xL and Bcl-2, genes that are down-regulated in GCs, show that blocking apoptosis promotes the survival of low-affinity Ag-experienced B cells that enter the Bmem compartment (53, 54). Our finding that low-affinity Tg κ GC B cells produce Bmem indicates that the initiation of a Bmem differentiation program requires low BCR affinity. Thus, the breadth of humoral immunity provides the host with a spectrum of Ab protection, ranging from an immediate germline transitory response to an affinity-matured, enduring Ig response.
Acknowledgment
Kathryn A. Bennett is gratefully acknowledged for expert animal husbandry.
This work was supported by National Institutes of Health Grants AI-26296 and AI-42234 (to R.J.N.), National Center for Research Resources, Centers of Biomedical Research Excellence Grants AR-052902 and P20RR16437 (to L.D.E.), and grants from the Arthritis Foundation (to L.D.E.) and the American Federation of Aging Research and Pfizer (to L.A.V.).
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Abbreviations used in this paper: GC, germinal center; AID, activation-induced cytidine deaminase; BM, bone marrow; Bmem, memory B cell; CT, cycle threshold; HEL, hen egg lysozyme; int, intermediate; KLH, keyhole limpet hemocyanin; NIP, (4-hydroxy-5-iodo-3-nitrophenyl)acetyl; NP, (4-hydroxy-3-nitrophenyl)acetyl; PC, plasma cell; PCpre, PC precursor; PNA, peanut agglutinin; QM, quasi-monoclonal; SHM, somatic hypermutation; TD, T cell dependent; Tg, transgenic; 1°, primary; 2°, secondary.
Disclosures The authors have no financial conflict of interest.
References
- 1.MacLennan IC. Germinal centers. Annu. Rev. Immunol. 1994;12:117–139. doi: 10.1146/annurev.iy.12.040194.001001. [DOI] [PubMed] [Google Scholar]
- 2.Kelsoe G. In situ studies of the germinal center reaction. Adv. Immunol. 1995;60:267–288. doi: 10.1016/s0065-2776(08)60587-8. [DOI] [PubMed] [Google Scholar]
- 3.Gray D, Siepmann K, van Essen D, Poudrier J, Wykes M, Jainandunsing S, Bergthorsdottir S, Dullforce P. B-T lymphocyte interactions in the generation and survival of memory cells. Immunol. Rev. 1996;150:45–61. doi: 10.1111/j.1600-065x.1996.tb00695.x. [DOI] [PubMed] [Google Scholar]
- 4.Dal Porto JM, Haberman AM, Shlomchik MJ, Kelsoe G. Antigen drives very low affinity B cells to become plasmacytes and enter germinal centers. J. Immunol. 1998;161:5373–5381. [PubMed] [Google Scholar]
- 5.Cumano A, Rajewsky K. Clonal recruitment and somatic mutation in the generation of immunological memory to the hapten NP. EMBO J. 1986;5:2459–2468. doi: 10.1002/j.1460-2075.1986.tb04522.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berek C, Berger A, Apel M. Maturation of the immune response in germinal centers. Cell. 1991;67:1121–1129. doi: 10.1016/0092-8674(91)90289-b. [DOI] [PubMed] [Google Scholar]
- 7.Clarke SH, Staudt LM, Kavaler J, Schwartz D, Gerhard WU, Weigert MG. V region gene usage and somatic mutation in the primary and secondary responses to influenza virus hemagglutinin. J. Immunol. 1990;144:2795–2801. [PubMed] [Google Scholar]
- 8.Jacob J, Przylepa J, Miller C, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. III. The kinetics of V region mutation and selection in germinal center B cells. J. Exp. Med. 1993;178:1293–1307. doi: 10.1084/jem.178.4.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takahashi Y, Dutta PR, Cerasoli DM, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. V. Affinity maturation develops in two stages of clonal selection. J. Exp. Med. 1998;187:885–895. doi: 10.1084/jem.187.6.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith KG, Light A, Nossal GJ, Tarlinton DM. The extent of affinity maturation differs between the memory and antibody-forming cell compartments in the primary immune response. EMBO J. 1997;16:2996–3006. doi: 10.1093/emboj/16.11.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shih TA, Meffre E, Roederer M, Nussenzweig MC. Role of BCR affinity in T cell dependent antibody responses in vivo. Nat. Immunol. 2002;3:570–575. doi: 10.1038/ni803. [DOI] [PubMed] [Google Scholar]
- 12.Kouskoff V, Famiglietti S, Lacaud G, Lang P, Rider JE, Kay BK, Cambier JC, Nemazee D. Antigens varying in affinity for the B cell receptor induce differential B lymphocyte responses. J. Exp. Med. 1998;188:1453–1464. doi: 10.1084/jem.188.8.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dal Porto JM, Haberman AM, Kelsoe G, Shlomchik MJ. Very low affinity B cells form germinal centers, become memory B cells, and participate in secondary immune responses when higher affinity competition is reduced. J. Exp. Med. 2002;195:1215–1221. doi: 10.1084/jem.20011550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Erickson LD, Vogel LA, Cascalho M, Wong J, Wabl M, Durell BG, Noelle RJ. B cell immunopoiesis: visualizing the impact of CD40 engagement on the course of T cell-independent immune responses in an Ig transgenic system. Eur. J. Immunol. 2000;30:3121–3131. doi: 10.1002/1521-4141(200011)30:11<3121::AID-IMMU3121>3.0.CO;2-M. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Erickson LD, Durell BG, Vogel LA, O'Connor BP, Cascalho M, Yasui T, Kikutani H, Noelle RJ. Short-circuiting long-lived humoral immunity by the heightened engagement of CD40. J. Clin. Invest. 2002;109:613–620. doi: 10.1172/JCI14110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cascalho M, Ma A, Lee S, Masat L, Wabl M. A quasi-monoclonal mouse. Science. 1996;272:1649–1652. doi: 10.1126/science.272.5268.1649. [DOI] [PubMed] [Google Scholar]
- 17.O'Connor BP, Cascalho M, Noelle RJ. Short-lived and long-lived bone marrow plasma cells are derived from a novel precursor population. J. Exp. Med. 2002;195:737–745. doi: 10.1084/jem.20011626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nieto A, Gaya A, Jansa M, Moreno C, Vives J. Direct measurement of antibody affinity distribution by hapten-inhibition enzyme immunoassay. Mol. Immunol. 1984;21:537–543. doi: 10.1016/0161-5890(84)90070-1. [DOI] [PubMed] [Google Scholar]
- 19.Kanayama N, Cascalho M, Ohmori H. Analysis of marginal zone B cell development in the mouse with limited B cell diversity: role of the antigen receptor signals in the recruitment of B cells to the marginal zone. J. Immunol. 2005;174:1438–1445. doi: 10.4049/jimmunol.174.3.1438. [DOI] [PubMed] [Google Scholar]
- 20.Oliver AM, Martin F, Gartland GL, Carter RH, Kearney JF. Marginal zone B cells exhibit unique activation, proliferative and immunoglobulin secretory responses. Eur. J. Immunol. 1997;27:2366–2374. doi: 10.1002/eji.1830270935. [DOI] [PubMed] [Google Scholar]
- 21.Kanayama N, Kimoto T, Todo K, Nishikawa Y, Hikida M, Magari M, Cascalho M, Ohmori H. B cell selection and affinity maturation during an antibody response in the mouse with limited B cell diversity. J. Immunol. 2002;169:6865–6874. doi: 10.4049/jimmunol.169.12.6865. [DOI] [PubMed] [Google Scholar]
- 22.Allman DM, Ferguson SE, Lentz VM, Cancro MP. Peripheral B cell maturation II. Heat-stable antigen hi splenic B cells are an immature developmental intermediate in the production of long-lived marrow-derived B cells. J. Immunol. 1993;151:4431–4444. [PubMed] [Google Scholar]
- 23.Nutt SL, Heavey B, Rolink AG, Busslinger M. Commitment to the B-lymphoid lineage depends on the transcription factor Pax5. Nature. 1999;401:556–562. doi: 10.1038/44076. [DOI] [PubMed] [Google Scholar]
- 24.Mikkola I, Heavey B, Horcher M, Busslinger M. Reversion of B cell commitment upon loss of Pax5 expression. Science. 2002;297:110–113. doi: 10.1126/science.1067518. [DOI] [PubMed] [Google Scholar]
- 25.Dent AL, Shaffer AL, Yu X, Allman D, Staudt LM. Control of inflammation, cytokine expression, and germinal center formation by BCL-6. Science. 1997;276:589–592. doi: 10.1126/science.276.5312.589. [DOI] [PubMed] [Google Scholar]
- 26.Ye BH, Cattoretti G, Shen Q, Zhang J, Hawe N, de Waard R, Leung C, Nouri-Shirazi M, Orazi A, Chaganti RS, et al. The BCL-6 proto-onco-gene controls germinal-centre formation and Th2-type inflammation. Nat. Genet. 1997;16:161–170. doi: 10.1038/ng0697-161. [DOI] [PubMed] [Google Scholar]
- 27.Fukuda T, Yoshida T, Okada S, Hatano M, Miki T, Ishibashi K, Okabe S, Koseki H, Hirosawa S, Taniguchi M, et al. Disruption of the Bcl6 gene results in an impaired germinal center formation. J. Exp. Med. 1997;186:439–448. doi: 10.1084/jem.186.3.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muto A, Tashiro S, Nakajima O, Hoshino H, Takahashi S, Sakoda E, Ikebe D, Yamamoto M, Igarashi K. The transcriptional programme of antibody class switching involves the repressor Bach2. Nature. 2004;429:566–571. doi: 10.1038/nature02596. [DOI] [PubMed] [Google Scholar]
- 29.Shaffer AL, Lin KI, Kuo TC, Yu X, Hurt EM, Rosenwald A, Giltnane JM, Yang L, Zhao H, Calame K, Staudt LM. Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity. 2002;17:51–62. doi: 10.1016/s1074-7613(02)00335-7. [DOI] [PubMed] [Google Scholar]
- 30.Shapiro-Shelef M, Lin KI, McHeyzer-Williams LJ, Liao J, McHeyzer-Williams MG, Calame K. Blimp-1 is required for the formation of immunoglobulin secreting plasma cells and preplasma memory B cells. Immunity. 2003;19:607–620. doi: 10.1016/s1074-7613(03)00267-x. [DOI] [PubMed] [Google Scholar]
- 31.Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM, Friend D, Grusby MJ, Alt F, Glimcher LH. Plasma cell differentiation requires the transcription factor XBP-1. Nature. 2001;412:300–307. doi: 10.1038/35085509. [DOI] [PubMed] [Google Scholar]
- 32.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 33.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 34.Toyama H, Okada S, Hatano M, Takahashi Y, Takeda N, Ichii H, Takemori T, Kuroda Y, Tokuhisa T. Memory B cells without somatic hypermutation are generated from Bcl6-deficient B cells. Immunity. 2002;17:329–339. doi: 10.1016/s1074-7613(02)00387-4. [DOI] [PubMed] [Google Scholar]
- 35.Billadeau D, Ahmann G, Greipp P, Van Ness B. The bone marrow of multiple myeloma patients contains B cell populations at different stages of differentiation that are clonally related to the malignant plasma cell. J. Exp. Med. 1993;178:1023–1031. doi: 10.1084/jem.178.3.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yaccoby S, Epstein J. The proliferative potential of myeloma plasma cells manifest in the SCID-hu host. Blood. 1999;94:3576–3582. [PubMed] [Google Scholar]
- 37.Pilarski LM, Belch AR. Clonotypic myeloma cells able to xeno-graft myeloma to nonobese diabetic severe combined immunodeficient mice co-purify with CD34+ hematopoietic progenitors. Clin. Cancer Res. 2002;8:3198–3204. [PubMed] [Google Scholar]
- 38.Jacob J, Kelsoe G, Rajewsky K, Weiss U. Intraclonal generation of antibody mutants in germinal centres. Nature. 1991;354:389–392. doi: 10.1038/354389a0. [DOI] [PubMed] [Google Scholar]
- 39.Jacob J, Kassir R, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. I. The architecture and dynamics of responding cell populations. J. Exp. Med. 1991;173:1165–1175. doi: 10.1084/jem.173.5.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Toellner KM, Jenkinson WE, Taylor DR, Khan M, Sze DM, Sansom DM, Vinuesa CG, MacLennan IC. Low-level hypermutation in T cell-independent germinal centers compared with high mutation rates associated with T cell-dependent germinal centers. J. Exp. Med. 2002;195:383–389. doi: 10.1084/jem.20011112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Griffiths GM, Berek C, Kaartinen M, Milstein C. Somatic mutations and maturation of the immune response to z-phenyl oxazolone. Nature. 1984;312:271–275. doi: 10.1038/312271a0. [DOI] [PubMed] [Google Scholar]
- 42.McKean D, Huppi K, Bell M, Staudt L, Gerhard W, Weigert M. Generation of antibody diversity in the immune response of BALB/c mice to influenza virus hemagglutinin. Proc. Natl. Acad. Sci. USA. 1984;81:3180–3184. doi: 10.1073/pnas.81.10.3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martin F, Oliver AM, Kearney JF. Marginal zone and B1 B cells unite in the early response against T-independent blood-borne particulate antigens. Immunity. 2001;14:617–629. doi: 10.1016/s1074-7613(01)00129-7. [DOI] [PubMed] [Google Scholar]
- 44.Tarlinton DM, Smith KG. Dissecting affinity maturation: a model explaining selection of antibody-forming cells and memory B cells in the germinal centre. Immunol. Today. 2000;21:436–441. doi: 10.1016/s0167-5699(00)01687-x. [DOI] [PubMed] [Google Scholar]
- 45.Paus D, Phan TG, Chan TD, Gardam S, Basten A, Brink R. Antigen recognition strength regulates the choice between extrafollicular plasma cell and germinal center B cell differentiation. J. Exp. Med. 2006;203:1081–1091. doi: 10.1084/jem.20060087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shih TA, Roederer M, Nussenzweig MC. Role of antigen receptor affinity in T cell-independent antibody responses in vivo. Nat. Immunol. 2002;3:399–406. doi: 10.1038/ni776. [DOI] [PubMed] [Google Scholar]
- 47.Calame KL. Plasma cells: finding new light at the end of B cell development. Nat. Immunol. 2001;2:1103–1108. doi: 10.1038/ni1201-1103. [DOI] [PubMed] [Google Scholar]
- 48.Falini B, Fizzotti M, Pucciarini A, Bigerna B, Marafioti T, Gambacorta M, Pacini R, Alunni C, Natali-Tanci L, Ugolini B, et al. A monoclonal antibody (MUM1p) detects expression of the MUM1/IRF4 protein in a subset of germinal center B cells, plasma cells, and activated T cells. Blood. 2000;95:2084–2092. [PubMed] [Google Scholar]
- 49.Shanmugam A, Shi MJ, Yauch L, Stavnezer J, Kenter AL. Evidence for class-specific factors in immunoglobulin isotype switching. J. Exp. Med. 2000;191:1365–1380. doi: 10.1084/jem.191.8.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zelazowski P, Carrasco D, Rosas FR, Moorman MA, Bravo R, Snapper CM. B cells genetically deficient in the c-Rel transactivation domain have selective defects in germline CH transcription and Ig class switching. J. Immunol. 1997;159:3133–3139. [PubMed] [Google Scholar]
- 51.Ma L, Wortis HH, Kenter AL. Two new isotype-specific switching activities detected for Ig class switching. J. Immunol. 2002;168:2835–2846. doi: 10.4049/jimmunol.168.6.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Snapper C, Zelazowski P, Rosas F, Kehry F, Tian M, Baltimore D, Sha W. B cells from p50/NFκB knockout mice have selective defects in proliferation, differentiation, germline CH transcription and Ig class switching. J. Immunol. 1996;156:183–191. [PubMed] [Google Scholar]
- 53.Takahashi Y, Cerasoli DM, Dal Porto JM, Shimoda M, Freund R, Fang W, Telander DG, Malvey EN, Mueller DL, Behrens TW, Kelsoe G. Relaxed negative selection in germinal centers and impaired affinity maturation in bcl-xL transgenic mice. J. Exp. Med. 1999;190:399–410. doi: 10.1084/jem.190.3.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith KG, Light A, O'Reilly LA, Ang SM, Strasser A, Tarlinton D. Bcl-2 transgene expression inhibits apoptosis in the germinal center and reveals differences in the selection of memory B cells and bone marrow antibody-forming cells. J. Exp. Med. 2000;191:475–484. doi: 10.1084/jem.191.3.475. [DOI] [PMC free article] [PubMed] [Google Scholar]